International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 22; 2026
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(10):1225-1235. doi:10.7150/ijbs.15154 This issue Cite
Research Paper
Deficiency of DJ-1 Ameliorates Liver Fibrosis through Inhibition of Hepatic ROS Production and Inflammation
Yingxue Yu1,3*, Xuehua Sun2*, Jinyang Gu1*, Chang Yu1,3, Yankai Wen1,3, Yueqiu Gao2, Qiang Xia1 ![]() , Xiaoni Kong1
, Xiaoni Kong1 ![]()
1. Department of Liver Surgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China.
2. Department of liver diseases, Shuguang Hospital affiliated to Shanghai University of Chinese Traditional Medicine, Shanghai, China.
3. School of Biomedical Engineering, Shanghai Jiao Tong University, Shanghai, China.
*These authors contributed equally to this work.
Received 2016-1-31; Accepted 2016-8-8; Published 2016-9-15
Citation:
Yu Y, Sun X, Gu J, Yu C, Wen Y, Gao Y, Xia Q, Kong X. Deficiency of DJ-1 Ameliorates Liver Fibrosis through Inhibition of Hepatic ROS Production and Inflammation. Int J Biol Sci 2016; 12(10):1225-1235. doi:10.7150/ijbs.15154. https://www.ijbs.com/v12p1225.htm
Other stylesAbstract

Liver fibrosis is a global health problem and previous studies have demonstrated that reactive oxygen species (ROS) play important roles in fibrogenesis. Parkinson disease (autosomal recessive, early onset) 7 (Park7) also called DJ-1 has an essential role in modulating cellular ROS levels. DJ-1 therefore may play functions in liver fibrogenesis and modulation of DJ-1 may be a promising therapeutic approach. Here, wild-type (WT) and DJ-1 knockout (DJ-1 KO) mice were administrated with carbon tetrachloride (CCl4) to induce liver fibrosis or acute liver injury. Results showed that DJ-1 depletion significantly blunted liver fibrosis, accompanied by marked reductions in liver injury and ROS production. In the acute CCl4 model, deficiency of DJ-1 showed hepatic protective functions as evidenced by decreased hepatic damage, reduced ROS levels, diminished hepatic inflammation and hepatocyte proliferation compared to WT mice. In vitro hepatic stellate cells (HSCs) activation assays indicated that DJ-1 has no direct effect on the activation of HSCs in the context of with or without TGFβ treatment. Thus our present study demonstrates that in CCl4-induced liver fibrosis, DJ-1 deficiency attenuates mice fibrosis by inhibiting ROS production and liver injury, and further indirectly affecting the activation of HSCs. These results are in line with previous studies that ROS promote HSC activation and fibrosis development, and suggest the therapeutic value of DJ-1 in treatment of liver fibrosis.
Keywords: DJ-1, liver fibrosis, liver injury, ROS, inflammation, lipid peroxidation.
Introduction
Liver fibrosis is a major health problem worldwide. Without appropriate treatment, liver fibrosis can develop from mild to extensive, eventually to irreversible stages, cirrhosis or hepatocellular carcinoma [1]. Under persistent cycles of chronic liver injury and repair, the balance between extracellular matrix (ECM) synthesis and degradation is disrupted, which directly leads to the excessive accumulation of ECM, a typical characteristic of liver fibrosis [2]. There are tremendous studies focusing on the crosstalk between inflammation and liver fibrosis. It has been shown that macrophages play a critical role in determining fibrotic scarring or resolution [3]. Additionally, studies have validated that liver fibrosis results from a series of events triggered by inflammation [4]. Briefly, in majority of the chronic liver diseases, exogenous or endogenous insults can lead to necrosis and/or excessive apoptosis of parenchymal cells, resulting in the initiation of inflammation, HSCs activation, and liver fibrosis [1].
Some other efforts over the past decades have focused on the role of reactive oxygen species (ROS) in fibrogenesis. Previous studies indicate that ROS derived from cytochrome p450 2E1 (Cyp2e1) of hepatocytes can induce alpha 2 collagen type I (Col1α2) gene expression in HSCs [5, 6]. In addition, ROS derived from phagocytic NADPH oxidase of Kupffer cells and non-phagocytic NADPH oxidase of HSC can respectively contribute to liver fibrosis by activating HSCs [7, 8]. Furthermore, increased ROS levels can exacerbate the inflammatory responses by activating several pro-inflammatory signal pathways, such as MAPK, NF-κb and JAK-STAT [9-12]. Therefore, ROS can promote liver fibrogenesis either by directly activating HSCs resulting in increased collagen production and deposition or by aggravating the inflammatory responses resulting in indirect enhancement of liver fibrosis.
DJ-1, a conserved ubiquitous protein, is encoded by Park7 gene. Loss-of-function mutations of DJ-1 contribute to the early onset of Parkinson's disease (PD). Many previous studies have shown that DJ-1 functions as an important scavenger in ROS elimination. Without DJ-1, dopaminergic neurons are more sensitive to oxidative stress [13]. Furthermore, DJ-1 has a protective role in preventing cell death from oxidative stress [14, 15]. In addition, DJ-1 can also modulate the expression of superoxide dismutase1 (SOD1), a well-known enzyme for dismutating superoxide, through Erk1/2-Elk1 pathway [16]. These findings strongly indicate that DJ-1 is involved in intracellular defense responses of ROS. In contrast to the well-defined antioxidant role of DJ-1, recent studies indicate that DJ-1 may also determine the ROS generation. Vasseur et al. have demonstrated that DJ-1 knockdown lead to reduced ROS accumulation in MEF cells [17]. Our recent study has shown that DJ-1 is involved in ROS production in a NADPH oxidase-dependent manner in macrophages [18]. Therefore, DJ-1 seems to play a dual role in buffering cellular ROS levels: DJ-1 functions as a scavenger in high ROS levels, whereas helps ROS production when essential ROS are required.
Given that both ROS and inflammation contribute to liver fibrogenesis and that ROS can exacerbate the inflammation, we hypothesized that DJ-1 plays a pivotal role in liver fibrosis by modulating the cellular redox state. In present study, we demonstrate much less liver fibrosis in DJ-1 KO mice than that in WT mice in a CCl4-induced fibrosis model. This alleviated liver fibrosis is mainly attributed to the reduction of ROS production, lipid peroxidation, hepatic damage and inflammation response in DJ-1 KO mice. These findings suggest the therapeutic value of DJ-1 in the treatment of liver fibrosis.
Materials and Methods
Patient samples
Liver tissues of cirrhosis patients were obtained during liver surgery from Renji Hospital. The human tissues experiment was approved by the institutional review board at the Renji Hospital. All participants provided written informed consent. About 10 paraffin-embedded cirrhosis samples and 5 normal tissues were used. Patient information was provided in Supplementary Table 1.
Animal models
DJ-1 knockout mice (stock#006577) were purchased from the Jackson Laboratory (Bar Harbor, Maine, USA). C57BL/6 mice were purchased from SLAC laboratory (Shanghai, China). All mice were bred and maintained at an animal facility under specific pathogen free condition. Age- and sex-matched mice were used in all experiments. For the chronic CCl4 model to induce liver fibrosis, mice with C57BL/6 background were intraperitoneally injected with CCl4 (10% in olive oil, 2 ml/kg body weight) three times per week for 4 or 8 weeks. Animals were sacrificed at 24h after the final injection. For the acute CCl4 model to induce liver injury, mice received a single intraperitoneal injected with CCl4 (10% in olive oil, 2 ml/kg body weight) and sacrificed at indicated time points. All animal experiments were approved by Bioethics Committee of Shanghai Jiao Tong University.
Hepatocytes, Kupffer cells and Hepatic satellite cells isolation and culture
Primary HSCs were obtained by using perfusion method. In brief, liver was perfused via portal vein with EGTA buffer (5.4mM KCl, 0.44mM KH2PO4, 0.338mM Na2HPO4, 137mM NaCl, 25mM Tricine and 0.5mM EGTA) 40ml for 5 min, followed by another perfusion with Gey's balanced salt solution (GBSS) (2mM CaCl2, 1mM MgCl2, 0.285mM MgSO4, 5mM KCl, 0.20mM KH2PO4, 27mM NaHCO3, 120mM NaCl, 0.8mM Na2HPO4, 5.6mM D-Glucose) containing 0.075% collagenase IV (Sigma) 40ml for 20min. After that, liver was dissociated with digestion GBSS buffer with collagenase IV (0.008%) for 30 min at 37℃ and filtered with a 70-μm Nylon cell strainer. After centrifuged at 500 rpm for 5 min, the hepatocyte pellets were suspended in DMEM with 10% FBS and seeded at 24-well plates for further experiments. The supernatant was performed for density gradient centrifugation with a 17% or 12.5% OptiPrep Gradient (sigma) for isolation of Kupffer cells or HSCs respectively. Kupffer cells or HSCs were counted and respectively cultured in RPMI 1640 medium supplemented with 10% FBS (Gibco) or 10% FBS (Gibco) plus 10% horse serum (Gibco). Cell lysates of HSCs were collected at 1, 4, 7 days for mRNA analysis.
TGFβ-induced activation of HSCs
After primary HSCs were isolated from WT mice and DJ-1 KO mice, cells were cultured with RPMI 1640 culture medium in 12-well plates at a density of 100,000 cells/well for 2 days. Then the medium was replaced by serum-free medium. After 24h starvation, cells were treated with 5ng/ml TGFβ or with solvent (4mM HCl containing 0.1% BSA). Cell lysates were collected for qPCR analysis after 24h of treatment.
Biochemical analysis
Blood samples were obtained from mice injected with CCl4, and serum was acquired after centrifugation. Serum alanine transaminase (ALT) and aspartate aminotransferase (AST) levels were assessed according to the manufacturer's instructions from Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
Western Blotting
Liver samples were homogenized with RIPA buffer (Thermo Scientific, Rockford, IL) containing a protease inhibitor cocktail (Calbiochem, Raleigh, NC). Protein quantification was performed with a BCA protein assay kit (Thermo Scientific, Rockford, IL). Protein samples were separated by 10% SDS-PAGE. The gel was transferred to a nitrocellulose membrane and probed with antibodies against indicated antigens, followed by incubation with corresponding secondary antibodies coupled with horseradish peroxidase. Blotting signals were detected and quantified by using the ChemiDocTM XRS+ System with Image LabTM Software (Bio-Rad). Primary antibodies were rabbit anti-α-SMA, rabbit anti-DJ-1, mouse anti-GAPDH (Abcam, Cambridge, MA), rabbit anti-Smad2/3, rabbit anti-Phospho-Smad2, rabbit anti-Phospho-Smad3 (Cell Signaling, Boston, MA).
Histological analysis
4% paraformaldehyde-fixed liver tissues were embedded with paraffin, and 5-μm-thick sections were collected and stained with hematoxylin-eosin (HE) and Sirius red staining (Sigma) to detect liver injury and liver fibrosis respectively. For immunohistochemistry staining, paraffin liver sections were dewaxed, rehydrated and subjected to epitope retrieval in citric acid solution, Proteinase K or Tris-EDTA buffer (PH 9.0). Endogenous peroxidase activity was blocked with 3% hydrogen peroxide. Sections were then blocked with 10% bovine serum albumin and incubated with primary antibodies of DJ-1 (1:200, Abcam, Cambridge, MA), α-SMA (1:200, Abcam, Cambridge, MA), F4/80 (1:100, AbD Serotec, Oxford, UK), Ki67 (1:100, Abcam, Cambridge, MA), PCNA (1:100 Cell Signaling, Boston, MA), Cleaved-caspase 3 (1:300, Cell Signaling, Boston, MA), CD11b (1:400, Abcam, Cambridge, MA) and MPO (BioCare Medical, Concord, CA) at 4℃ overnight. Corresponding second antibodies conjugated with horseradish peroxidase were used and hematoxylin counterstaining was performed. Six to ten images from random fields in each section were taken with a light microscope (Axio Imager A1; Zeiss).
Quantitative RT-PCR analysis
Total RNA was extracted by using RNeasy Mini Kit (Qiagen, Valencia, CA), according to the manufacturer's instructions. Total RNA (1μg) was used for cDNA synthesis (Thermo Scientific, Rockford, IL). Gene expression levels were detected by SYBR Green based Quantitative RT-PCR (TaKaRa Biotechnology, Dalian, China) with gene specific primers listed below. Primers: Mouse α-SMA primers, forward: TGCCATGTATGTGGCTATTCA; reverse: ACCAGTTGTACGTCCAGAAGC, Mouse TIMP-1 primers, forward: GGCATCCTCTTGTTGCTATCACTG; reverse: GTCATCTTGATCTCATAACGCTGG, Mouse COL1A1 primers, forward: GACATGTTCAGCTTTGTGGAC; reverse: GCAGCTGACTTCAGGGATG, Mouse COL3A1 primers, forward: GCCCACAGCCTTCTACAC; reverse: CCAGGGTCACCATTTCTC, Mouse TIMP-2 primers, forward: CACCCAGAAGAAGAGCCTGA; reverse: GGGAGGAGATGTAGCAAGGG, Mouse MMP-2 primers, forward: ACCCAGATGTGGCCAACTAC; reverse: TCATTTTAAGGCCCGAGCAA, Mouse TGFβ primers, forward: CACGATCATGTTGGACAACTGCTCC; reverse: CTTCAGCTCCACAGAGAAGAACTGC, Mouse CCL4 primers, forward: GAAACAGCAGGAAGTGGGAG; reverse: CATGAAGCTCTGCGTGTCTG, Mouse CCL3 primers, forward: TTCTCTGTACCATGACACTCTGC; reverse: CGTGGAATCTTCCGGCTGTAG, Mouse MMP-9 primers, forward: GTTTTTGATGCTATTGCTGAGATCCA; reverse: CCCACATTTGACGTCCAGAGAAGAA, Mouse CXCL2 primers, forward: CAGAGCTTGAGTGTGACG; reverse: TCGTACCTGATGTGCCTC, Mouse ICAM-1 primers, forward: CAATTTCTCATGCCGCACAG; reverse: AGCTGGAAGATCGAAAGTCCG, Mouse DJ-1 primers, forward: GTGCAGTGTAGCCGTGATGT; reverse: CCTCCTGGAAGAACCACCAC, Mouse IL-6 primers, forward: TGTTCTCTGGGAAATCGTGGA; reverse: TTTCTGCAAGTGCATCATCGT, Mouse TNF-α primers, forward: TTCTATGGCCCAGACCCTCA; reverse: TTTGCTACGACGTGGGCTAC, Mouse CYP2E1 primers, forward: GGGAATGGGGAAACAGGGTA; reverse: GGTGCACAGCCAATCAGAAA, Mouse HMOX-1 primers, forward: AAGCCGAGAATGCTGAGTTCA; reverse: GCCGTGTAGATATGGTACAAGGA, Mouse MCP-1 primers, forward: CCCTAAGGTCTTCAGCACCT; reverse: ACTGTCACACTGGTCACTCC, Mouse CCL5 primers, forward: ATATGGCTCGGACACCACTC; reverse: GTGACAAACACGACTGCAAGA, Mouse GAPDH primers, forward: AGGCCGGTGCTGAGTATGTC; reverse: TGCCTGCTTCACCACCTTCT.
ROS production determination
We examined the intracellular ROS levels with the 2', 7'-Dichlorofluorescin diacetate (DCFH-DA, sigma) probe in the frozen sections of liver tissues from either the CCl4 induced liver fibrosis model or the acute liver injury model. Briefly, liver tissues were embedded with OCT-Freeze medium (Sakura Tissue-Tek; Torrance, CA). After frozen, tissues were sliced into 8-μm-thick sections and washed with ice-cold phosphate buffer (PH 7.4), subsequently incubated with the fluorescent probe DCFH-DA (25μM) at 37℃ for 30min. Fluorescence intensity was determined with an excitation wavelength of 488 nm and an emission wavelength of 526 nm. Images were captured with the fluorescence microscope (Leica).
In vitro ROS detection in primary Kupffer cells and hepatocytes
Isolated primary Kupffer cells and hepatocytes were seed into 24-well plates at a density of 50,000 cells/well with RPMI 1640 medium or DMEM medium respectively. After adherence, cells were incubated with DCFH-DA probe (20μM) at 37℃ for 30min. Fluorescence intensity was detected with 488 nm excitation and 526 nm emission wavelengths. Images were captured with the fluorescence microscope (Leica).
MDA and GSH detection
For MDA and GSH detection, commercial kits were used according to the manufacturer's instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China). Liver tissues were homogenized with ice-cold phosphate buffer (PH 7.4), then centrifuged at 13,000rpm for 15min at 4℃. The supernatant was used to detect the content of MDA and GSH [19]. Total protein content was determined by BCA protein assay kit (Thermo Scientific, Rockford, IL).
Statistical Analysis
Results were presented as mean ± SEM from at least 3 independent biological replicated experiments. Data were analyzed using unpaired or paired Student's t-tests. *P<0.05; **P<0.01; ***P<0.001.
Results
DJ-1 deficiency ameliorates CCl4-induced experimental liver fibrosis in vivo
Carbon tetrachloride (CCl4) induced liver fibrosis is a common and classical mouse fibrosis model. On one hand, CCl4 can directly impair hepatocytes by changing the permeability of plasma, lysosome and mitochondrial membranes, resulting in cell apoptosis or/and necrosis; on the other hand, CCl4 can be metabolized into trichloromethyl radicals by CYP2E1 enzyme and lead to ROS production and intracellular lipid peroxidation, resulting in the activation of hepatic stellate cells [20, 21]. To address whether DJ-1 plays a role in development of hepatic fibrosis, the mouse chronic liver injury process was mimicked by weekly injection of the hepatotoxic chemical CCl4. Increased DJ-1 expression was found in the liver tissues of WT mice 8 weeks after CCl4 treatment (Supplementary Figure 3). In fact, the increased DJ-1 levels were also observed in the liver sections of cirrhosis patients (Supplementary Figure 3), which is consistent with our recent study [22]. These findings suggest the association of DJ-1 with the progression of liver fibrosis. Collagen deposition and HSC activation, the two key markers of liver fibrosis, were examined after 8 weeks of CCl4 injection. Compared to WT mice, DJ-1 KO mice showed less collagen deposition in ECM and HSC activation as demonstrated by Sirius Red and α-SMA staining respectively (Fig. 1A). Consistent with the IHC results, both mRNA and protein levels of α-SMA were decreased in DJ-1 KO mice compared to their WT counterparts (Figure 1B and C). Additionally, the expression of other activated HSC markers, such as TIMP-1, Col1α1, Col3α1, TIMP-2 and MMP-2 was examined in liver tissues of both WT and DJ-1 KO mice. In line with decreased HSC activation, the hepatic mRNA levels of those activated HSC markers were lower in DJ-1 KO mice compared to WT mice after 8 weeks of CCl4 exposure (Figure 1B). Given that TGFβ is a key cytokine for HSC activation, we next examined the hepatic TGFβ levels by qPCR after 4 or 8 weeks of CCl4 treatment. As shown in Figure 1D, significantly decreased TGFβ mRNA levels were detected in DJ-1 KO mice at both time points. In response to decreased TGFβ level, the phosphorylation of Smad2 and Smad3 was subsequently downregulated in DJ-1 KO mice at 8 weeks after CCl4 injection (Figure 1E and Supplementary Figure 4). Collectively, our data indicate that DJ-1 deficiency has an antifibrotic role in the CCl4-induced experimental model.
Figure 1
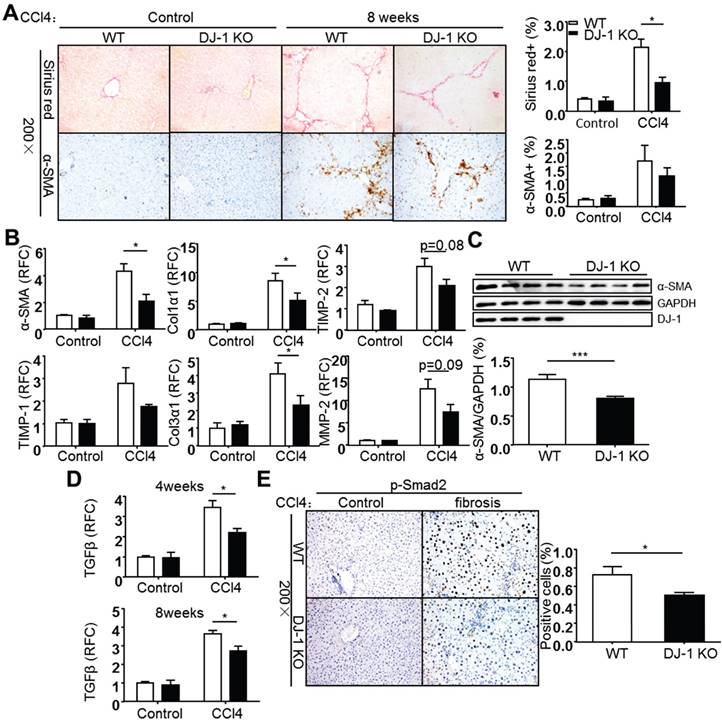
DJ-1 deficiency attenuates CCl4-induced liver fibrosis in Vivo. (A) Representative Sirius red staining and α-SMA immunohistochemistry of livers from WT mice and DJ-1 KO mice after CCl4 injection for 8 weeks (200× magnification). Bar graphs show quantification of them (n = 6-9). The data are shown as the mean ± SEM. *P<0.05. (B) α-SMA, TIMP-1, Col1α1, Col3α1, TIMP-2, MMP-2 mRNA expression in livers was analyzed by qPCR after CCl4 injection for 8 weeks (n = 6-9). All gene expression levels were normalized to housekeeping control GAPDH. *P<0.05. (C) Protein expression of α-SMA in whole livers from mice treated with CCl4 for 8 weeks was analyzed by immunoblotting. Bar graph shows quantification of it (n = 4). ***P<0.001. (D) TGFβ mRNA expression in livers of WT and DJ-1 KO mice after CCl4 injection for 4 weeks or 8 weeks was analyzed by qPCR (n = 6-9). The data are shown as the mean ± SEM. *P<0.05. RFC: relative fold change. (E) Phosphorylation levels of Smad2 in livers from WT mice and DJ-1 KO mice treated with CCl4 for 8 weeks were analyzed by immunohistochemistry. Bar graph shows quantification of it (n = 7). *P<0.05.
DJ-1 has no direct effect on the activation of HSCs
Previous reports have demonstrated that activated HSCs are the predominant ECM-producers in the course of hepatic fibrosis [23]. Although we found less liver fibrosis and HSC activation in DJ-1 KO mice and less liver injury seems attributable to this antifibrotic phenotype in DJ-1 KO mice, we can not rule out the possibility that DJ-1 may affect HSC activation directly. To address this concern, we isolated primary HSCs from both WT and DJ-1 KO mice and examined their culture activation for 1d, 4d, or 7d. As shown in Figure 2A, the expression of HSC activation markers, such as TIMP-1, α-SMA and Col1α1, was increased with time in both WT and DJ-1 KO mice and HSC activation showed no difference in both genotypes. Given that TGFβ can enhance HSC activation in vitro and in vivo, we next examined whether DJ-1 affects TGFβ mediated HSC activation in vitro. Although TGFβ treatment augmented the HSC activation as demonstrated by increased expression of TIMP-1, α-SMA and Col1α1, the induction of those genes were comparable between WT and DJ-1 KO mice (Figure 2B). Therefore, these findings indicate that DJ-1 has no direct effect on HSC activation.
Figure 2
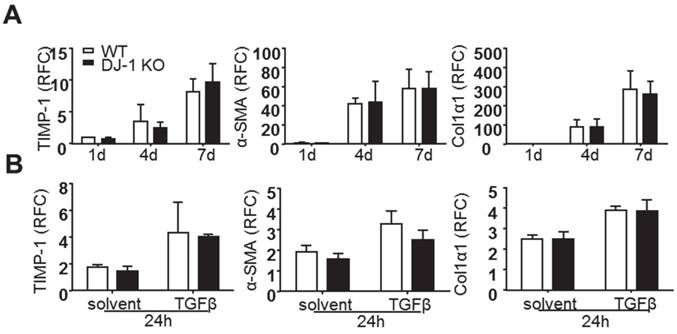
DJ-1 has no direct effect on primary HSCs activation in vitro. (A) mRNA levels of activated HSCs markers were analyzed by qPCR after isolated HSCs cultured for 1d, 4d and 7d (n = 4-5). (B) mRNA levels of TIMP-1, α-SMA, Col1α1 in isolated HSCs of the two groups were analyzed after TGFβ treatment for 24h. The data are shown as the mean ± SEM. RFC: relative fold change.
DJ-1 KO mice are protected from CCl4-induced acute liver injury
As liver fibrosis is a pathological reaction to liver injury by depositing ECM to injury sites [2], we then employed an acute CCl4 injury model to further explore the role of DJ-1 in liver injury. After a single CCl4 (2ml/kg BW) injection, mice from both genotypes were sacrificed and their livers were harvested at indicated time points. Liver injury was evaluated by HE staining and serum ALT levels. As shown in Figure 3A, DJ-1 deficiency resulted in significant hepatic protection as demonstrated by decreased liver injury areas and ALT levels. The decreased liver injury could be due to less apoptosis levels in DJ-1 KO mice as detected by cleaved-caspase 3 staining (Figure 3C). Corresponding to the decreased liver injury, less hepatic immune cells were detected in DJ-1 KO mice as demonstrated by F4/80 (a macrophage marker) and MPO (a neutrophil marker) staining (Fig. 3B). Meanwhile, pro-inflammatory cytokines IL-6 and TNF-α were also decreased when DJ-1 was depleted (Fig. 3D). Given that the induction of inflammatory cytokines can generally feedback to hepatocytes resulting in increased proliferation. We then measured the proliferation changes in both WT and DJ-1 KO mice. Compared to WT mice, DJ-1 KO mice showed greatly reduced PCNA and Ki67 staining signals 36h and 48h after CCl4 injection respectively (Fig. 3E). Since a previous study indicates that ROS mainly contribute to CCl4 induced acute liver injury [24], we examined whether DJ-1 can affect CCl4 directed liver injury by modulating ROS levels in an acute model. ROS levels were visualized by the DCFH-DA fluorescence probe in frozen liver sections from WT mice and DJ-1 KO mice 24h after CCl4 treatment. As shown in Figure 3F, greater ROS levels were observed in WT mice than those in DJ-1 KO mice 24h after a single CCl4 challenge. As excessive ROS can cause lipid peroxidation resulting in second round of liver damage and Malondialdehyde (MDA), one of the endogenous end products during lipid peroxidation and a parameter of oxidative stress [25], we then measured the MDA levels in the acute liver injury model. In parallel with the ROS levels, decreased MDA levels were also detected in the liver homogenates of DJ-1 KO mice compared to WT mice (Figure 3G). Reduced ROS levels are unlikely due to antioxidant increase in DJ-1 KO mice because the levels of GSH, a major antioxidant, are same in WT and DJ-1 KO mice (Supplementary Figure 1). Previous reports demonstrate that trichloromethyl free radical (CCl3•) and trichloromethyl peroxy radical (CCl3OO•), generated by CYP2E1 enzyme of hepatocytes post CCl4 injection, cause lipid peroxidation and liver damage [25, 26]. Therefore, the different levels of ROS and liver injury between WT and DJ-1 KO mice maybe possibly due to differential expression of CYP2E1 in WT and DJ-1 KO mice. To examine this possibility, we detected CYP2E1 expression by qPCR in both WT and DJ-1 KO mice 24h and 36h after CCl4 injection. qPCR results showed that the CYP2E1 mRNA levels are comparable in the liver tissues of WT and DJ-1 KO mice (Figure 3H), suggesting that the attenuated lipid peroxidation and liver damage in DJ-1 KO mice are not caused by CYP2E1. These results indicate that DJ-1 deficiency is protective for CCl4-induced acute liver injury.
Figure 3
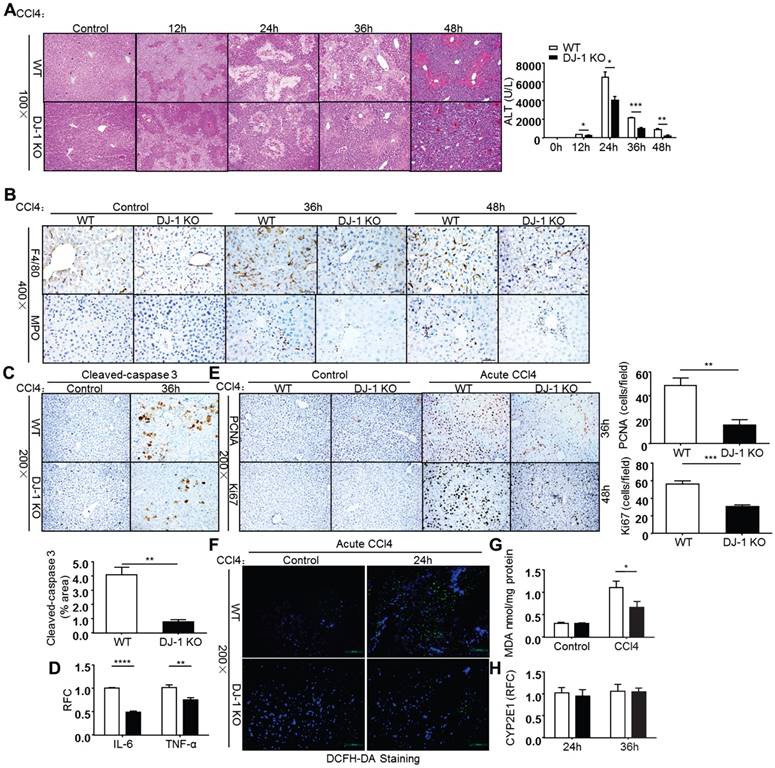
Deficiency of DJ-1 attenuates liver damage and ROS generation in CCl4-induced acute liver injury. (A) HE staining and serum ALT levels were displayed at different time points after CCl4 injection (n = 4-7). *P<0.05, **P<0.01, ***P<0.001. (B) F4/80 and MPO Immunochemistry were performed in liver sections of WT mice and DJ-1 KO mice after CCl4 injection for 36h and 48h (400× magnification). (C) Cleaved-caspase 3 in liver sections from WT mice and DJ-1 KO mice after CCl4 injection for 36h was shown (n = 3) (200× magnification). Quantification of the positive areas was also performed (n = 3). **P<0.01. (D) mRNA levels of IL-6, TNF-α cytokines were determined by qPCR 36h post CCl4 injection. **P<0.01, ***P<0.001. (E) Immunochemistry for PCNA and Ki67 were performed after CCl4 injection for 36h and 48h respectively (200× magnification). Bar graphs show quantification of them (n = 5). **P<0.01, ***P<0.001. (F) ROS production was visualized by DCFH-DA probe in frozen liver sections from WT mice and DJ-1 KO mice 24h after CCl4 treatment (200× magnification). (G) MDA content in liver tissue homogenates was evaluated between WT mice and DJ-1 KO mice 24h after CCl4 injection. *P<0.05. (H) Gene expression of CYP2E1 in two groups was displayed 24h and 36h after CCl4 injection.
DJ-1 deficiency has a hepatic protective role to CCl4-induced chronic liver injury
As liver fibrosis develops from chronic and reiterative injury-repair cycles, we then sought to examine whether DJ-1 deficiency affects CCl4-induced chronic liver injury. Liver injury was assessed by measuring the serum ALT/AST levels 8 weeks after CCl4 treatment. As shown in Figure 4A, ALT/AST levels were decreased in DJ-1 KO mice compared to WT mice, suggesting the protective role of DJ-1 deficiency in CCl4-induced chronic liver injury. In agreement with less liver injury, although the overall apoptosis levels were greatly reduced in the chronic CCl4 model compared to the acute one, lower cleaved-caspase 3 signals were observed in DJ-1 KO mice compared to WT mice (Figure 4B). Meanwhile, we did not get any positive Ki67 and PCNA signals in the liver sections of mice from the chronic CCl4 model (data not shown), despite abundant those positive signals observed in the acute CCl4 model (Figure 3E). This finding is consistent with a previous report showing rare Ki67 signals in the liver after a chronic CCl4 treatment [27]. In addition, hepatic neutrophils and macrophages were also examined in both WT and DJ-1 KO mice 8 weeks after CCl4 exposure.
Figure 4
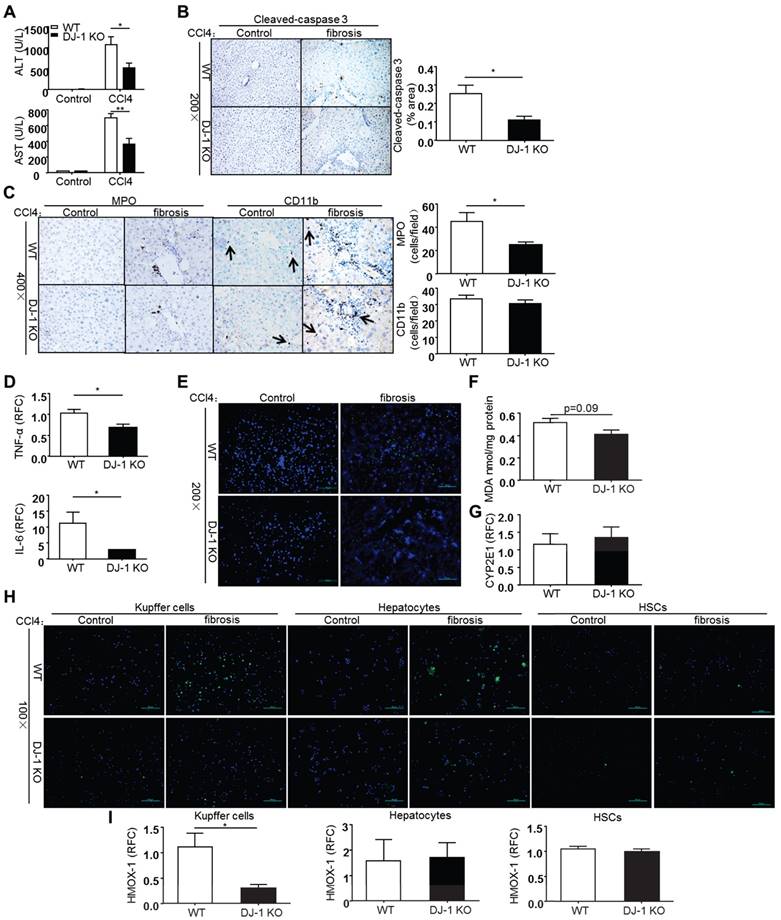
DJ-1 deletion suppresses ROS production and lipid peroxidation in CCl4-induced chronic liver injury. (A) Serum ALT and AST levels were determined in WT mice and DJ-1 KO mice after CCl4 injection for 8 weeks (n = 6-9). *P<0.05, **P<0.01. (B) Cleaved-caspase 3 in liver sections from WT mice and DJ-1 KO mice after CCl4 injection for 8 weeks was shown (n = 3) (200× magnification). Quantification of the positive areas was also performed (n = 3). *P<0.05. (C) Immunochemistry for MPO and CD11b were performed in liver sections of WT mice and DJ-1 KO mice after CCl4 injection for 8 weeks (400× magnification). Bar graphs show quantification of them, *P<0.05. (D) Gene expression of cytokines IL-6, TNF-α in the whole liver tissues was analyzed by qPCR after CCl4 injection for 8 weeks. *P<0.05. RFC: relative fold change. (E) ROS production was visualized in frozen liver sections and (F) MDA content was evaluated in liver tissue homogenates from WT mice and DJ-1 KO mice treated with CCl4 for 4 weeks (200× magnification). (G) Gene expression of CYP2E1 in two groups was displayed after 8 weeks CCl4 injection. (H) ROS production in isolated Kupffer cells, hepatocytes, HSCs was shown with mice treated with CCl4 for 4 weeks (100× magnification). (I) mRNA levels of HMOX-1 in cultured Kupffer cells, hepatocytes, HSCs were analyzed by qPCR with mice treated with CCl4 for 4 weeks. *P<0.05. RFC: relative fold change.
As shown in Figure 4C, less hepatic neutrophils but not macrophages was observed in DJ-1 KO mice compared to WT ones. However, the increased neutrophil seems not associated with the changes of traditional chemokines, because the expression of various chemokines, including CCL4, CCL3, CXCL2 and ICAM-1, had no change between WT and DJ-1 KO mice except that of MCP-1 and CCL5, which are not related to neutrophil infiltration (Supplementary Figure 2). Interestingly, the pro-inflammatory cytokines, TNF-α and IL-6, which are mainly produced by activated macrophages, were decreased in DJ-1 KO mice compared to WT mice (Figure 4D), although the hepatic macrophage was comparable in both genotypes. We then evaluated the ROS levels in this chronic CCl4 model. As shown in Figure 4E and 4F, similar to the results in the acute model, higher ROS levels were found in WT mice detected by DCFH-DA fluorescent staining and MDA measurement respectively. The CYP2E1 expression levels were same in both genotypes (Figure 4G), suggesting that CYP2E1 does not contribute to the different ROS levels in this chronic model. To confirm the cell source of ROS production in our model, HSCs, hepatocytes and Kupffer cells were isolated from WT and DJ-1 KO mice after 4 weeks of CCl4 treatment and followed with a ROS detection using the DCFH-DA probe. The purities of those cell types were more than 90% (Supplementary Figure 5). Our results showed very few signals in both HSCs and hepatocytes but abundant ROS signals were observed in Kupffer cells (Figure 4H). In addition, compared to WT mice, DJ-1 KO mice showed dramatically decreased ROS levels in Kupffer cells (Figure 4H). To further confirm these ROS results, we measured the levels of HMOX-1, an early ROS response gene, in WT and DJ-1 KO mice. In line with the decreased ROS levels observed in Kupffer cells of DJ-1 KO mice, the HMOX-1 mRNA levels of Kupffer cells were significantly lower in DJ-1 KO mice compared to WT ones. Additionally, the HMOX-1 levels in either HSCs or hepatocytes were comparable in both genotypes (Figure 4I). Collectively, these findings support the hepatic protective role of DJ-1 deficiency in CCl4-induced chronic liver injury.
Discussion
In present study, we find elevated DJ-1 expression in the CCl4 chronic experimental model and in the liver tissues of cirrhosis patients (Supplementary Figure 3), suggesting the possible involvement of DJ-1 in the pathogenesis of liver fibrosis. We further demonstrate that CCl4 induced liver fibrosis is severely attenuated in DJ-1 KO mice. Given that HSC activation plays a major role in liver fibrosis, we then performed the experiments of in-culture HSC activation to determine whether DJ-1 can directly regulate this process. Our results showed similar activation rate of HSCs derived from WT and DJ-1 KO mice either in the presence or in the absence of TGFβ (Figure 2), suggesting that this disadvantage of liver fibrogenesis in DJ-1 KO mice is not due to the direct effect of DJ-1 on HSCs. Through CCl4 induced acute liver injury and chronic fibrosis models, we find that DJ-1 deficiency leads to a liver protective phenomenon as evidenced by marked reductions in liver injury, ROS production and inflammation in response to CCl4 exposure (Figure 3 and 4).
It is well known that liver fibrosis is a complicated process involving various pathological factors. Among them, ROS is one of the most potent pro-fibrogenic factors in the pathology of hepatic fibrosis, by activating the HSCs [28]. Reduction of ROS decreases collagen accumulation in experimental liver fibrosis models [29]. A previous study demonstrates that NADPH oxidase derived ROS play a critical role in liver fibrogenesis because depletion of p47phox, a key subunit of NADPH oxidase, leads to attenuated liver injury and fibrosis [8].
It is well known that DJ-1 functions as an antioxidant in the context of high ROS levels to protect cells from ROS induced damage. However, our recent study demonstrates that, by interacting with p47phox, DJ-1 is required for NADPH oxidase derived ROS production in macrophages [18]. Based on the studies of our and other groups, we therefore hypothesize that DJ-1 has a dual role in maintaining cellular ROS levels. On one hand, DJ-1 limits the cellular oxidant stress when the levels of ROS are high; on the other hand, DJ-1 is essential for ROS induction in response to cellular insults from outside or inside. According to the fundamental role of DJ-1 in regulating ROS, it is conceivable that DJ-1 can affect fibrogenesis by modulating ROS levels. In fact, we observed much less ROS levels in DJ-1 KO mice compared to WT mice in both acute and chronic models, suggesting that the attenuated liver fibrosis in DJ-1 KO mice may at least partially attributed to the reduced ROS levels.
Previous studies demonstrate that there are three main cell sources of ROS production during liver fibrosis, including hepatocytes, Kupffer cells (hepatic resident macrophages) and HSCs. For example, CYP2E1 mediated hepatocytic ROS production induces proliferation, transdifferentiation and collagen synthesis in HSCs and subsequently contributes to fibrogenesis [30], and ROS derived from phagocytic NADPH oxidase of Kupffer cells and non-phagocytic NADPH oxidase of HSCs can respectively contribute to liver fibrosis by activating HSCs [7, 8]. Given that there is no difference in the in vitro HSC activation from both WT and DJ-1 KO groups (Figure 2), we think that HSCs are not the major cell source of ROS in our present study. This presumption has been confirmed by very few ROS signals detected in HSCs after CCl4 treatment (Figure 4H). By comparing the ROS levels in the hepatocytes and Kupffer cells, we found that Kupffer cells are the major cell source for ROS production and that Kupffer cells in WT mice produce greater amount of ROS than those in DJ-1 KO mice (Figure 4H). Given that Kupffer cells are liver resident macrophages, this finding is consistent with our previous study that DJ-1 depletion impairs ROS production in macrophages [18]. Therefore, DJ-1 may specifically affect the ROS production from phagocytic NADPH oxidase.
In present study, we find increased neutrophil infiltration in WT mice compared to DJ-1 KO mice in both acute and chronic models (Figure 3 and 4). However, the expression of various chemokines is comparable between WT and DJ-1 KO mice except MCP-1 and CCL5, which is not responsible for neutrophil infiltration (Supplementary Figure 2). Therefore, the infiltration of neutrophil in present study may not depend on the chemokines. Given that certain products of lipid peroxidation can serve as potent chemotactic factors for neutrophils [31], the increased neutrophil infiltration is very likely due to higher lipid oxidation levels in WT mice.
In summary, our present study demonstrates that DJ-1 depletion significantly attenuated liver fibrosis in a CCl4-induced experimental model. DJ-1 depletion shows a hepatic protective role in both CCl4-induced acute and chronic liver injury models as evidenced by less liver injury, ROS production, and inflammation. Mechanistically, we believe that the less ROS production at least partially contributes to amelioration of liver fibrosis in DJ-1 KO mice in the CCl4 mouse model. Our findings highly suggest the therapeutic potential of DJ-1 in liver fibrosis.
Abbreviations
ROS: reactive oxygen species; CCl4: carbon tetrachloride; HSCs: hepatic stellate cells; PD: Parkinson's disease; ECM: extracellular matrix; TIMP: tissue inhibitor of metalloproteinase; MMP: matrix metallopeptidase; MDA: malondialdehyde; HMOX-1: heme oxygenase 1; CYP2E1: cytochrome p450 2E1; α-SMA: alpha smooth muscle actin; Col1α1: alpha 1 collagen type I; Col3α1: alpha 1 collagen type III; TGFβ: transform growth factor β; Smad: mothers against decapentaplegic homolog; CD11b: cluster of differentiation molecule 11b; ICAM-1: intercellular adhesion molecules 1; CCL: Chemokine ligand; MCP-1: monocyte chemotactic protein 1; IL: interleukin; TNF-α: tumor necrosis factor α; GSH: glutathione; ALT: alanine transaminase; AST: aspartate aminotransferase; PCNA: proliferating cell nuclear antigen; MPO: myeloperoxidase; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; RFC: relative fold change; SEM: standard error of mean.
Supplementary Material
Supplementary Table and Figures.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31300742 to X Kong, 81372233 to H. Wu, 8147224 to Q Xia, 81473629 to X Sun and 81473629 to Y Gao), and the Shanghai Education Committee (Eastern Scholar Program) and program from the Shanghai Key Laboratory of hepatocellular and cholangiocellular carcinomas (13DZ2261100) to X Kong, grants from the Committee of Science and Technology of Shanghai Municipal Government (13401902900) to Y Gao, the Shanghai Health Bureau Key Joint Efforts Foundation (2013ZYJB001) to Q Xia.
Competing interests
The authors have declared that no competing interest exists.
References
1. Bataller R, Brenner DA. Liver fibrosis. The Journal of clinical investigation. 2005;115:209-18
2. Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annual review of pathology. 2011;6:425-56
3. Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. The Journal of clinical investigation. 2008;118:3522-30
4. Marra F. Chemokines in liver inflammation and fibrosis. Frontiers in bioscience: a journal and virtual library. 2002;7:d1899-914
5. Nieto N, Friedman SL, Cederbaum AI. Cytochrome P450 2E1-derived reactive oxygen species mediate paracrine stimulation of collagen I protein synthesis by hepatic stellate cells. The Journal of biological chemistry. 2002;277:9853-64
6. Nieto N, Friedman SL, Greenwel P, Cederbaum AI. CYP2E1-mediated oxidative stress induces collagen type I expression in rat hepatic stellate cells. Hepatology (Baltimore, Md). 1999;30:987-96
7. De Minicis S, Brenner DA. NOX in liver fibrosis. Archives of biochemistry and biophysics. 2007;462:266-72
8. Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J. et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. The Journal of clinical investigation. 2003;112:1383-94
9. Reuter S, Gupta SC, Chaturvedi MM, Aggarwal BB. Oxidative stress, inflammation, and cancer: how are they linked? Free radical biology & medicine. 2010;49:1603-16
10. McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxidants & redox signaling. 2006;8:1775-89
11. Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S, Aggarwal BB. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. The Journal of biological chemistry. 2003;278:24233-41
12. Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim KY. et al. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). The Journal of experimental medicine. 2011;208:519-33
13. Martinat C, Shendelman S, Jonason A, Leete T, Beal MF, Yang L. et al. Sensitivity to oxidative stress in DJ-1-deficient dopamine neurons: an ES- derived cell model of primary Parkinsonism. PLoS biology. 2004;2:e327
14. Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO reports. 2004;5:213-8
15. Yokota T, Sugawara K, Ito K, Takahashi R, Ariga H, Mizusawa H. Down regulation of DJ-1 enhances cell death by oxidative stress, ER stress, and proteasome inhibition. Biochemical and biophysical research communications. 2003;312:1342-8
16. Wang Z, Liu J, Chen S, Wang Y, Cao L, Zhang Y. et al. DJ-1 modulates the expression of Cu/Zn-superoxide dismutase-1 through the Erk1/2-Elk1 pathway in neuroprotection. Annals of neurology. 2011;70:591-9
17. Vasseur S, Afzal S, Tomasini R, Guillaumond F, Tardivel-Lacombe J, Mak TW. et al. Consequences of DJ-1 upregulation following p53 loss and cell transformation. Oncogene. 2012;31:664-70
18. Liu W, Wu H, Chen L, Wen Y, Kong X, Gao WQ. Park7 interacts with p47(phox) to direct NADPH oxidase-dependent ROS production and protect against sepsis. Cell research. 2015;25:691-706
19. Wu Y, Li L, Wen T, Li YQ. Protective effects of echinacoside on carbon tetrachloride-induced hepatotoxicity in rats. Toxicology. 2007;232:50-6
20. Knockaert L, Berson A, Ribault C, Prost PE, Fautrel A, Pajaud J. et al. Carbon tetrachloride-mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Laboratory investigation; a journal of technical methods and pathology. 2012;92:396-410
21. Weiler-Normann C, Herkel J, Lohse AW. Mouse models of liver fibrosis. Zeitschrift fur Gastroenterologie. 2007;45:43-50
22. Chen L, Luo M, Sun X, Qin J, Yu C, Wen Y. et al. DJ-1 deficiency attenuates expansion of liver progenitor cells through modulating the inflammatory and fibrogenic niches. Cell death & disease. 2016;7:e2257
23. Friedman SL. Evolving challenges in hepatic fibrosis. Nature reviews Gastroenterology & hepatology. 2010;7:425-36
24. Taniguchi M, Takeuchi T, Nakatsuka R, Watanabe T, Sato K. Molecular process in acute liver injury and regeneration induced by carbon tetrachloride. Life sciences. 2004;75:1539-49
25. Basu S. Carbon tetrachloride-induced lipid peroxidation: eicosanoid formation and their regulation by antioxidant nutrients. Toxicology. 2003;189:113-27
26. Tien YC, Liao JC, Chiu CS, Huang TH, Huang CY, Chang WT. et al. Esculetin ameliorates carbon tetrachloride-mediated hepatic apoptosis in rats. International journal of molecular sciences. 2011;12:4053-67
27. Huang B, Cheng X, Wang H, Huang W, la Ga Hu Z, Wang D. et al. Mesenchymal stem cells and their secreted molecules predominantly ameliorate fulminant hepatic failure and chronic liver fibrosis in mice respectively. Journal of translational medicine. 2016;14:45
28. Richter K, Konzack A, Pihlajaniemi T, Heljasvaara R, Kietzmann T. Redox-fibrosis: Impact of TGFbeta1 on ROS generators, mediators and functional consequences. Redox biology. 2015;6:344-52
29. Refik Mas M, Comert B, Oncu K, Vural SA, Akay C, Tasci I. et al. The effect of taurine treatment on oxidative stress in experimental liver fibrosis. Hepatology research: the official journal of the Japan Society of Hepatology. 2004;28:207-15
30. Nieto N, Friedman SL, Cederbaum AI. Stimulation and proliferation of primary rat hepatic stellate cells by cytochrome P450 2E1-derived reactive oxygen species. Hepatology (Baltimore, Md). 2002;35:62-73
31. Curzio M, Esterbauer H, Di Mauro C, Cecchini G, Dianzani MU. Chemotactic activity of the lipid peroxidation product 4-hydroxynonenal and homologous hydroxyalkenals. Biological chemistry Hoppe-Seyler. 1986;367:321-9
Author contact
![]() Corresponding authors: Xiaoni Kong, PhD., Department of Liver Surgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 160 Pujian Road, Shanghai, China. 200127. Telephone: +862168383283; E-mail: xiaonikongedu.cn; xiaoni-kongcom or Qiang Xia, MD., E-mail: xiaqiangcom.cn or Yueqiu Gao, MD., Email: gaoyueqiucom.
Corresponding authors: Xiaoni Kong, PhD., Department of Liver Surgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 160 Pujian Road, Shanghai, China. 200127. Telephone: +862168383283; E-mail: xiaonikongedu.cn; xiaoni-kongcom or Qiang Xia, MD., E-mail: xiaqiangcom.cn or Yueqiu Gao, MD., Email: gaoyueqiucom.
Citation styles
APA
Yu, Y., Sun, X., Gu, J., Yu, C., Wen, Y., Gao, Y., Xia, Q., Kong, X. (2016). Deficiency of DJ-1 Ameliorates Liver Fibrosis through Inhibition of Hepatic ROS Production and Inflammation. International Journal of Biological Sciences, 12(10), 1225-1235. https://doi.org/10.7150/ijbs.15154.
ACS
Yu, Y.; Sun, X.; Gu, J.; Yu, C.; Wen, Y.; Gao, Y.; Xia, Q.; Kong, X. Deficiency of DJ-1 Ameliorates Liver Fibrosis through Inhibition of Hepatic ROS Production and Inflammation. Int. J. Biol. Sci. 2016, 12 (10), 1225-1235. DOI: 10.7150/ijbs.15154.
NLM
Yu Y, Sun X, Gu J, Yu C, Wen Y, Gao Y, Xia Q, Kong X. Deficiency of DJ-1 Ameliorates Liver Fibrosis through Inhibition of Hepatic ROS Production and Inflammation. Int J Biol Sci 2016; 12(10):1225-1235. doi:10.7150/ijbs.15154. https://www.ijbs.com/v12p1225.htm
CSE
Yu Y, Sun X, Gu J, Yu C, Wen Y, Gao Y, Xia Q, Kong X. 2016. Deficiency of DJ-1 Ameliorates Liver Fibrosis through Inhibition of Hepatic ROS Production and Inflammation. Int J Biol Sci. 12(10):1225-1235.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.