ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2016; 12(9):1063-1073. doi:10.7150/ijbs.16430 This issue Cite
Research Paper
Gamma-secretase Inhibitor Prevents Proliferation and Migration of Ductus Arteriosus Smooth Muscle Cells through the Notch3-HES1/2/5 Pathway
Jiunn-Ren Wu1,2,3,*, Jwu-Lai Yeh1,4,*, Shu-Fen Liou5, Zen-Kong Dai1,2,3, Bin-Nan Wu4, Jong-Hau Hsu1,2,3, ![]()
1. Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
2. Department of Pediatrics, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan
3. Department of Pediatrics, Faculty of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
4. Department and Graduate Institute of Pharmacology, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
5. Department of Pharmacy, Chia-Nan University of Pharmacy and Science, Tainan, Taiwan
* These authors contributed equally to this work.
Abstract

Patent ductus arteriosus (PDA) can cause morbidity and mortality in neonates. Vascular remodeling, characterized by proliferation and migration of smooth muscle cells (SMCs), is an essential process for postnatal DA closure. Notch signaling is an important mediator of vascular remodelling but its role in DA is unkonwn. We investigated the effects and underlying mechanisms of γ-secretase inhibitor DAPT, a Notch signaling inhibitor on angiotensin II (Ang II)-induced proliferation and migration of DASMCs. Proliferation and migration of DASMCs cultured from neonatal Wistar rats were induced by Ang II, with or without DAPT pre-treatment. In addition, potential underlying mechanisms including cell cycle progression, Ca2+ influx, reactive oxygen species (ROS) production, signal transduction of MAPK and Akt, and Notch receptor with its target gene pathway were examined. We found that DAPT inhibited Ang II-induced DASMCs proliferation and migration dose dependently. DAPT also arrested the cell cycle progression in the G0/G1-phase, and attenuated calcium overload and ROS production caused by Ang II. Moreover, DAPT inhibited nuclear translocation of Notch3 receptor intracellular domain, with decreased expression of its down-stream genes including HES1, HES2 and HES5. Finally, Ang II-activated ERK1/2, JNK and Akt were also counteracted by DAPT. In conclusion, DAPT inhibits Ang II-induced DASMCs proliferation and migration. These effects are potentially mediated by decreased calcium influx, reduced ROS production, and down-regulation of ERK1/2, JNK and Akt, through the Notch3-HES1/2/5 pathway. Therefore, Notch signaling has a role in DA remodeling and may provide a target pathway for therapeutic intervention of PDA.
Keywords: Ductus arteriosus, Notch signaling, remodeling
Introduction
The ductus arteriosus (DA), a unique blood vessel connecting the pulmonary artery and the aorta, has an essential role in the fetal circulation. During fetal life, the DA directs blood flow away from the pulmonary circulation, bypassing the non-ventilated lungs and towards the placenta. After birth, the DA closes spontaneously within 48 h [1, 2]. Postnatal persistence of a patent DA (PDA) is a major cause of morbidity and mortality in preterm neonates, leading to pulmonary congestion, ventricular dysfunction, and respiratory failure that require medical or surgical interventions [3, 4]. On the other hand, DA patency is required for infants with certain obstructive congenital heart defects to maintain systemic blood flow. Therefore, elucidating the molecular mechanisms of DA closure is not only important for vascular biology but also for clinical problems in pediatrics.
It is well established that DA closure consists of two processes, functional and anatomical closures [5, 6]. Functional closure, an acute response to increase oxygen tension and declined circulating prostaglandin E2 at birth, is accomplished by oxygen-induced DA vasoconstriction. Anatomical closure, a constitutive process of luminal occlusion, is characterized by progressive vascular structural remodeling during the perinatal period, and thus results in permanent DA closure. While current clinical approach for medical closure of PDA is mainly targeted on vasoconstriction by cyclooxygenases inhibitors such as indomethacin or ibuprofen, these agents failed to close the DA in about 30% of premature infants [3, 7]. Therefore, expanding research efforts are directed toward novel pharmacologic strategy by exploring mechanisms underlying DA remodeling.
DA remodeling is characterized by neointimal formation caused by smooth muscle cell (SMC) proliferation and migration of from vascular media into the endothelial layer. Intriguingly, this physiologic process in DA closely resembles the pathologic process of pulmonary vascular remodeling in pulmonary artery hypertension (PAH) and neointimal formation after vascular injury. The mechanisms involved in vascular remodeling are complex and not fully elucidated, but emerging evidence has demonstrated a role for Notch signaling. The Notch system is highly expressed in human vasculature and regulates cell behavior including proliferation, migration and angiogenesis [8]. The Notch system consists of 4 receptors (Notch1-4), and five ligands (Jagged 1 and 2, Delta-like 1-3) [9, 10]. Notch receptors are trans-membranous proteins with extracellular and intracellular domains (ICDs). After ligand binding, Notch receptors undergo proteolytic events through γ-secretase-dependent cleavage, resulting in release of cleaved ICDs with subsequent nuclear translocation. In the nucleus, the ICD further leads to transcriptional activations of down-stream genes, most notably HES (Hairy/Enhancer of Split) and Hey (HES-related transcriptional factor), which regulate cell fate [10, 11].
Recent evidence demonstrates a critical role of Notch signaling in the pathogenesis of vascular remodeling. For example, up-regulated Notch 3 signaling has been recently found correlated with severity of human PAH [12], and inhibition of Notch signaling by the γ-secretase inhibitor (DAPT) prevents murine PAH by prevention of PASMC proliferation, migration and intimal thickening [12-14]. In addition, in mice with carotid arterial injury, Notch1 signaling has been found crucial for aortic SMCs proliferation and neointimal formation [15]. As the biologic response to Notch receptor activation varies with cell type and organ context [16], information regarding the role of Notch signaling in DA remodeling is important but still lacking.
In this context, we examined if γ-secretase inhibitor (DAPT), an inhibitor of Notch signaling, can prevent DASMC proliferation and migration induced by angiotensin-II (Ang II), a mediator implicated in DA remodeling [17-19], and further explored the potential mechanisms underlying these effects.
Materials and Methods
Materials and Reagents
N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester (DAPT), a γ-secretase inhibitor, Ang II, α-actin and α-smooth muscle actin antibodies, and 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) were obtained from Sigma-Aldrich, Inc. (St. Louis, MO). 20,70-Dichlorodihydrofluorescein diacetate (DCFH-DA) was purchased from Molecular Probes (Eugene, OR). Antibodies of ERK1/2, p38, Akt, JNK and phosphorylated JNK were obtained from Upstate Biotechnology (Lake Placid, NY) while antibodies of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were obtained from Santa Cruz Biotech (Santa Cruz, CA). Antibodies of phosphorylated Akt, p38 and ERK1/2 were obtained from Cell Signaling Technology (Beverly, MA). Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin, streptomycin, and all other tissue culture reagents were obtained from GIBCO BRL Life Technologies (Grand Island, NY). The primers of HES1, HES2, HES5 and GAPDH for PCR amplification were purchased from Applied Biosystems Inc. (Foster City, CA).
Primary Culture of rat DASMCs
Time-pregnant rats were purchased from Lasco, Inc. (Taipei, Taiwan). This study was approved by the Animal Care and Use Committee of the Kaohsiung Medical University. Animals were cared in accordance with Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health. DASMCs in primary culture were all obtained from DAs of newborn Wistar rats. In brief, immediately (within 10 min) after spontaneous delivery of the pregnant rat on day 21 (full term) of gestation, the newborn rats were sacrificed by decapitation followed by opening of the thoracic cavity. After removal of surrounding adhesive tissues, DAs were identified and removed under dissecting microscope. The outer sphere of the DA was peeled and the endothecia were shaved lightly two to three times to remove endothelial cells. DAs were then plated onto Petri dishes (Corning, NY, USA) and cultured in DMEM supplemented with 10% FBS, 100 U/ml of penicillin and 100 μg/ml streptomycin, maintained in a humidified incubator (37°C, 5% CO2, 95% air). When the cultures reach confluence, cells were sub-cultured using 0.5% trypsin. Culture media were changed every 3 days and primary cultures of two passages were used in the experiments. The purity of DASMC was confirmed under confocal microscope using immunofluorescence staining for α-smooth muscle actin (>95% of cells stained positive), with typical “hill and valley” appearance and lack of staining for vimentin, as we previously described [20]. Serum starvation before experiments was achieved by 1% FBS for 48 h to synchronize cells into quiescence.
Cell proliferation assay
To assess DASMCs proliferation, MTT assay was first performed as we previously described [21]. In brief, after serum starvation for 48 h, proliferation of DASMCs was induced by Ang II (10 nM) for 48 h in DMEM supplemented with 1% FBS in 96-well plates at a density of 2 x 104 cells per well, with or without pre-treatment (1 h) and co-treatment (48 h) of DAPT (0.1-10 μM). After induction for 48h, MTT (0.5 mg/ ml) was added in the medium for 4 hr. The culture medium was then removed and the cells were dissolved in isopropanol and shaken for 10 min. The amount of MTT formazan was quantified at absorbance of 540 and 630 nm using an ELISA reader (DYNEX Technologies, Denkendorf, Germany).
DNA synthesis assay
To investigate effects of DAPT on Ang II-induced cell growth, we also examined DNA synthesis as assayed by the incorporation of 5-bromo-2′-deoxyuridine (BrdU) into nuclei as we previously described [22]. In brief, confluent DASMCs were trypsinized, suspended in DMEM supplemented with 10% FBS, and seeded into 96-well plates at 1 × 103 cells/ well. After serum starvation, DASMCs were incubated in DMEM with 1% FBS and Ang II (10 nM) for 48 h, with or without pre-treatment (1 h) and co-treatment (48 h) of DAPT (0.1-10 μM), followed by incubation in the same medium containing BrdU (10 μM) for 4 h. Cells were harvested for the detection of DNA synthesis by incorporation of BrdU using a cell proliferation ELISA (Roche Molecular Biochemicals, Germany).
Cell Migration Assay
Migration of DASMCs was assessed on transwell polyethylene terephthalate cell culture inserts with 8-μm pores as we previously described [21]. After serum starvation for 48 h, DASMCs (2 x 104 cells) were loaded into the upper compartment and incubated for 48 h at 37°C (5% CO2, 95% air). Meanwhile, in the lower compartment, Ang II (10 nM) was dissolved in DMEM, with or without pre-treatment (1 h) and co-treatment (48 h) of DAPT (0.1-10 μM). After 48 h, the non-migrated cells on the upper membrane surface were removed and those on the lower surface were fixed in methanol and stained with Giemsa. The numbers of cells per six high-power fields (200 x HPF) were counted and the mean numbers of cells were used to express migration activity.
Assessment of cell cycle by flow cytometry
DASMCs were synchronized at the G0-phase by serum starvation for 48 h. After replenishment with fresh DMEM supplemented with 1% FBS, and then Ang II (10 nM) was added to allow progression of the cell cycle for 48 h, with or without pre-treatment (1 h) and co-treatment (48 h) of DAPT (0.1-10 μM). Then cells were trypsinized, centrifuged at 1,250 × g for 5 min, washed twice with cold PBS, and treated with RNase A (10 μg/ml). DNA was stained with propidium iodide (50 μg/ml) for 30 min at 37℃, and 5 × 103 cells were then analyzed by flow cytometry as we previously described [20].
Measurement of intracellular free calcium concentration
Intracellular calcium concentration ([Ca2+]i) was measured as previously described [21, 22]. Trypsinized cells (1 × 106 cells/ml) were loaded with 2 μM of the ester form of fura-2 (fura-2/acetoxy methyl) for 30 min at 25℃ in DMEM. After loading, the cells were kept in a balanced salt solution (BSS, mM: 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 5 glucose, pH 7.4). Fura-2 fluorescence measurements were performed in a water-jacketed cuvette with continuous stirring. The cuvette contained 1 ml of BSS and 5 × 105 cells. The DASMCs were pretreated with or without DAPT (0.1-10 μM) for 1h before stimulation with Ang II (10 nM) for 9 min in Ca2+-containing buffer. Fluorescence was monitored with a Shimadzu RF-5301PC spectrofluorophotometer (Shimadzu, Kyoto, Japan) by recording excitation signals at 340 and 380 nm, and emission signals at 510 nm in two-second intervals.
Determination of intracellular reactive oxygen species
In order to measure the production of reactive oxygen species (ROS), an assay using 2', 7'-dichlorodihydrofluorescein diacetate (DCFH-DA, Molecular Probes) was performed as we previously described [22]. After serum starvation for 48 h, DASMCs (1×105 cells) were stimulated by Ang II (10 nM) for 48 h with or without pre-treatment (1 h) and co-treatment (48 h) of DAPT (0.1-10 μM). In order to measure ROS production, cells were stained with 10 μM of DCFH-DA for 30 min at 37℃, detached with trypsin/EDTA, washed, re-suspended in PBS, and then immediately analyzed by flow cytometry. A FACScan flow cytometer was used to measure ROS generation by the fluorescence intensity (FL-1, 525 nm) of 10,000 cells as we previously described [23].
Immunocytochemistry for nuclear translocation of Notch1 ICD and Notch3 ICD
The effects of DAPT on Ang II-induced nuclear translocation of Notch1 ICD and Notch3 ICD were assessed by the confocal microscope (Zeiss LSM 700). In brief, DASMCs were treated with Ang II (10 nM) with or without co-treatment of DAPT (10 μM) for 48 h. Fixation was performed in 10% formaldehyde for 30 min in 4°C. After overnight incubation with primary antibody by anti-Notch1 ICD (Val-1744, Cell Signaling Technology) and anti-Notch3 ICD (sc-7424, Santa Cruz Biotechnology, INC) in 4°C, slides are incubated for 2 h with the rhodamine-conjugated secondary antibody. This was followed by staining of cytoplasmic α-smooth muscle actin (SMA) with anti-SMA antibody (Sigma-Aldrich) and counterstaining of cell nuclei by Fluoromount-G with 4,6-diamidino-2-phenylindole (DAPI, Molecular Probes). The slides were imaged under constant conditions for all experimental groups, and appropriate controls are performed in all cases.
Reverse transcription-polymerase chain reaction (RT-PCR)
Expressions of down-stream target genes of Notch signaling were further investigated by RT-PCR. In brief, DASMCs were cultured in 6-well plates and co-treated with Ang II (10 nM) and DAPT (0.1-10 μM) for 48 h. Total RNA was isolated from rat DASMCs using the SV total RNA Isolation System (Promega, Madison, WI, USA) according to the manufacturer's recommendations. Reverse transcription (RT) reactions were performed on 2 μg of total RNA with the Reverse Transcription System (Promega). RT-generated cDNAs encoding HES1, HES2, HES5 and GAPDH were amplified using PCR. Sequences of the HES1 primers are 5′- AGCTGGAGAAGGCAGACATT-3′ (forward) and 5′- CGTTGATCTGGGTCATGCAG -3 (reverse). Primers for HES2 are 5′- TTCCCGCAGAACATGAGACT -3′ (forward) and 5′- CGGGAAGTCTCTGCTCCTAG-3 (reverse). Primers for HES5 are 5′- GCTCAGTCCCAAGGAGAAAA-3 (forward) and 5′- GTCGGGGT CTCCTTGACAG -3 (reverse). For GAPDH, the primer was 5′- GTGAGGTGACCGCATCTTCT -3′ (forward) and 5′- CTTGCCGTGGGTAGAGTCAT -3 (reverse).
PCR was performed in a final volume of 20 μl containing master mix, oligonucleotide primers, and RT products. Briefly, the PCR was amplified by 32 cycles for HES1, HES2, HES5 and 25 cycles for GAPDH at 95 ℃ for 30 s, annealing at 60℃ for 45 s, and extension at 72 ℃ for 60 s. During the first cycle, the 95℃ step was extended to10 min, and on the final cycle the 72℃ step was extended to 7 min. PCR products were separated on 2% agarose gels containing 0.5 mg/ml ethidium bromide and visualized by UV transillumination. GAPDH served as an internal control for sample loading and mRNA integrity.
Western blot analysis
The protein samples were extracted from the whole cells with the procedures as we previously described [22, 23]. All of the extracted protein solutions were stored at -80℃ until analysis. Equivalent amount of protein was resolved by SDS-polyacrylamide gel electrophoresis (PAGE) (10-14%) and transferred to polyvinylidene difluoride membranes. After blocking for 1 h in 5% non-fat dry milk and Tris-buffered saline, the membrane was incubated with the desired primary antibody for 2 h. The membrane was then treated with appropriate horseradish peroxidase conjugated secondary antibody (Chemicon Inc., Temecula, CA, USA), and the immuno-reactive bands were detected by chemiluminescence (ECL) reagents (PerkinElmer Life Sciences Inc. Waltham, MA, USA). Proteins labeled with specific primary and secondary antibodies were visualized with enhanced chemiluminescence. β-Actin was probed as a control to ensure equal protein loading.
Statistical analysis
Results are expressed as the mean ± SEM. The statistical significance of the differences was determined by analysis of variance (ANOVA) with Dunnett post-hoc testing. A P < 0.05 was considered statistically significant.
Results
Effects of DAPT on Ang II-Induced proliferation of DASMCs
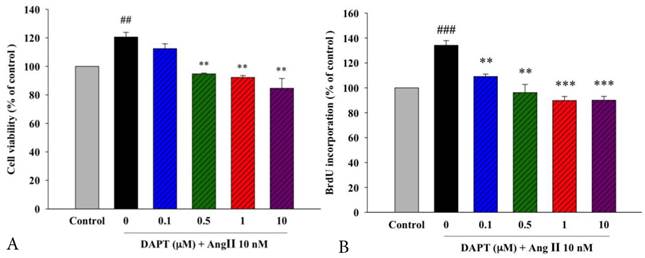
Ang II has been implicated as a mediator in the processes of DA remodeling due to its proliferative effects [17-19]. To determine whether DAPT attenuates Ang II-induced DASMC proliferation, we first examined the viability of PASMCs by the MTT assay. Our results showed that DAPT attenuated Ang II-induced proliferation of DASMCs in the concentration-dependent manner (Figure 1A).
Effects of DAPT on Ang II-Induced DNA synthesis of DASMCs
In addition to the MTT assay, another indicator of cell proliferation, quantitative colorimetric assay for DNA synthesis based on BrdU incorporation, was carried out to examine the anti-proliferative effects of DAPT. In parallel, we found that DAPT attenuated Ang II-induced DNA synthesis of DASMCs dose-dependently (Figure 1B).
Effects of DAPT on Ang II-induced DASMC migration
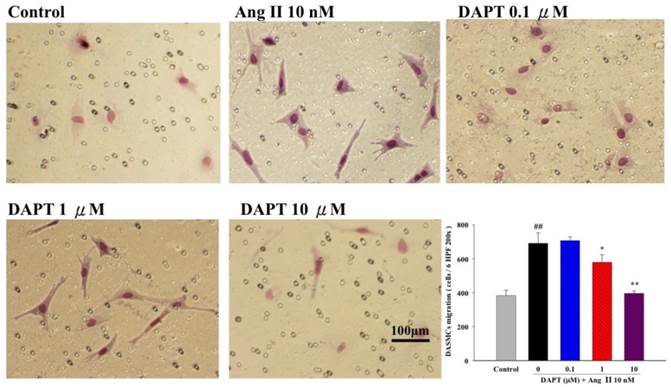
Progressive DASMC migration from vascular media into the endothelial layer is an important vascular remodeling process of the DA at birth [24]. Therefore, we further determined the effects of DAPT on DASMCs migration induced by Ang II (10 nM) were evaluated by the Boyden chamber assay. We found that Ang II potently stimulated PASMCs but co-treatment of DAPT inhibited Ang II-stimulated migration in a concentration-dependent manner (Figure 2).
Effects of DAPT on Ang II-induced proliferation and DNA synthesis of DASMCs. (A). The cell viability of DASMCs was increased after incubation with Ang II for 48 h. DAPT attenuated Ang II-induced proliferation in a concentration-dependent manner. (B). BrdU incorporation of DASMCs was increased after incubation with Ang II for 48 h. DAPT attenuated Ang II-induced BrdU incorporation in a concentration-dependent manner. Values represent mean ± SEM. N=6. Control: DASMCs were placed in medium with 1% FBS. ##P < 0.01 and ### P < 0.001 vs. control group; **P < 0.01 and ***P < 0.001 vs. cells exposed to Ang II alone.
Effects of DAPT on Ang II-induced migration of DASMCs. Ang II induced migration of DASMCs into the upper chamber from the lower chamber at 48 h. DAPT inhibited Ang II-induced migration dose-dependently. The bar graph shows migration activities assayed for the number of cells observed in six high-power fields. Values represent mean ± SEM. N=6. ##P < 0.01 vs. control group; *P < 0.05 and **P < 0.01 vs. cells exposed to Ang II alone.
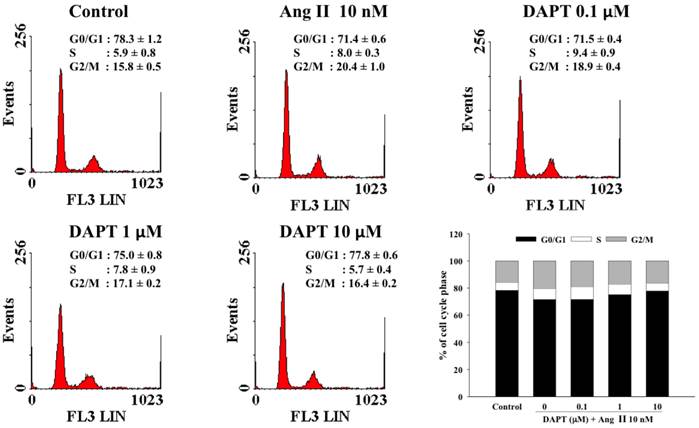
Effects of DAPT on cell cycle progression affected by Ang II. Treatment of Ang II for 48 h significantly decreased the percentage of cells in the G0/G1-phase from 78.3 ± 1.2% to 71.4 ± 0.6% (P < 0.05). DAPT counteracted this inhibitory effect and increased the percentage of cells in the G0/G1-phase dose-dependently (P < 0.05). Values represent mean ± SEM. N=6.
Effect of DAPT on Ang II-stimulated cell arrest in the G0/G1-phase
The effects of DAPT on cell cycle progression influenced by Ang II have been illustrated in Figure 3. In this study, 78.3 ± 1.2% of the cells arrested in the G0/G1-phase of the cell cycle after cells were rendered quiescent by serum starvation. After treatment with Ang II for 48 h, the percentage of cells in the G0/G1-phase significantly decreased to 71.4 ± 0.6% (P < 0.05). However, DAPT attenuated the Ang II-induced decrease in G0/G1-phase dose-dependently (P < 0.05). Accordingly, DAPT could arrest Ang II-stimulated DASMCs in the G0/G1-phase.
Effects of DAPT on Ang II-induced Ca2+ overload
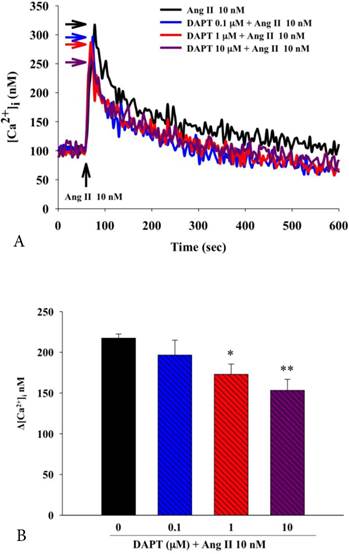
Calcium overload plays an important role in proliferation and migration of SMCs. We therefore further examined the effects of DAPT on Ang II-induced Ca2+ overload in DASMCs. We found that Ang II caused Ca2+ overload in DASMCs as shown by increased intracellular calcium concentration ([Ca2+]i) (Figure 4A and 4B), and pretreatment of DAPT attenuated Ang II-induced increase of [Ca2+]i dose-dependently.
Effects of DAPT on Ang II-induced elevation of intracellular calcium concentration ([Ca2+]i) in DASMCs. (A) In calcium-containing buffer, Ang II increased [Ca2+]i, and pretreatment with DAPT attenuated these effects in a dose-dependent fashion. (B) These effects were quantified by the bar graph. Values represent mean ± SEM. N=6. *P < 0.05 and **P < 0.01 vs. cells exposed to Ang II alone.
Effects of DAPT on Ang II-induced production of ROS
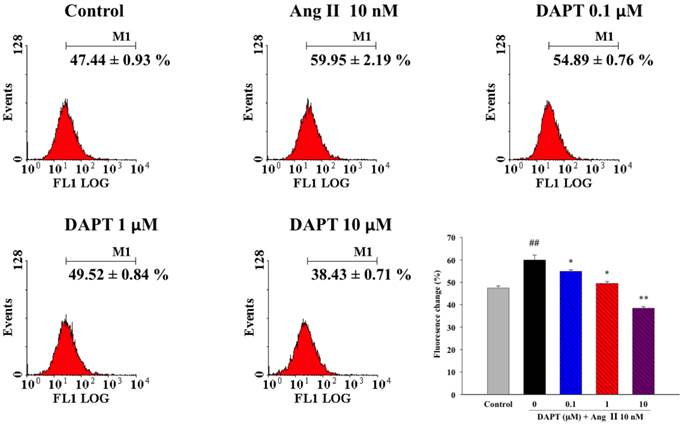
We further determine if the effects of DAPT on Ang II-induced production of reactive oxygen species (ROS), a key mediator of vascular remodeling. We found that Ang II induced significant ROS production in DASMCs as shown by oxidation of intracellular DCFH-DA to fluorescent DCFH determined by flow cytometry (Figure 5). DAPT significantly attenuated the production of ROS induced by Ang II in a concentration-dependent manner.
Effects of DAPT on Ang II-induced nuclear translocation of Notch1 ICD and Notch3 ICDs
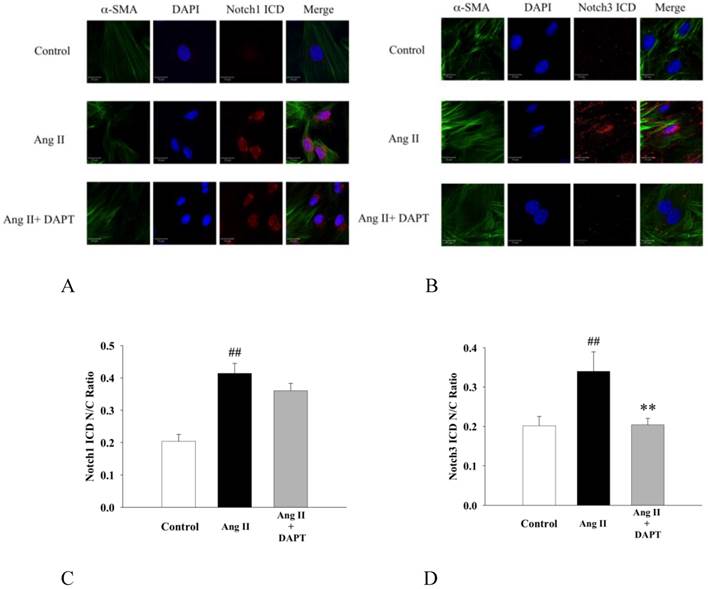
It has been previously shown that Notch1 and Notch3 promoted proliferation, migration and aggregation of SMCs [12, 15]. Since nuclear translocation of ICD after cleavage of trans-membrane receptor is an integral process of Notch signaling activation, we further investigated the inhibitory effects of DAPT on nuclear translocation of Notch ICDs by immunofluorescence. As shown in Fig 6, Ang II enhanced the nuclear translocation of Notch1 ICD and Notch3 ICD in DASMCs, while DAPT co-treatment inhibited nuclear translocation of Notch3 ICD but not Notch1 ICD.
Effects of DAPT on Ang II-induced ROS production. Ang II induced significant ROS production in DASMCs as shown by oxidation of intracellular DCFH-DA to fluorescent DCFH determined by flow cytometry. DAPT significantly attenuated the production of ROS induced by Ang II in a concentration-dependent manner. Values represent mean ± SEM. N=6. ##P < 0.01 vs. control group; *P < 0.05 and **P < 0.01 vs. cells exposed to Ang II alone.
Effects of DAPT on expressions of Notch down-stream genes
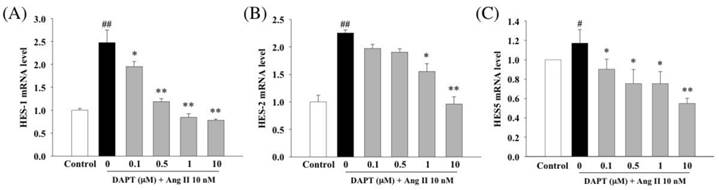
In the next set of experiments by RT-PCR, we further investigated effects of DAPT on expressions of major target genes of Notch signaling, HES1, HES2 and HES5. Our results demonstrated that DAPT attenuated Ang II-induced up-regulations of HES1, HES2 and HES5 in a dose-dependent manner (Fig 7A-C).
Effects of DAPT on Ang II-induced phosphorylations of MAPKs and Akt
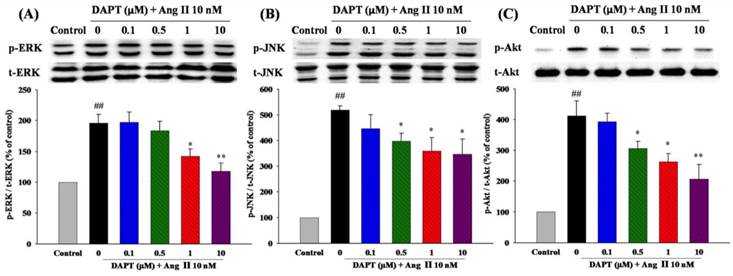
We finally investigated the effects of DAPT on signaling pathways of MAPK and Akt, which play important roles in cell proliferation and migration. As shown in Fig 8A and 8B, we found that DAPT inhibited Ang II-induced phosphorylations of ERK1/2 and JNK dose-dependently, respectively. However, there were no effects on p38 (data not shown). Furthermore, DAPT attenuated Ang II-induced phosphorylations of Akt in a dose-dependent manner (Fig. 8C).
Discussion
While many previous studies have identified signaling pathways and genetic factors that are able to govern DA constriction, many mysteries remain as to mechanisms that underlie DA remodeling. The Notch signaling pathway is a key mediator of vascular development, and its role has been recently highlighted in proliferative vascular diseases, such as atherosclerosis, pulmonary arterial hypertension or neointimal formation after vascular injury [12-15, 25]. However, its role on DA remodeling, a process mainly characterized by DASMC proliferation and migration, has not been explored.
The present study demonstrates for the first time in DASMCs the effects and mechanisms of anti-proliferative and anti-migratory effects conveyed by γ-secretase inhibitor DAPT, a Notch inhibitor. Our results indicates that these effects are mediated by the Notch3-HES1/2/5 pathway with associated decreased calcium influx, reduced ROS production, and attenuated signal transductions of ERK1/2, JNK and Akt. Therefore, this study further broadens the role of Notch system as a mediator of vascular remodeling from pulmonary and systemic vasculature into DA, a vessel connecting these two systems, and thus sheds some lights in development of novel therapeutic strategy in management of PDA.
DAPT inhibited Ang II -induced nuclear translocation of Notch3 ICD but not Notch1 ICD. (A and B) Representative photomicrographs by a confocal microscope showing that DASMCs were unstimulated (control), or stimulated with Ang II 10 nM (Ang II) alone or with DAPT 10 μM (Ang II + DAPT) for 48 h. Cells were stained for α-smooth muscle actin (α-SMA, green) and Notch1/3 ICD (red). DAPI (blue) was used as a nuclear marker. 'Merge' indicates the merged images of α-SMA, DAPI and Notch1/3 ICD. Scale bars: 20μm. (C and D) The bar graphs represent nuclear/cytoplasmic (N/C) ratios of Notch1/3 ICD fluorescent intensities derived from individual nuclei and cytoplasm for each fluorescent channel. Values represent mean ± SEM. N=6. ##P < 0.01 vs. control; **P < 0.01 vs. cells exposed to Ang II alone.
Effects of DAPT on Ang II-induced gene expressions of HES1, HES2 and HES5. Ang II was tested alone or in combination with DAPT for 48 h. RT-PCR analysis demonstrated that Ang II induced relative gene expressions of HES1 (A), HES2 (B) and HES5 (C). DAPT down-regulated expressions of all these genes dose-dependently. Values represent mean ± SEM. N=6. #P < 0.05 and ##P < 0.01 vs. control; *P < 0.05 and **P < 0.01 vs. cells exposed to Ang II alone.
Effects of DAPT on Ang II-induced ERK1/2, JNK, and Akt demonstrated by Western blotting. Ang II was tested alone or in combination with DAPT for 48 h. (A and B) DAPT down-regulated Ang II-induced ERK1/2 and JNK expression dose-dependently. (C) DAPT down-regulated Ang II-induced phosphorylation of Akt dose-dependently. Densitometric results represent mean ± SEM. N = 6. ##P < 0.01 vs. control; *P < 0.05 and **P < 0.01 vs. cells exposed to Ang II alone.
In this study we demonstrated that in DASMCs, DAPT could not only inhibit proliferation by promoting cell cycle arrest in the G0/G1-phase, but also attenuate migration induced by Ang II. Even though the mechanisms involved in DASMC proliferation and migration are not fully elucidated, a growing body of evidence suggests that calcium influx has an important role. For example, it has been demonstrated that both T- and L-type voltage-dependent calcium channels are involved in proliferation of rat DASMCs [26]. In addition, calcium influx via type-T calcium channel not only promotes oxygen-induced DA constriction but also DA remodeling as evidenced by enhanced DASMC migration [27]. Furthermore, a recent study regarding interaction between Notch signaling and calcium hemostasis revealed that Notch signaling promoted calcium influx in human pulmonary arterial SMCs (PASMCs) [28]. Extending these notions further, our novel results demonstrated that DAPT prevented Ang II-induced increase of [Ca2+]i in DASMCs dose-dependently, and thus implicate calcium regulation as an important mechanism underlying effects of Notch signaling on DA remodeling.
Even though little is known regarding the role of ROS in DA remodeling, it is well established that ROS has an important role in proliferation and migration of aortic SMCs or PASMCs. For example, our recent study has demonstrated that through reducing ROS production, a new generation calcium channel blocker can inhibit proliferation and migration of aortic vascular SMCs [18]. We also found that anti-proliferative and anti-migratory effects of B-type natriuretic peptide on PASMCs are associated with reduced ROS production by mitochondria and NADPH oxidase [17]. How Notch signaling affects the redox state has not been well understood. Intriguingly, a recent study indicates that the Notch1 signaling pathway can induce ROS production by mitochondria in macrophages [29]. Accordingly, we speculate that the anti-oxidative effects conveyed by DAPT in DASMCs partly account for its anti-proliferation and anti-migration in our study.
The function of each Notch receptor is complex and not fully understood. Increasing evidence demonstrates that Notch1 and Notch3 promote SMC proliferation, migration and aggregation, with indispensable roles in the pathogenesis of vascular remodeling. For example, in the animal model of vascular injury induced by carotid artery ligation, mice with a heterozygous deletion of Notch1 in SMCs exhibit a 70% decrease in neointimal formation [15]. Similarly, a recent study shows that Notch1-HES1 signaling is activated in patients and an animal model of abdominal aortic aneurysm with vascular remodeling [30]. Furthermore, a crucial role of the Notch3-HES5 pathway has been demonstrated in the development of PAH [12]. Although these studies suggest the requirement of Notch1 and Notch3 in vascular remodeling, to our knowledge their roles in DASMCs have not been reported. We therefore further investigated the effects of DAPT on nuclear translocation of Notch1 ICD and Notch3 ICD by immunofluorescence. We found that DAPT inhibited Ang II-induced nuclear translocation of Notch3 ICD but not Notch1 ICD. We further investigated expressions of downstream target genes in Notch signaling, including HES1, HES2 and HES5. We found that DAPT inhibited HES1/2/5 gene expression dose-dependently. Taken together, these findings suggest that Notch3-HES1/2/5 is a unique pathway regulating DA remodeling, which is not fully identical with those regulating systemic or pulmonary vasculatures.
We finally investigated MAPK family and Akt, two signaling transduction pathways associated with cell proliferation and migration. Activation of MAPK family, a down-stream mediator of ROS, is found in diseases associated with vascular remodeling, such as atherosclerosis and hypertension. [31, 32] Akt, which may be regulated by ROS and calcium in SMCs, [33] is another transduction mechanism implicated in the control of cell proliferation and migration. [34] The MAPK family includes three major groups: ERK, p38, and JNK. In general, ERK1/2 is involved in cell growth regulation, while p38 MAPK is regarded to regulate cell death. JNK has been suggested to have not only essential roles in inflammation and apoptosis, but also in cell migration and proliferation. [35-37] In line with these notions, the present study demonstrated that DAPT inhibited activations of ERK1/2, JNK and Akt induced by Ang II, which reasoned potential mechanisms underlying anti-proliferative and anti-migratory effects by Notch inhibition in DASMCs.
In conclusion, this present study indicates that Notch inhibition by DAPT inhibits Ang II-induced DASMCs proliferation and migration. These effects are potentially mediated by attenuated calcium overload, reduced ROS production, and deactivations of ERK1/2, JNK and Akt signal transduction, through the Notch3-HES1/2/5 pathway (Figure S1). Therefore, Notch signaling has a role in DA remodeling and may provide a target pathway for therapeutic intervention of PDA. Further animal and clinical studies are needed to substantiate these important findings.
Supplementary Material
Supplementary figure S1.
Acknowledgements
This research was supported by the grant from the National Science Council of Taiwan (NSC-101-2314-B-037-012-MY2), the Cologne-Kaohsiung Alliance (CKA) Research Fund, Kaohsiung Medical University, and grants from Kaohsiung Medical University Hospital, Taiwan (KMUH101-1R32, KMUH102-2R27).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Smith GC. The pharmacology of the ductus arteriosus. Pharmacol Rev. 1998;50:35-58
2. Hermes-DeSantis ER, Clyman RI. Patent ductus arteriosus: pathophysiology and management. J Perinatol. 2006;26(Suppl 1):S14-S18
3. Van Overmeire B, Smets K, Lecoutere D, Van de Broek H, Weyler J, Degroote K. et al. A comparison of ibuprofen and indomethacin for closure of patent ductus arteriosus. N Engl J Med. 2000;343:674-81
4. Little DC, Pratt TC, Blalock SE, Krauss DR, Cooney DR, Custer MD. Patent ductus arteriosus in micropreemies and full-term infants: the relative merits of surgical ligation versus indomethacin treatment. J Pediatr Surg. 2003;38:492-6
5. Coceani F, Baragatti B. Mechanisms for ductus arteriosus closure. Semin Perinatol. 2012;36:92-7
6. Bokenkamp R, DeRuiter MC, van Munsteren C, Gittenberger-de Groot AC. Insights into the pathogenesis and genetic background of patency of the ductus arteriosus. Neonatology. 2010;98:6-17
7. Hsu JH, Yang SN, Chen HL, Tseng HI, Dai ZK, Wu JR. B-type natriuretic peptide predicts responses to indomethacin in premature neonates with patent ductus arteriosus. J Pediatr. 2010;157:79-84
8. Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216-33
9. Yamamizu K, Matsunaga T, Uosaki H, Fukushima H, Katayama S, Hiraoka-Kanie M. et al. Convergence of Notch and beta-catenin signaling induces arterial fate in vascular progenitors. J Cell Biol. 2010;189:325-38
10. Miele L. Notch signaling. Cancer Res. 2006;12:1074-9
11. Ehebauer M, Hayward P, Arias AM. Notch, a universal arbiter of cell fate decisions. Science. 2006;314:1414-5
12. Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P. et al. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat Med. 2009;15:1289-97
13. Yu YR, Mao L, Piantadosi CA, Gunn MD. CCR2 deficiency, dysregulation of Notch signaling, and spontaneous pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2013;48:647-54
14. Qiao L, Xie L, Shi K, Zhou T, Hua Y, Liu H. Notch signaling change in pulmonary vascular remodeling in rats with pulmonary hypertension and its implication for therapeutic intervention. PLoS one. 2012;7:e51514
15. Li Y, Takeshita K, Liu PY, Satoh M, Oyama N, Mukai Y. et al. Smooth muscle Notch1 mediates neointimal formation after vascular injury. Circulation. 2009;119:2686-92
16. Morrow D, Guha S, Sweeney C, Birney Y, Walshe T, O'Brien C. et al. Notch and vascular smooth muscle cell phenotype. Circ Res. 2008;103:1370-82
17. Takizawa T, Kawahata M, Ikeda Y, Yamamoto M, Arishima K, Muto M. et al. Smooth muscle cell proliferation in the ductus arteriosus and the descending aorta, and effects of enalapril on SMC proliferation in perinatal rats. J Vet Med Sci. 1999;61:1215-8
18. Costa M, Barogi S, Socci ND, Angeloni D, Maffei M, Baragatti B. et al. Gene expression in ductus arteriosus and aorta: comparison of birth and oxygen effects. Physiol Genomics. 2006;25:250-62
19. Campos AH, Zhao Y, Pollman MJ, Gibbons GH. DNA microarray profiling to identify angiotensin-responsive genes in vascular smooth muscle cells: potential mediators of vascular disease. Circ Res. 2003;92:111-8
20. Hsu JH, Liou SF, Yang SN, Wu BN, Dai ZK, Chen IJ. et al. B-type natriuretic peptide inhibits angiotensin II-induced proliferation and migration of pulmonary arterial smooth muscle cells. Pediatr Pulmonol. 2014;49:734-44
21. Hsu JH, Wu JR, Liou SF, Chen HM, Dai ZK, Chen IJ. et al. Labedipinedilol-A prevents lysophosphatidylcholine-induced vascular smooth muscle cell death through reducing reactive oxygen species production and anti-apoptosis. Atherosclerosis. 2011;217:379-86
22. Liou SF, Yeh JL, Liang JC, Chiu CC, Lin YT, Chen IJ. Inhibition of mitogen-mediated proliferation of rat vascular smooth muscle cells by labedipinedilol-A through PKC and ERK 1/2 pathway. J Cardiovasc Pharmaco. 2004;44:539-51
23. Yeh JL, Liou SF, Chang YP, Lin SW, Liu TS, Wu BN. et al. Isoeugenodilol inhibits smooth muscle cell proliferation and neointimal thickening after balloon injury via inactivation of ERK1/2 pathway. J Biomed Sci. 2008;15:375-89
24. Rabinovitch M. Cell-extracellular matrix interactions in the ductus arteriosus and perinatal pulmonary circulation. Semin Perinatol. 1996;20:531-41
25. Fukuda D, Aikawa E, Swirski FK, Novobrantseva TI, Kotelianski V, Gorgun CZ. et al. Notch ligand delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc Natl Acad Sci U S A. 2012;109:E1868-77
26. Yokoyama U, Minamisawa S, Adachi-Akahane S, Akaike T, Naguro I, Funakoshi K. et al. Multiple transcripts of Ca2+ channel alpha1-subunits and a novel spliced variant of the alpha1C-subunit in rat ductus arteriosus. Am J Physiol Heart Circ Physiol. 2006;290:H1660-70
27. Akaike T, Jin MH, Yokoyama U, Izumi-Nakaseko H, Jiao Q, Iwasaki S. et al. T-type Ca2+ channels promote oxygenation-induced closure of the rat ductus arteriosus not only by vasoconstriction but also by neointima formation. J Biol Chem. 2009;284:24025-34
28. Yamamura H, Yamamura A, Ko EA, Pohl NM, Smith KA, Zeifman A. et al. Activation of Notch signaling by short-term treatment with Jagged-1 enhances store-operated Ca(2+) entry in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol. 2014;306:C871-8
29. Xu J, Chi F, Guo T, Punj V, Lee WN, French SW. et al. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J Clin Invest. 2015;125:1579-90
30. Zheng YH, Li FD, Tian C, Ren HL, Du J, Li HH. Notch gamma-secretase inhibitor dibenzazepine attenuates angiotensin II-induced abdominal aortic aneurysm in ApoE knockout mice by multiple mechanisms. PLoS one. 2013;8:e83310
31. Wu JR, Liou SF, Lin SW, Chai CY, Dai ZK, Liang JC. et al. Lercanidipine inhibits vascular smooth muscle cell proliferation and neointimal formation via reducing intracellular reactive oxygen species and inactivating Ras-ERK1/2 signaling. Pharmacol Res. 2009;59:48-56
32. Griendling KK, Sorescu D, Lassegue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175-83
33. Kaimoto T, Yasuda O, Ohishi M, Mogi M, Takemura Y, Suhara T. et al. Nifedipine inhibits vascular smooth muscle cell dedifferentiation via downregulation of Akt signaling. Hypertension. 2010;56:247-52
34. Oudit GY, Sun H, Kerfant BG, Crackower MA, Penninger JM, Backx PH. The role of phosphoinositide-3 kinase and PTEN in cardiovascular physiology and disease. J Mol Cell Cardiol. 2004;37:449-71
35. Gururajan M, Chui R, Karuppannan AK, Ke J, Jennings CD, Bondada S. c-Jun N-terminal kinase (JNK) is required for survival and proliferation of B-lymphoma cells. Blood. 2005;106:1382-91
36. Kang YJ, Jeon ES, Song HY, Woo JS, Jung JS, Kim YK. et al. Role of c-Jun N-terminal kinase in the PDGF-induced proliferation and migration of human adipose tissue-derived mesenchymal stem cells. J Cell Biochem. 2005;95:1135-45
37. Wang W, Prince CZ, Mou Y, Pollman MJ. Notch3 signaling in vascular smooth muscle cells induces c-FLIP expression via ERK/MAPK activation. Resistance to Fas ligand-induced apoptosis. J Cell Biochem. 2002;277:21723-9
Author contact
![]() Corresponding author: Jong-Hau Hsu, MD, PhD, Department of Pediatrics, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, 80708, Taiwan. Tel: 886-7-3121101-6510; Email: jhh936com.tw.
Corresponding author: Jong-Hau Hsu, MD, PhD, Department of Pediatrics, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung, 80708, Taiwan. Tel: 886-7-3121101-6510; Email: jhh936com.tw.
Received 2016-6-9
Accepted 2016-6-28
Published 2016-7-18