Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(9):1114-1120. doi:10.7150/ijbs.13774 This issue Cite
Research Paper
Novel FOXC2 Mutation in Hereditary Distichiasis Impairs DNA-Binding Activity and Transcriptional Activation
Leilei Zhang1*, Jie He1*, Bing Han2*, Linna Lu1, Jiayan Fan1, He Zhang1, Shengfang Ge1, Yixiong Zhou1 ![]() , Renbing Jia1
, Renbing Jia1 ![]() , Xianqun Fan1
, Xianqun Fan1 ![]()
1. Department of Ophthalmology, Ninth People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China
2. Department of endocrinology, Ninth People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China.
* These authors contributed equally.
Received 2015-9-6; Accepted 2016-7-6; Published 2016-8-6
Abstract

Distichiasis presents as double rows of eyelashes arising from aberrant differentiation of the meibomian glands of the eyelids, and it may be sporadic or hereditary. FOXC2 gene mutations in hereditary distichiasis are rarely reported. Here, we examined two generations of a Chinese family with hereditary distichiasis but without lymphedema or other features of LD syndrome. The FOXC2 gene was amplified and sequenced in all family members. Subcellular localization and luciferase assays were performed to assess the activity of the mutant FOXC2 protein. Clinical examinations showed distichiasis, lower eyelid ectropion, congenital ptosis and photophobia in all affected individuals. Sequence analysis revealed a novel frameshift mutation, c.964_965insG, in the coding region of the FOXC2 gene. This mutation caused protein truncation due to the presence of a premature stop codon. A fluorescence assay showed that this mutation did not change the nuclear localization of the protein. However, it impaired DNA-binding activity and decreased transcriptional activation. This is the first report of a FOXC2 mutation in hereditary distichiasis in the Chinese population. The findings of our study expand the FOXC2 mutation spectrum and contribute to the understanding of the genotype-phenotype correlation of this disease.
Keywords: congenital distichiasis, FOXC2, mutation.
Introduction
Distichiasis manifests as double rows of eyelashes arising from aberrant differentiation of the meibomian glands of the eyelids. It is a row of excess eyelashes also called a secondary eyelash. These superfluous eyelashes often lean back then abrade the cornea, resulting in corneal stimulation even corneal epithelial defects and opacification. Distichiasis can be hereditary or sporadic. The hereditary distichiasis is a developmental abnormality of the eyelashes. Also, it can be independent or associated with lymphedema-distichiasis syndrome (LD, OMIM 153400).
FOXC2 is a member of the human Forkhead-box gene (FOX) family through encoding a regulatory transcription factor. It is a major contributor in embryogenesis, particularly in lymphatic and blood vascular development [1,2]. FOXC2 located in the long arm of chromosome 16 and contained only one single exon [3]. LD syndrome is a dominantly autosomal genetic disorder caused by mutations in FOXC2 with onset of distichiasis at birth and lower extremity lymphedema at or just after puberty. With the deepening of research, other complications associated with LD syndrome were identified including ptosis, congenital heart cardiac defects, cleft palate, spinal extradural cysts, uterine and renal anomalies and so on [1,4]. In 1999, Mangion et al. first reported LD syndrome in two unrelated families and mapped a gene in the long arm of chromosome 16 [3]. Since then, additional FOXC2 mutations have been found in families with LD syndrome [5]. Subsequently, more FOXC2 mutations were founded in LD.
Hereditary distichiasis (OMIM 126300) is an autosomal dominant inherited disease with high penetrance but variable expressivity [6]. Clinical manifestations of this disease mainly occur in the eyes. To date, rare cases of hereditary distichiasis have been reported worldwide, but the mechanisms underlying this disease have remained unknown [7]. A FOXC2 gene mutation in hereditary distichiasis has been reported in only two US families [6,7]. Furthermore, the molecular mechanism of FOXC2 in hereditary distichiasis has yet to be clarified.
In this study, we identified a novel FOXC2 mutation (c.964_965insG) in an isolated Chinese family with hereditary distichiasis, which manifested as distichiasis, lower eyelid ectropion, congenital ptosis and photophobia but without lymphedema or other symptoms. This mutation was detected in all affected family members. Further analysis demonstrated that this mutation caused aberrant FOXC2 function. To our knowledge, this is the first description of a FOXC2 mutation and hereditary distichiasis in the Chinese population.
Materials and methods
Subjects
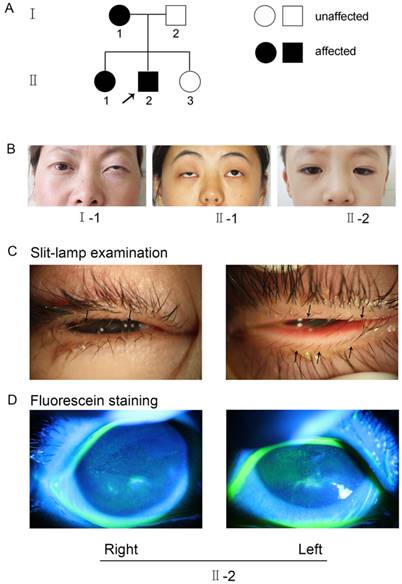
The research followed the tenets of the Declaration of Helsinki. The Institutional Ethics Committee of Shanghai Ninth Hospital, Ethics Committee of China, approved the study. Informed consent was obtained from each family member prior to participation in the study. The methods were carried out in accordance with the approved guidelines. A seven-year-old boy (II-2, Figure 1) sought treatment at our clinic for symptomatic distichiasis because of aberrant eyelashes, which were rubbing his cornea and causing recurrent corneal irritation (Figure 1C). Other distichiasis complications (e.g., ptosis and photophobia) were also found in affected individuals in his family (Figure 1B). A corneal injury with superficial ulceration was discovered by fluorescence staining (Figure 1D). The mother (I-1, Figure 1B) and older sister (II-1, Figure 1B) of the proband also suffered from distichiasis, with an age of onset of between 6 and 8 years. The father (I-2, Figure 1A) and the younger sister (II-3, Figure 1A) were normal. None of the patients had any other clinical manifestations, except for their eye symptoms.
(A) Pedigree of the family with distichiasis. Squares and circles represent males and females, respectively. The shaded symbols indicate the presence of distichiasis. Unaffected individuals are depicted by unshaded symbols. (B) Images of the three affected pedigree members. Both I-1 and II-1 had blepharoptosis, and II-2 had epicanthus. (C) Details of eyes with distichiasis. Note the double row of eyelashes originating from the inner margin of the eyelids, as indicated by the black arrows (II-2). (D) Images of the proband after fluorescein staining, as visualized using a slit lamp. The green fluorescence indicates the corneal injuries in the right and left eyes (II-2).
Mutation analysis
Mutation analysis of the FOXC2 (GenBank NG_012025.1) gene was performed. Genomic DNA was extracted from peripheral blood leukocytes of the patient and his family members. The FOXC2 exon was amplified as previously described [8]. Then, the PCR products were purified and sequenced. Once a mutation was identified, PCR fragments amplified from 100 normal subjects were also analyzed to exclude polymorphism. Informed consent for examination and DNA analysis was obtained from all subjects in accordance with the Shanghai Jiaotong University School of Medicine.
Plasmid construction, cell culture and transfection
Before the experiment, we attempted to clone cDNA from the proband and insert it into an expression vector, which failed. It had been reported that FOXC2 is highly expressed in heart, adipose, kidney and skeletal muscle tissues. However, there is little expression in the blood. Except for blood, we could not obtain sufficient tissue (or RNA from that tissue) from the seven-year-old male patient after surgery. Therefore, we created this mutation. Complementary DNA (cDNA) encoding the FOXC2 ORF was cloned and inserted into pLenti-CMV-EGFP-3FLAG-PGK-Puro, leading to the production of an EGFP-FOXC2-wild type plasmid. Then, site-directed mutagenesis was conducted to generate an EGFP-FOXC2-mutant plasmid. All constructs were sequenced to exclude additional mutations. This mutation simulated the mutation in the proband, which was then transcribed into cDNA and translated into protein, similarly to the process in patients. 293T cells were cultured and plated as described previously [8]. After cells were grown to 70-80% confluence, each well was transfected with EGFP-FOXC2-wild type and EGFP-FOXC2-mutant constructs separately with Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. At 48 hours after transfection, cells were harvested and prepared for the following experiments.
Detection of fluorescence in cells
293T cells were seeded on glass coverslips (VWR, West Chester, PA, USA) in 12-well plates and cultured overnight. After the cells were grown to 70-80% confluence, each well was transfected with EGFP-FOXC2-wild type and EGFP-FOXC2-mutant (500 ng) constructs separately with Lipofectamine 2000 reagent, which spontaneously emits green fluorescence. At 48 hours after transfection, the cells were fixed in 4% (w/v) paraformaldehyde (Sigma-Aldrich, St. Louis, MO, USA) for 15 min at room temperature. Subsequently, the plates were washed three times with PBS and incubated in PBS containing 10% (v/v) normal goat serum (Invitrogen, Carlsbad, CA, USA), 0.3% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA) and 0.1% NaN3 (Sigma-Aldrich, St. Louis, MO, USA) for 1 h. After an additional three washes with PBS, nuclei were counterstained with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA, USA) for 2 min at room temperature. Cells were imaged using a fluorescence microscope (Olympus BX51, Japan). For the plasmid that contained an EGFP (enhanced green fluorescent protein) tag, we observed green fluorescence at 488 nm, which indicated the presence of FOXC2 protein, while the nuclei could be seen as blue fluorescence.
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays (EMSAs) were performed as described previously [9]. The EMSA probe (5'-GTAAATAAA-3') was synthesized with two single-stranded oligonucleotide probes. After the probe was biotin labeled, it was mixed and annealed, forming a double-stranded oligonucleotide probe. The probe was labeled according to the instructions of the Biotin 3 End DNA Labeling Kit (89818, Thermo, Pierce, USA). EMSA assay was incubated with an in vitro-derived FOXC2 consensus oligonucleotide probe and an mononal anti-FOXC2 antibody (H00002303-M04, Abnova, Massachusetts, USA) [10]. The non-specific antibody isogenic IgG (Ab109489, California, USA) was used as the control. The top band marked the super-shift band, which contained the anti-FOXC2 antibody, the nuclear protein and the probe, which indicated the specificity of the EMSA. The shift band only contained the nuclear probe, which allowed it to move faster than the super-shift band. The negative control group was the lane that contained only the labeled probes. The positive control group was the lane containing both the samples and the labeled probes. Competitor 1, competitor 2 and competitor 3 represent increased concentrations (1 μM, 30 μM and 90 μM, respectively) of the unlabeled specific probe that were added, which were used to compete with the sample and binding of FOXC2 with DNA. The control lane indicated nuclear protein that was extracted from 293T samples, which were transfected blank plasmid. The wild type and the mutant group lanes indicated nuclear protein that was extracted from 293T samples, which were transfected with EGFP-FOXC2-wild type and EGFP-FOXC2-mutant expression vectors, respectively.
Dual luciferase assay
293T cells were cultured at a concentration of 3000 cells per well in 96-well plates. After 24 h, the cells were transfected with expression vector (0.01 μg/well) (wild type or mutant FOXC2 construct) and 0.5 μg of reporter plus the pcDNA3.1 expression vector. The PRL-TK vector constitutively expresses Renilla luciferase and thus served as an indicator for estimations of transfection efficiency. Luciferase assays were conducted according to the manufacturer's instructions using a Dual Luciferase Reporter System (Promega Benelux, Leiden, Netherlands) to measure luciferase activity, measured with a Lumimark luminometer (Bio-Rad Laboratories, Hercules, CA, USA). All transfections were performed in triplicate, and each experiment was performed three times.
Results
Genetic and sequence analyses
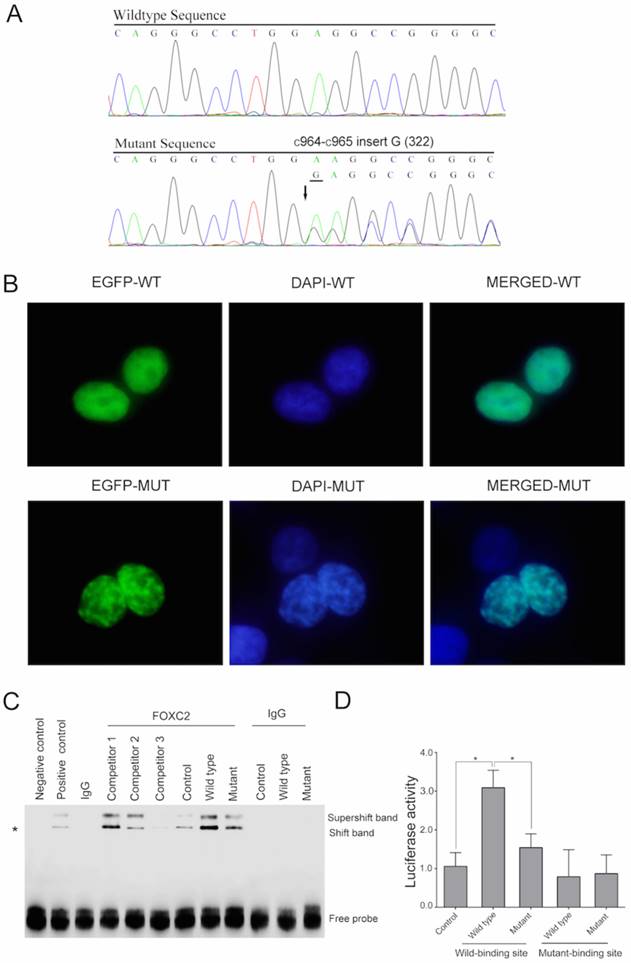
A novel heterozygous mutation (c.964_965insG) in the FOXC2 gene was detected in all affected individuals of this pedigree (Figure 2A).
(A) The normal and mutated sequences are shown separately and were detected by Sanger sequencing. (B) A fluorescence assay demonstrated that the mutant plasmid (the lower plate), but not the wild type plasmid (the upper plate), resulted in nuclear aggregation after transfection with EGFP tags. (C) The DNA-FOXC2 binding activity was detected by EMSA. DNA-FOXC2 protein complexes are indicated by asterisk, which mark the shift band. The non-specific antibody isogenic IgG was used as the control. It could not form the DNA protein complexes. The super-shift band contained the DNA probe, the nuclear protein and the anti-FOXC2 antibody, which represented the specificity of the EMSA. The negative control was the lane that only contained the labeled probes. The positive control was the lane that contained the samples and the labeled probes. Competitor 1, competitor 2 and competitor 3 (1 μM, 30 μM and 90 μM, respectively) represent increased concentrations of unlabeled specific probe, which were used to compete with the sample and with FOXC2-DNA binding. The bottom bands indicate free probe. (D) Transactivation assays were performed using 293T cells transfected with a luciferase reporter along with wild type and mutant FOXC2 constructs. Luciferase values were normalized to Renilla luciferase. Mean luciferase values from a representative experiment with transfection in triplicate are presented. The error bars correspond to the standard error of the mean (P<0.05).
This mutation was not present in 100 normal subjects or in the human SNP database (http://www.ncbi.nlm.nih.gov/SNP). The mutation led to a frame-shift in the gene following amino acid 322, which produced a truncated protein with 461 amino acids due to the introduction of a premature stop codon, whereas the full-length wild type FOXC2 encoded a 501 amino acid protein. (http://www.ncbi.nlm.nih.gov/protein/NP_005242.1). Conservative analysis indicates that this glutamic acid amino acid and the following amino acids are evolutionarily conserved in Homo sapiens, Equus caballus, Mus musculus and Rattus norvegicus (Figure 3).
Conservation analysis of FOXC2 indicated that the FOXC2 amino acid sequence is partly evolutionarily conserved in Homo sapiens, Equus caballus, Mus musculus and Rattus norvegicus. The arrow indicates the 322nd amino acid.

Molecular modeling
To further explore the molecular structures of the wild type and mutant proteins, we constructed three-dimensional computer models with the I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/) [11], which is an online platform for protein structure and function predictions. Wild type (residues 137-501) and mutant (residues 137-461) proteins were modeled based on multiple-threading alignments based on LOMETS and iterative template fragment assembly simulations. The local solid surface and electrostatic potential were completely changed in the mutant compared with the wild type FOXC2 (Supplemental Figure 1).
Subcellular localization
To investigate the effect of this mutation on the subcellular location of the FOXC2 protein, we constructed fusion plasmids with EGFP and performed localization studies using 293T cells. Wild type FOXC2 was uniformly distributed in the nucleus, which is consistent with its function as a transcription factor (Figure 2B-upper). In contrast, cells transfected with the mutant plasmid displayed uneven distribution (Figure 2B-lower).
DNA binding and transcriptional activity
To assess the impact of this mutation on the DNA binding capacity of the protein, EMSAs were performed. DNA-FOXC2 protein complexes are indicated by asterisk in Figure 2C. When equal amounts of protein were added (Supplemental Figure 2), enhanced binding to the DNA probe was observed for wild type FOXC2 and was decreased with the mutant (Figure 2C). However, when the probe was added without sample, no bands were detected (negative control). When control extract was added with additional unlabeled specific probe to compete with the sample, and when increased concentrations of this unlabeled specific probe were used, there was no specific binding when greater than 90 μM of the competitor probe was added (competitor 3). Additionally, the transactivation capacities of the wild type and mutant FOXC2 genes to activate a luciferase reporter containing six tandem copies of the FOXC2 consensus sequence (TCACAAAGTAAATAAACAACAGAGATCCAAGTAAATAAACAACAGA) were examined in 293T cells co-transfected with a reporter construct. Compared with a control plasmid, the wild type FOXC2 construct displayed significantly increased transcriptional activity, whereas the mutant showed reduced activity. However, there was no significant difference between the wild type and mutant constructs when the binding site was mutated (Figure 2D). These results indicated that this mutation impaired the transcriptional activity.
Discussion
FOXC2 is an evolutionarily conserved and functionally diverse. It is an important regulator in energy homeostasis, development and oncogenesis. FOXC2 belongs to the human Forkhead-box gene (FOX) family which consists of at least 43 members [12]. To date, except FOXC2, seven human FOX genes, including FOXC1, FOXE1, FOXE3, FOXL2, FOXN1, FOXP2 and FOXP3 have been identified as disease-causing mutations. They are always mutated in human congenital disorders [10,12,13]. FOXC1, FOXC2, FOXD1 and FOXE3 are involved in the development of the eye. Mutations in these genes can lead to the abnormal development of glaucoma, lens, retina and iris. Mutations in the FOXC1 resulted in Axenfeld-Rieger syndrome (ARS), a developmental disorder affecting structures of the anterior segment of the eye [14]. There were approximately 75% of ARS patients, who with FOXC1 mutations developed early-onset glaucoma [15,16]. Mutations in the FOXE3 have been reported associated with anterior segment ocular dysgenesis and cataracts [17,18]. Mutations in the FOXL2 contributed to Blepharophimosis-ptosis-epicanthus inversus syndrome and premature ovarian failure [19-21]. Finally, expression profiles, genetic alterations and epigenetic changes of FOX family genes as well as binding proteins of FOX family transcription factors should be comprehensively investigated. This plays an essential role in developing novel treatments and preventative strategies for human diseases.
FOXC2, formerly known as MFH-1, encodes a 1.7-kb transcript with a single exon-coding region and encodes a forkhead transcription factor. The protein encoded by FOX is characterized by a DNA-binding domain known as the forkhead domain (FHD), which is an evolutionarily conserved sequence of 110 amino acids in length, spanning approximately from amino acid 69 to 178, and it is composed of a variant of the helix-turn-helix motif known as the winged-helix motif [22]. Other reports showed the C-terminal domain was also the transactivation domain [23]. To date, approximately 70 FOXC2 mutations have been listed in the HGMD Database, with more than 57 mutations for LD syndrome and 8 for primary lymphedema (www.hgmd.cf.ac.uk). The most common types of FOXC2 gene mutations include missense and nonsense mutations, small deletions and insertions (www.hgmd.cf.ac.uk) [24]. Various types of mutations have been found throughout the coding sequence, with approximately 30% in the FHD and approximately 60% in the C-terminal domain, from amino acids 180 to 501. Nevertheless, FOXC2 mutations in hereditary distichiasis are quite rare, and only two have been previously reported [6,7].
In this study, we described a novel FOXC2 gene mutation (c.964_965insG) in a Chinese family with hereditary distichiasis. c.964_965insG is a frameshift mutation, with an insertion of one nucleotide (G) between c.964 and c.965, leading to protein truncation and resulting in a final protein of 461 amino acids in length due to the presence of a premature stop codon.
Distichiasis typically presents sporadically or in association with LD syndrome [10,25], and hereditary distichiasis is rarely observed [26]. Occasionally, this disease is asymptomatic because the lashes curl away from the cornea or due to corneal hypoesthesia. However, hereditary distichiasis is likely to lead to serious complications. Reported ophthalmologic complications of distichiasis (75% of cases) include photophobia, corneal irritation, recurrent conjunctivitis and styes, corneal ulceration and partial ectropion of the lateral third portion of the eyelashes [27]. In the pedigree in this report, congenital ptosis, photophobia and superficial ulceration were found to occur in addition to distichiasis, which was caused by abrasion of the second inner row of eyelashes. The mother and elder sister of the proband had mild or no corneal symptoms because their accessory eyelashes had been removed at an earlier age, although residual eyelashes were still present. LD syndrome with lymphedema usually occurs during puberty and typically involves the limbs. The age of onset in males is earlier than that in females, with disease development by 11 years of age, whereas it develops in females by their early 20s. The penetrance in this series appeared to be completed in the 40s [27]. However, lymphedema was not found in the 45-year-old mother. Neither the proband (7 years) nor the elder sister (16 years) had lymphedema or associated symptoms. No other abnormalities were observed in this pedigree, except for distichiasis and associated eye discomfort. Thus, hereditary distichiasis diagnoses were established.
Three-dimensional computer models of the wild type and mutant proteins were constructed. The C-terminus of wild type FOXC2 carried a negative charge, which was opposite of the charge of the nearby region. However, the mutation resulted in a positively charged C-terminus, the same as the adjacent region. A fluorescence assay demonstrated that the mutant FOXC2 was localized to the nucleus, indicating that it did not affect nuclear localization signal (NLS) functioning. However, granular nuclear aggregation was observed in cells transfected with mutant FOXC2 but not in wild type cells. EMSA and a dual luciferase assay demonstrated that this mutation greatly impaired the DNA-binding and transcriptional regulatory activities of the protein. Truncation of the protein caused impairment of the structure of the C-terminus, which is located outside of the FHD. To date, the function of the FOXC2 C-terminus is unclear. The data obtained from our study suggest that this region may participate in stabilization of the FOXC2 molecule.
In conclusion, we have identified a novel FOXC2 mutation in a Chinese hereditary distichiasis family. In vitro evidence showed that this mutation caused a loss of function of FOXC2. Our study expands the FOXC2 mutation spectrum as well as the understanding of the genotype-phenotype correlation of this disease.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank the participating patients and their families. The authors alone are responsible for the content and writing of the paper. This work was supported by the Scientific Research Program of National Health and Family Planning Commission of China (201402014), the National Natural Science Foundation of China grant (81372469, 81570884, 81402258, 81372909) and the Science and Technology Commission of Shanghai (14JC1404100, 14JC1404200, 14430723100).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Vreeburg M, Heitink MV, Damstra RJ. et al. Lymphedema-distichiasis syndrome: a distinct type of primary lymphedema caused by mutations in the FOXC2 gene. Int J Dermatol. 2008;47(Suppl 1):52-55
2. Mellor RH, Brice G, Stanton AW. et al. Mutations in FOXC2 are strongly associated with primary valve failure in veins of the lower limb. Circulation. 2007;115:1912-1920
3. Mangion J, Rahman N, Mansour S. et al. A gene for lymphedema-distichiasis maps to 16q24.3. Am J Hum Genet. 1999;65:427-432
4. Yabuki S, Kikuchi S, Ikegawa S. Spinal extradural arachnoid cysts associated with distichiasis and lymphedema. Am J Med Genet A. 2007;143A:884-887
5. Traboulsi EI, Al-Khayer K, Matsumoto M. et al. Lymphedema-distichiasis syndrome and FOXC2 gene mutation. Am J Ophthalmol. 2002;134:592-596
6. Patil BB, Bell R, Brice G. et al. Distichiasis without lymphoedema? Eye (Lond). 2004;18:1270-1272
7. Brooks BP, Dagenais SL, Nelson CC. et al. Mutation of the FOXC2 gene in familial distichiasis. J AAPOS. 2003;7:354-357
8. Zhang L, Jia R, Zhao J. et al. Novel mutations in the RB1 gene from Chinese families with a history of retinoblastoma. Tumour Biol. 2014;36:2409-2420
9. Saleem RA, Banerjee-Basu S, Berry FB. et al. Analyses of the effects that disease-causing missense mutations have on the structure and function of the winged-helix protein FOXC1. Am J Hum Genet. 2001;68:627-641
10. Berry FB, Tamimi Y, Carle MV. et al. The establishment of a predictive mutational model of the forkhead domain through the analyses of FOXC2 missense mutations identified in patients with hereditary lymphedema with distichiasis. Hum Mol Genet. 2005;14:2619-2627
11. Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725-738
12. Carlsson P, Mahlapuu M. Forkhead transcription factors: key players in development and metabolism. Dev Biol. 2002;250:1-23
13. Katoh M. Human FOX gene family (Review). Int J Oncol. 2004;25:1495-1500
14. Lines MA, Kozlowski K, Walter MA. Molecular genetics of Axenfeld-Rieger malformations. Hum Mol Genet. 2002;11:1177-1184
15. Ito YA, Goping IS, Berry F. et al. Dysfunction of the stress-responsive FOXC1 transcription factor contributes to the earlier-onset glaucoma observed in Axenfeld-Rieger syndrome patients. Cell Death Dis. 2014;5:e1069
16. Du RF, Huang H, Fan LL. et al. A novel mutation of FOXC1 (R127L) in an Axenfeld-Rieger syndrome family with glaucoma and multiple congenital heart diseases. Ophthalmic Genet. 2014 [Epub ahead of print]: 1-5
17. Semina EV, Brownell I, Mintz-Hittner HA. et al. Mutations in the human forkhead transcription factor FOXE3 associated with anterior segment ocular dysgenesis and cataracts. Hum Mol Genet. 2001;10:231-236
18. Islam L, Kelberman D, Williamson L. et al. Functional analysis of FOXE3 mutations causing dominant and recessive ocular anterior segment disease. Hum Mutat. 2014;36:296-300
19. Crisponi L, Deiana M, Loi A. et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet. 2001;27:159-166
20. Fan J, Zhou Y, Huang X. et al. The combination of polyalanine expansion mutation and a novel missense substitution in transcription factor FOXL2 leads to different ovarian phenotypes in blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) patients. Hum Reprod. 2012;27:3347-3357
21. Laissue P, Lakhal B, Benayoun BA. et al. Functional evidence implicating FOXL2 in non-syndromic premature ovarian failure and in the regulation of the transcription factor OSR2. J Med Genet. 2009;46:455-457
22. Berry FB, Saleem RA, Walter MA. FOXC1 transcriptional regulation is mediated by N- and C-terminal activation domains and contains a phosphorylated transcriptional inhibitory domain. J Biol Chem. 2002;277:10292-10297
23. Fujita H, Kang M, Eren M. et al. Foxc2 is a common mediator of insulin and transforming growth factor beta signaling to regulate plasminogen activator inhibitor type I gene expression. Circ Res. 2006;98:626-634
24. Sutkowska E. Novel mutation in the FOXC2 gene in three generations of a family with lymphoedema - distichiasis syndrome. Gene. 2012;498:96-99
25. van Steensel MA, Damstra RJ, Heitink MV. et al. Novel missense mutations in the FOXC2 gene alter transcriptional activity. Hum Mutat. 2009;30:E1002-1009
26. Jeffery S. How double eyelashes give you swollen legs. Br J Ophthalmol. 2002;86:1074
27. Brice G, Mansour S, Bell R. et al. Analysis of the phenotypic abnormalities in lymphoedema-distichiasis syndrome in 74 patients with FOXC2 mutations or linkage to 16q24. J Med Genet. 2002;39:478-483
Author contact
![]() Corresponding authors: Tel./fax: +86 21 63135606 fanxqnet (XQ. Fan) Ninth People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, 200025, P.R. China.
Corresponding authors: Tel./fax: +86 21 63135606 fanxqnet (XQ. Fan) Ninth People's Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, 200025, P.R. China.