International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(11):1298-1308. doi:10.7150/ijbs.16569 This issue Cite
Research Paper
Histone Deacetylase Inhibitor Trichostatin a Promotes the Apoptosis of Osteosarcoma Cells through p53 Signaling Pathway Activation
Zhantao Deng1,2,3,*, Xiaozhou Liu1,*, Jiewen Jin2,3, Haidong Xu1, Qian Gao2,3, Yong Wang2,3, ![]() , Jianning Zhao1,
, Jianning Zhao1, ![]()
1. Department of Orthopedics, Jinling Hospital, School of Medicine, Nanjing University, Nanjing, Jiangsu, China.
2. Center for Translational Medicine, Nanjing University Medical School, Nanjing, Jiangsu, PR China.
3. Jiangsu Key Laboratory for Molecular Medicine, Nanjing University Medical School, Nanjing, PR China.
* These authors contributed equally to this work.
Received 2016-6-22; Accepted 2016-8-20; Published 2016-10-18
Citation:
Deng Z, Liu X, Jin J, Xu H, Gao Q, Wang Y, Zhao J. Histone Deacetylase Inhibitor Trichostatin a Promotes the Apoptosis of Osteosarcoma Cells through p53 Signaling Pathway Activation. Int J Biol Sci 2016; 12(11):1298-1308. doi:10.7150/ijbs.16569. https://www.ijbs.com/v12p1298.htm
Other stylesAbstract

Purpose: The purpose of this study was to investigate the profile of histone deacetylase (HDAC) activity and expression in osteosarcoma cells and tissues from osteosarcoma patients and to examine the mechanism by which a histone deacetylase (HDAC) inhibitor, Trichostatin A (TSA), promotes the apoptosis of osteosarcoma cells.
Methods: HDAC activity and histone acetyltransferase (HAT) activity were determined in nuclear extracts of MG63 cells, hFOB 1.19 cells and tissues from 6 patients with primary osteosarcoma. The protein expression of Class I HDACs (1, 2, 3 and 8) and the activation of the p53 signaling pathway were examined by Western blot. Cell growth and apoptosis were determined by 3-(4, 5-dimethyl-2-thiazolyl)-2H-tetrazolium bromide (MTT) assay and flow cytometry, respectively.
Results: Nuclear HDAC activity and class I HDAC expression were significantly higher in MG63 cells than in hFOB 1.19 cells, and a similar trend was observed in the human osteosarcoma tissues compared with the paired adjacent non-cancerous tissues. TSA significantly inhibited the growth of MG63 cells and promoted apoptosis in a dose-dependent manner through p53 signaling pathway activation.
Conclusion: Class I HDACs play a central role in the pathogenesis of osteosarcoma, and HDAC inhibitors may thus have promise as new therapeutic agents against osteosarcoma.
Keywords: osteosarcoma, HDAC, Trichostatin A, apoptosis, p53
Introduction
Osteosarcoma is the most common primary bone cancer, occurring most frequently in adolescents with a second incidence peak among individuals aged over 60 years[1, 2]. Despite great efforts that have been made in improving surgical techniques and new adjuvant chemotherapy, more than thirty percent of patients still die from pulmonary metastases within 5 years, and the prognosis remains poor [3-7]. Similar to other neoplasms, genetic factors play a fundamental role in the development of osteosarcoma. Epigenetic regulation of gene transcription, including acetylation, methylation and ubiquitination, takes part in the characteristic pathogenesis of osteosarcoma cells, such as anti-apoptosis, tumor cell proliferation, persistent recruitment and activation[8-10]. Among these epigenetic mechanisms, histone modification through reversible acetylation is a crucial event in gene expression[11]. Histone acetylation is controlled by two enzymes, histone acetyltransferase (HAT) and histone deacetylase (HDAC) [12-14]. Mammalian HDACs are classified into four classes, including class I HDACs (HDACs 1, 2, 3 and 8), class II HDACs (HDACs 4 to 7,9 and 10), class III HDACs (Sirtuin 1-7) and class IV HDAC (HDAC11) [14]. Class I HDACs are the only one who are exclusively in the nucleus and can directly interact with chromatin. Other classes of HDACs translocate to nucleus after activation and interact with class I HDACs to produce biological effect. Gene regulation by HDACs/HATs is complex because the inhibition of HDAC activity results both in the induction and repression of gene expression, depending on the cell types and cell lines [15-19]. Acetylation and deacetylation of histones can alter chromatin structure by influencing histone-DNA interactions[20]. Deacetylated histones have been found to be associated with cell growth, whereas hyperacetylated histones are associated with cell growth arrest, differentiation and/or apoptosis. The aberrant recruitment of the HDAC complex that represses the transcription of specific genes, such as tumor suppressor genes, may lead to transcriptional regulation[21].
Apoptosis is a fundamental cellular process that eliminates damaged or aged cells during the maintenance of cellular homeostasis and is central to embryonic development and organogenesis. The protein p53 plays a central role in cellular apoptosis [22, 23]. Functional inactivation of p53 is a fundamental step in tumorigenesis, and p53 acetylated at lysine382 is one of the activated forms of the p53 protein. When p53 protein is deacetylated by HDACs, its function to mediate apoptosis in damaged fcell is reduced, and its DNA binding activity and inspection of DNA damage is impaired[24]. Bax, a Bcl-2 family protein that resides in the cytosol and translocates to mitochondria after activation of apoptosis signaling, can be upregulated and transcriptionally activated by the p53 protein[25, 26]. The p53-Bax pathway is important in cellular apoptosis and proliferation.
HDAC inhibitors have been shown to have a curative effect in various cancer cells, including human breast, prostate, lung, ovary and colon cancer cells, which can be explained in part by their effect on cellular growth arrest, differentiation and apoptosis [27-30]. Trichostatin A (TSA), a specific HDAC inhibitor, was initially characterized as an anti-fungal drug and was later found to inhibit HDAC activity at nanomolar concentrations[31]. TSA has been shown to block the catalytic reaction and to induce a biological effect by chelating a zinc ion in the active site pocket through its hydroxamic acid group[32]. The anti-proliferative effect of TSA has been demonstrated in some malignancies. In osteosarcoma, it has been reported that TSA could promote cell apoptosis in vitro[33]. However, the mechanism by which TSA promotes apoptosis of osteosarcoma cells has not been well studied.
Therefore, in the present study, we aimed to investigate the HDAC/HAT situation in osteosarcoma cells and human osteosarcoma tissues and to explore the mechanism by which TSA promotes apoptosis of osteosarcoma cells.
Methods and Materials
Reagents
Bovine serum albumin (BSA, A4161) and protease inhibitor cocktail (P8340) were purchased from Sigma-Aldrich Co. Ltd. Fetal bovine serum (FBS, 10099-141), TSA (S1045) and Pifithrin-α (PFTα) (S2929) were purchased from Selleck Co. Ltd. RIPA lysis buffer (P0013B) was obtained from Beyotime Co. Ltd.
Cell Culture
The human osteosarcoma MG63 cell line (CRL-1427) and the human osteoblastic cell line hFOB 1.19 (CRL-11372) were purchased from the American Type Culture Collection (ATCC). MG63 cells were maintained in an atmosphere of 5% CO2 at 37°C in DMEM containing 4 mM L-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose, 1.0 mM sodium pyruvate, 10% FBS and 1% penicillin/ streptomycin. Human osteoblastic cells hFOB 1.19 were maintained in an atmosphere of 5% CO2 at 34°C in a 1:1 mixture of Ham's F12 Medium and DMEM, with 2.5 mM L-glutamine (without phenol red), 0.3 mg/ml G418 and FBS to a final concentration of 10%.
Patients and Tumor Samples
Paired osteosarcoma tumor tissues and adjacent normal tissues surgically resected for subsequent research were collected from 6 primary osteosarcoma patients during operations at Jinling Hospital (Nanjing, China). Surgically removed tissues were quickly frozen in liquid nitrogen prior to analysis. The research protocol was approved by the ethics committee of Nanjing University, Nanjing, China; all study participants provided written informed consent, and all experiments strictly followed the Declaration of Helsinki principles.
Preparation of nuclear extracts
Nuclear extracts from MG63 cells, hFOB 1.19 cells and tissues from osteosarcoma patients were obtained using the Nuclear Extract kit (Beyotime, P0027) according to the manufacturer's instructions. The protein concentration of each sample was measured using a BCA protein assay kit (Beyotime, P0011).
Measurement of HAT and HDAC activity
HAT and HDAC activity were measured using a HAT Activity Assay Kit (Biovision, K332-100) and a HDAC Colorimetric Assay Kit (Biovision, K331-100)). All procedures were performed according to the manufacturer's instructions. Absorbance was then measured using a micro-titer plate reader (Labsystems Multiskan, MS, Finland) at 440 nm for HAT activity and at 405 nm for HDAC activity. The HDAC activity was expressed as arbitrary fluorescence units. HAT and HDAC activity are expressed as the relative O.D. value per 50 µg protein.
Cell proliferation and cytotoxicity assay
Cells were seeded in 96-well plates and cultured with the indicated reagents. Subsequently, cell proliferation and cytotoxicity was determined using an MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] cell viability/cytotoxicity assay kit (Beyotime, C0009) according to the manufacturer's instructions. Absorbance was then measured using a micro-titer plate reader (Labsystems Multiskan, MS, Finland) at 570 nm.
Flow cytometry analysis
Cells were seeded into 6-well plates and cultured with indicated reagents. Subsequently, the cells were collected and stained with the Annexin V (ANXA 5)-FITC apoptosis detection kit (4A Biotech, FXP018-100) according to the manufacturer's instructions; cells were then analyzed using flow cytometry (FACS Calibur, BD Biosciences, San Jose, CA, USA).
Western blotting
The cells and tissues were prepared for Western blot following lysis in RIPA lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, and 1 mM PMSF) with a protein inhibitor cocktail for 30 min on ice. The lysates were centrifuged at 12,000 g for 10 min at 4℃. The supernatants were collected, and the protein concentrations were determined using a BCA protein assay kit (Beyotime, P0011). Equal amounts of total protein were separated using 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene fluoride membrane (Pall Co., East Hills, NY, USA). Western blotting was performed using the following primary antibodies: anti-HDAC1 (Cell Signaling Technology, 5356), anti-HDAC2 (Cell Signaling Technology, 2540), anti-HDAC3 (Cell Signaling Technology, 2632), anti-HDAC8 (Abcam, ab187139), anti-β-actin (Bioworld Technology, AP0733), anti-Bax (Cell Signaling Technology, 2772), anti-p53 (Cell Signaling Technology, 9282), and anti-p53 Acetyl 382 (Cell Signaling Technology, 2525). Subsequently, the following secondary antibodies were applied: horseradish peroxidase-conjugated anti-rabbit IgG (Bioworld Technology, BS13278) and horseradish peroxidase-conjugated anti-mouse IgG (Bioworld Technology, BS12478). After probing with a specific primary antibody and incubating with a horseradish peroxidase-conjugated secondary antibody, the protein bands were detected using a chemiluminescence detection system (Syngene, Cambridge, UK). Band density was analyzed using Gene Tools (Syngene, Cambridge, UK).
Statistical analysis
Data are presented as the mean ± standard error of the means (SEM). The differences between groups were analyzed by Brown-Forsythe test and, if appropriate, by one-way ANOVA followed by Dunnett's test or Bonferroni correction. A p value of less than 0.05 was considered significant. Statistical analysis of the data was performed using SPSS17.0 software (SPSS, Chicago, IL).
Results
Upregulation of HDAC activity in osteosarcoma cells and human osteosarcoma tissues
Increased levels of HDAC activity have been associated with multiple tumors, but osteosarcoma has not been investigated [34-36]. In this context, we investigated the HAT and HDAC activity in MG63, a human osteosarcoma cell line, compared with a normal human osteoblastic cell line, hFOB 1.19. The HDAC activity in the nuclear extracts of MG63 cells was 3.6-fold higher than that in hFOB 1.19 cells (Figure 1B). However, no significant difference was observed in the HAT activity (Figure 1A), thereby decreasing the HAT/HDAC activity ratio by 69.9% (Figure 1C). Interestingly, as shown in Figure 1D~F, a similar trend was observed in the 6 samples of human osteosarcoma tissues compared with that in the paired adjacent non-cancerous tissues (clinical characteristics are shown in Table 1). The HAT/HDAC activity ratio was 49.1% lower in the osteosarcoma tissues than that in the adjacent normal tissues (Figure 1F). Collectively, dramatic increase in HDAC activity could be observed in the nuclei of osteosarcoma cells, compared with that observed in the normal osteoblasts, which may influence the status of acetylation in the pathogenesis of osteosarcoma.
Upregulation of class I HDAC protein levels in osteosarcoma cells and human osteosarcoma tissues
Because HDAC activity in the nuclei of MG63 cells was dramatically higher than that in hFOB 1.19 cells because and class I HDACs are found exclusively in the nuclei, we next investigated class I HDAC protein expression in osteosarcoma cells and human osteosarcoma tissues. As shown in Figure 2A and B, HDAC1 and HDAC2 protein levels in MG63 cells were 2.31-fold and 1.76-fold higher, respectively, than that in hFOB 1.19 cells. The most significantly changed class I HDAC was HDAC3, with a protein level 3.64-fold higher in MG63 cells compared with that in hFOB 1.19 cells, whereas no significant difference was observed in HDAC8. We then investigated the class I HDAC protein levels in 6 osteosarcoma patients, whose clinical characteristics are shown in Table 1. Similar to the osteosarcoma cells, the class I HDAC protein levels of the human osteosarcoma tissues were overexpressed compared with those of the paired adjacent normal tissues. Although individual differences could be observed, HDAC1, HDAC2 and HDAC3 were still 2.66-, 1.73- and 3.82-fold higher than that observed in the adjacent normal tissues (Figure 2C, D). Collectively, upregulation of Class I HDAC protein levels (except HDAC8) could be observed in osteosarcoma cells and human osteosarcoma tissues, compared with normal osteoblasts and adjacent normal tissues, respectively.
Figure 1
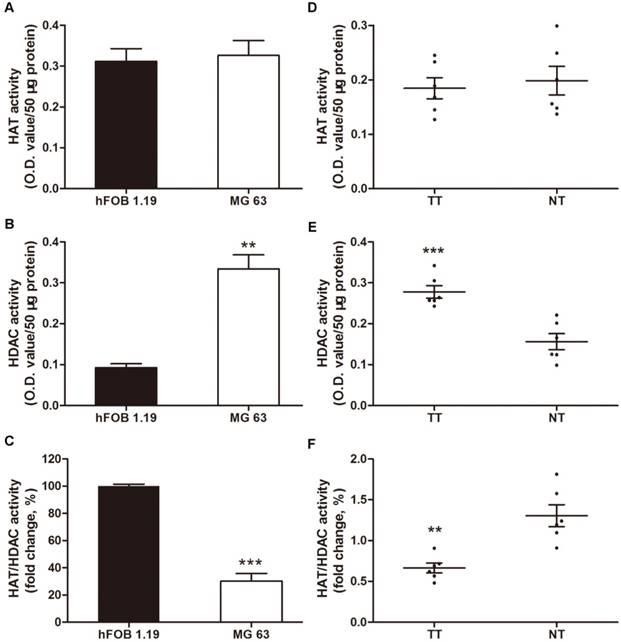
Total nuclear HDAC and HAT activity in osteosarcoma cells and human osteosarcoma tissues. (A) ~ (C) HAT, HDAC activity and ratio of HAT / HDAC activity in MG63 cell line compared with hFOB 1.19 cell line. Data are presented as means ± S.E.M. from 3 independent experiments. **, P < 0.01 vs. hFOB 1.19; ***, P < 0.001 vs. hFOB 1.19. (D) ~ (F) HAT, HDAC activity and ratio of HAT / HDAC activity in human osteosarcoma tissues compared with adjacent normal tissues. Data are presented as means ± S.E.M. N=6 patients. **, P < 0.01 vs. NT; ***, P<0.001vs. NT. TT, tumor tissues; NT, normal tissues.
Table 1
Clinical characteristics of all the 6 patients with osteosarcoma
| Case | Karnofsky performance status score | Gender | Age/year | Tumor site | Tumor size(cm) | Enneking's surgical staging | Pathological fracture | Surgical procedure | Neoadjuvant chemotherapy | Local recurrence | Metastasis |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient1 | 40 | F | 11 | Extremities | 6.5*13.5 | III | no | limb salvage | yes | no | yes |
| Patient2 | 90 | M | 17 | Extremities | 8.8*6.1 | II | no | limb salvage | yes | no | no |
| Patient3 | 50 | F | 16 | Extremities | 8.8*13 | II | no | limb salvage | yes | no | no |
| Patient4 | 90 | F | 33 | Extremities | 11*4.8*3.7 | II | no | limb salvage | yes | no | no |
| Patient5 | 50 | F | 8 | Extremities | 6.3*10.5 | III | no | limb salvage | yes | no | yes |
| Patient6 | 90 | F | 53 | Extremities | 5*3.9*5.3 | II | no | limb salvage | yes | no | no |
F=female, M=male
Figure 2
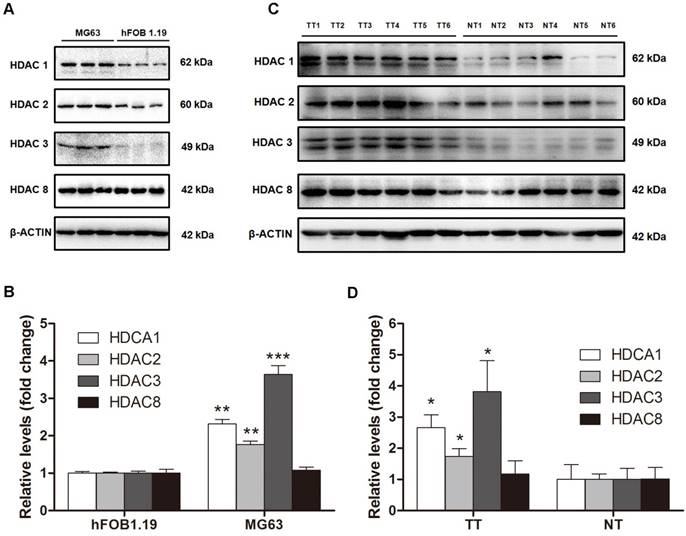
The protein levels of class I HDACs in osteosarcoma cells and human osteosarcoma tissues. (A) The protein levels of class I HDACs in MG63 cell line comparied with that in hFOB 1.19 cell line. (B) The density of the western blots bands shown in (A) was quantified using Gene Tools software. Data are presented as means ± S.E.M. from 3 independent experiments. **, P < 0.01 vs. hFOB 1.19; ***, P < 0.001 vs. hFOB 1.19. (C) The protein levels of class I HDACs in human osteosarcoma tissues comparied with that in adjacent normal tissues. (D) The density of the western blots bands shown in (C) was quantified using Gene Tools software. N=6 patients. *, P < 0.05 vs. NT. TT, tumor tissue; NT, normal tissue.
TSA promotes the apoptosis of osteosarcoma cells in a dose-dependent manner
According to the results above, we inferred that HDACs play a critical role in the pathogenesis of osteosarcoma, and we hypothesized that an HDAC inhibitor might have curable effects on osteosarcoma. To study the effect of TSA on osteosarcoma cells, we firstly examined the cell viability of MG63 cells treated with various concentrations of TSA (0, 50, 100 and 200 nM). The results demonstrated that TSA could dramatically decrease the expression and activity of Class I HDACs in osteosarcoma cells (Figure 3A, B, E). However, since the baseline of Class I HDACs expression and activity in normal osteoblast was much lower than that in osteosarcoma cells, TSA showed minor HDAC suppression effect on normal osteosarcoma (Figure C, D, F). It was also demonstrated that TSA decreased cell viability in a dose-dependent manner. The largest decrease in cell viability, 45.8%, was observed with the addition of 200 nM TSA (Figure 4A). To determine whether the apoptosis of osteosarcoma cells was involved in the TSA-induced decrease in cell viability, we examined the effect of TSA treatment on MG63 cell apoptosis using flow cytometry. Figure 4B and C illustrate that TSA could dramatically increase the apoptotic rate of cells in a dose-dependent manner. The apoptotic rate in cells treated with 200 nM TSA for 24 h was 34.9% higher than that observed in control cells.
TSA promotes the apoptosis of osteosarcoma cells through p53 activation
Because p53 is not only one of the downstream targets of HDACs but also plays a critical role in apoptosis, we next hypothesized that TSA promotes osteosarcoma cell apoptosis through p53 signaling pathway activation. Western blot analysis demonstrated that TSA treatment apparently upregulated the levels of Bax and acetyl-p53, but had only a slight effect on the expression of total p53, thereby significantly raising the acetyl-p53/total p53 ratio (Figure 5A, B). Interestingly, we found that when the TSA concentration was increased from 50nM to 100nM, the expression of Bax increased much more than acetyl-p53. Although the expression of p53-aacetyl 382 could be mainly control by HDACs, the expression of Bax could be regulated by other signaling pathway except HDACs signaling pathway, such as PI3K / Akt signaling pathway[37, 38].
Figure 3
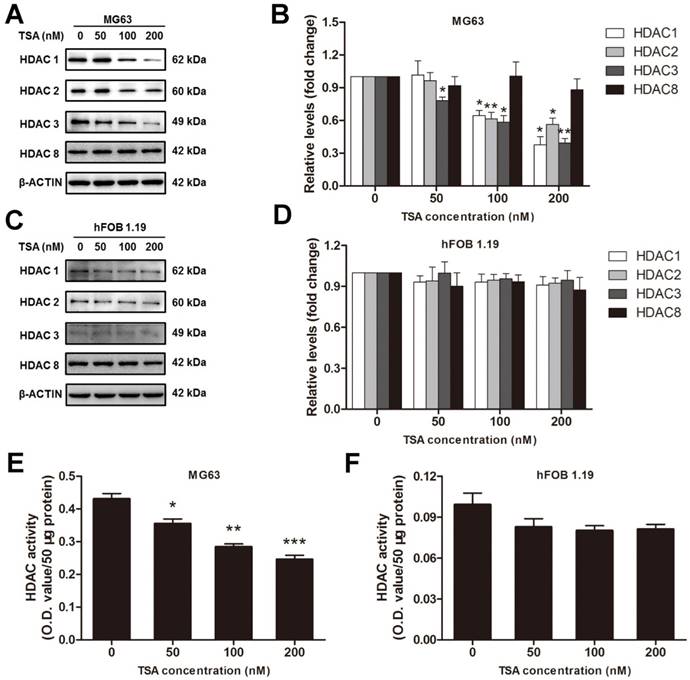
The HDACs inhibition effects of TSA on osteosarcoma cells and normal osteoblasts. (A) (C) The expression of Class I HDACs after MG63 cells or hFOB 1.19 cells were exposed to TSA at various concentrations (0, 50, 100, 200 nM) for 24h. (B) (D) The density of the western blots bands shown in (A) (C) were quantified using Gene Tools software. Data are presented as means ± S.E.M. from 3 independent experiments. *, P < 0.05; **, P < 0.01 vs. concentration 0. (E) (F) The activity of HDACs in nuclei after MG63 cells or hFOB 1.19 cells were exposed to TSA at various concentrations (0, 50, 100, 200 nM) for 24h.
Figure 4
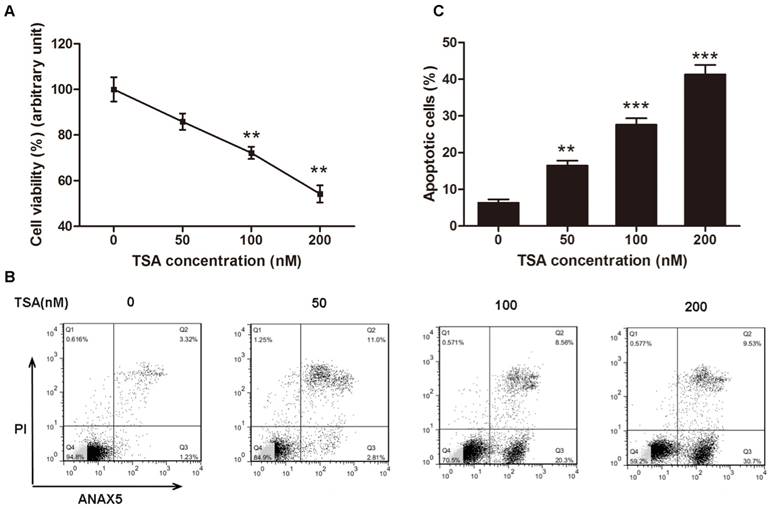
TSA promoted osteosarcoma cell apoptosis in dose-dependent manner. (A) Cell viability after TSA treatment. MG63 cells were exposed to TSA at various concentrations (0, 50, 100, 200 nM) for 24h. Data are presented as means ± S.E.M. from 3 independent experiments. **, P < 0.01 vs. 0 nM. (B) Flow cytometry analysis of ANXA5 and propidiumiodide staining of MG63 cells cultured with various concentrations (0, 50, 100, 200 nM) of TSA for 24h. (C) Quantification analysis of apoptotic cells in (B) (both upper- and lower- right quadrants in representative dot plots as shown). Data are presented as means ± S.E.M. from 3 independent experiments. **, P < 0.01 vs. 0 nM; ***, P < 0.001 vs. 0 nM.
To further confirm that the p53 signaling pathway was involved in the TSA-induced apoptosis of osteosarcoma cells, we used PFTα, a p53 inhibitor, to suppress p53-related cell apoptosis. The results indicated that cell viability was increased by 19.3% and 34.8% with 5μM and 10μM PFTα co-treatment, respectively, compared with that observed in MG63 cells with TSA treatment alone (Figure 5C). Similarly, PFTα reversed the pro-apoptotic effect of TSA, and the observed osteosarcoma cell apoptosis rate decreased by 15.8% and 20.3% with PFTα co-treatment, compared to the rate with TSA treatment alone (Figure 5D, E).
Collectively, these results demonstrate that TSA promotes the apoptosis of osteosarcoma cells through p53 signaling pathway activation.
Discussion
The acetylation level of histones is dynamically regulated by HATs and HDACs[39]. An inappropriate histone acetylation state can alter the pattern of cell death and lead to abnormal outgrowth, which promotes neoplastic transformation[40]. It has been shown that histone acetylation modulates the expression of angiogenic factors and subsequent angiogenesis[41]. The upregulation of HDAC1 increases vascular endothelial growth factor (VEGF) expression and suppresses p53 and Von Hippel-Lindau (VHL) gene expression both in vitro and in vivo, which results in the induction of angiogenesis[42]. These findings suggest that HDACs play an important role in angiogenesis, which is closely related to carcinogenesis. As an effective HDAC inhibitor, many other studies have been reported positive effects of TSA on normal osteoblasts, especially in promoting osteoblast differentiation and maturation [43-47]. Moreover, TSA could promoted alkaline phosphatase expression, which is vital in the function of osteoblast[48]. On the other hand, TSA has been reported to inhibit cell growth and induce apoptosis of various cancer cells, including osteosarcoma[33]. However, little is known about the mechanism by which TSA inhibits osteosarcoma cell growth and induces apoptosis, and few studies focus on acetylation state in the pathogenesis of osteosarcoma. Our study demonstrates that class I HDACs play a critical role in the pathogenesis of osteosarcoma in vivo and in vitro and that TSA promotes osteosarcoma cell apoptosis through p53 signaling pathway activation. These findings reveal that acetylation, which is mainly controlled by the balance between HDACs and HATs, might be a critical regulatory mechanism in the pathogenesis of osteosarcoma, along with other forms of epigenetic regulation, such as methylation and microRNA regulation [49-52].
TSA has also been shown to disturb the tumor cell cycle, primarily through growth arrest in the G1 and/or G2/M phase and the induction of cancer cell apoptosis[33, 53, 54]. The arrest in G1 phase after TSA treatment is thought to result from the increased expression of p21/WAF1[55]. Although TSA markedly reduced the osteosarcoma cells in G1 phase and concomitantly increased those in G2/M phase, the precise mechanism causing arrest in the G2/M phase following TSA treatment in cancer cells still remains unclear[33].
Figure 5
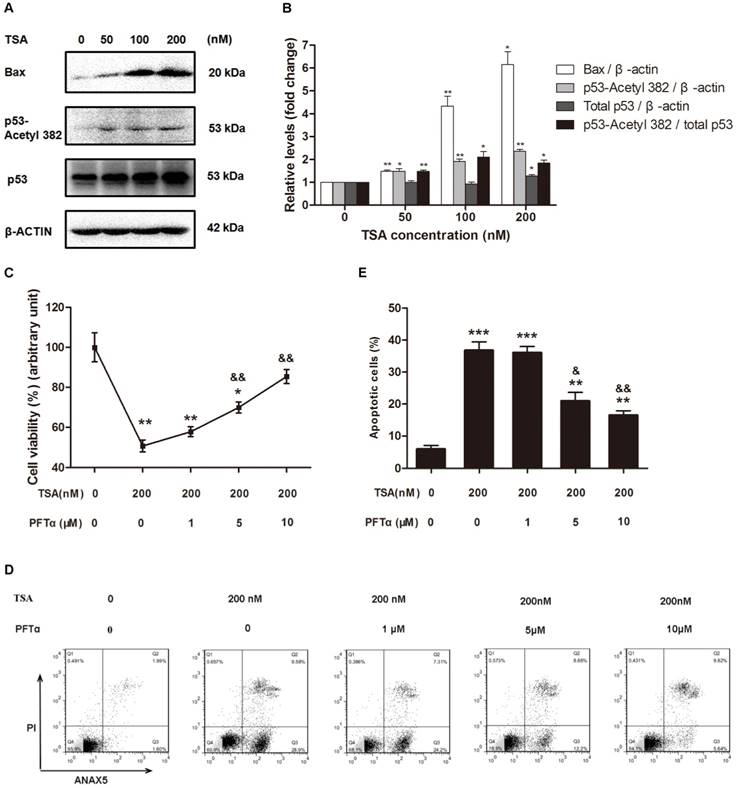
TSA promoted osteosarcoma cell apoptosis through p53 signal pathway activation. (A) Western blots performed after MG63 cells were incubated with TSA at various concentrations (0, 50, 100, 200 nM) for 24h. (B) The density of the western blots bands shown in (A) was quantified using Gene Tools software. Data are presented as means ± S.E.M. from 3 independent experiments. *, P < 0.05 vs. 0 nM; **, P < 0.01 vs. 0 nM. (C) Cell viability was measured after TSA and PFTα co-treatment. MG63 were exposed to TSA and various concentrations of PFTα (1, 5, 10μM) co-treatment compared with TSA treatment alone for 24h. Data are presented as means ± S.E.M. from 3 independent experiments. *, P < 0.05 vs. MG63 cells without TSA or PFTα treatment; **, P < 0.01 vs. MG 63 cells without TSA or PFTα treatment; &&, P < 0.01 vs. MG63 cells with TSA treatment alone. (D) Flow cytometry analysis of ANXA5 and propidiumiodide staining of MG63 cells with TSA and various concentrations of PFTα (1, 5, 10μM) co-treatment, compared with TSA treatment alone. (E) Quantification analysis of apoptotic cells in (D) (both upper- and lower- right quadrants in representative dot plots as shown). Data are presented as means ± S.E.M. from 3 independent experiments. **, P < 0.01 vs. MG63 cells without TSA or PFTα treatment; ***, P < 0.001 vs. MG63 cells without TSA or PFTα treatment; &, P < 0.05 vs. MG63 cells with TSA treatment alone. &&, P < 0.01 vs. MG63 cells with TSA treatment alone.
Along with class I HDACs, it was also important to examine the function of other HDACs in the pathological mechanisms of osteosarcoma. HDACs are divided into four groups based on their structural and functional similarities; class I includes HDACs 1, 2, 3 and 8; class II includes HDACs 4-7, 9 and 10; class III includes SIRTs 1-7; and class IV includes only HDAC11[56-58]. Among these HDACs, class I HDACs are studied the most widely, whereas the second most-studied HDAC class is the SIRT family. It has been reported that microRNA-204 could inhibit proliferation, migration, invasion and epithelial-mesenchymal transition in osteosarcoma cells via targeting SIRT1[59]. In addition, antitumor therapies such as melatonin and emodin have shown antitumor effects by regulating SIRT1[60, 61]. Moreover, two specific activator of SIRT1, resveratrol and isonicotinamide, both showed the ability to promote pro-apoptosis effect on osteosarcoma cells, which is similar to the effects of TSA [62], Although SIRT1 is also an important deacetylase, but it seems to be have opposite effect to Class I HDACs, which needs further studies to understand the network between Class I HDACs and SIRT1. However, Similar to class I HDACs, SIRT3 could modulate Bak- and Bax-dependent apoptosis in various cells[63]. In conclusion, it appears that different classes of HDACs construct a complex network to regulate gene expression and disease processes, and further studies should pay closer attention to the connection between different HDACs.
In the present study, we also investigated the expression of class I HDACs in 6 paired human osteosarcoma tissues and the adjacent normal tissues, whereas most studies focus only on in vitro experiments. Similar to the results in MG63 cells compared with hFOB 1.19 cells, HDACs 1, 2 and 3 were overexpressed in the human osteosarcoma tissues compared with the paired adjacent normal tissues. We also show the clinical characteristics of all 6 patients in Table 1. It has been reported that invasiveness, inflammation and other disease outcomes are related to HDAC levels and activity [39, 64-66]. Invasiveness is one of the vital characteristics of osteosarcoma and it was reported to be related to matrix metalloproteinases-2 (MMP-2) and matrix metalloproteinases-9 (MMP-9)[67, 68]. Moreover, MMPs have been indicated that it could be regulated by HDACs. Previous study also demonstrated that TSA repressed the activity of MMP-2 and thereby alleviated the invasive ability of tumor cells[69]. However, another study shows that increase in HDAC activity could suppress the invasiveness of cancer cells through deacetylating histone H3 and cooperating with matrix metalloproteinase 9 (MMP-9). Collectively, there is still controversial that whether HDAC inhibition could repress the invasion of osteosarcoma cells[70]. Moreover, in some cases, SIRT1 has been used as a molecular marker in diagnosis and detection of osteosarcoma metastasis[71]. Although our sample size was not large enough to evaluate the relationship between class I HDAC levels and other clinical characteristics, it is still interesting to consider that HDAC levels may be related to Karnofsky performance status score, tumor size, local recurrence and other outcomes. An understanding of the correlation between HDAC levels and these characteristics requires further studies with larger sample sizes.
Collectively, our findings demonstrated that class I HDACs were overexpressed both in human osteosarcoma tissues and in an osteosarcoma cell line and that an HDAC inhibitor, TSA, promoted osteosarcoma cell apoptosis through p53 signaling pathway activation. Our findings also indicate that epigenetic mechanisms, particularly acetylation, play a critical role in the pathogenesis of osteosarcoma. However, further in vivo studies are required to confirm the effectiveness and safety of TSA in the treatment of human osteosarcoma.
Conclusion
Our study demonstrated that Class I HDACs play a vital role in the pathogenesis of osteosarcoma. Moreover, HDAC inhibitors could promote the apoptosis of osteosarcoma cells and may thus have promise as new therapeutic agents against osteosarcoma.
Acknowledgements
We thank all of the osteosarcoma patients and family members who participated in the study. And this study was supported by Clinical Science and Technology Project Foundation of Jiangsu Province (BL2012002), Scientific Research Project of Nanjing Province (201402007), Natural Science Foundation of Jiangsu Province (BK20161385), Foundation of Jinling Hospital (2015003), P. R. China.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mirabello L, Scd R J T, † S A S M. Osteosarcoma incidence and survival rates from 1973 to 2004. Cancer. 2009;115:1531-43
2. Ottaviani G, Jaffe N. The Epidemiology of Osteosarcoma. Cancer Treatment & Research. 2009;152:3-13
3. Lewis V O. What's new in musculoskeletal oncology. Journal of Bone & Joint Surgery American Volume. 2009;91:1546-56
4. Meyers P A, Schwartz C L, Mark K, Kleinerman E S, Donna B, Bernstein M L. et al. Osteosarcoma: a randomized, prospective trial of the addition of ifosfamide and/or muramyl tripeptide to cisplatin, doxorubicin, and high-dose methotrexate. Journal of Clinical Oncology Official Journal of the American Society of Clinical Oncology. 2005;23:2004-11
5. Cho Y, Jung G H, Chung S H. et al. Long-Term Survivals of Stage IIB Osteosarcoma: A 20-Year Experience in a Single Institution. Clinics in Orthopedic Surgery. 2011;3:48-54
6. Tsuchiya H, Tomita K, Mori Y, Asada N, Morinaga T, Kitano S. et al. Caffeine-assisted chemotherapy and minimized tumor excision for nonmetastatic osteosarcoma. Anticancer Research. 1998;18:657-66
7. Bölling T, Schüller P, Distelmaier B, Schuck A, Ernst I, Gosheger G. et al. Perioperative high-dose rate brachytherapy using a bendy applicator (flab): treatment results of 74 patients. Anticancer Research. 2008;28:3885-90
8. Kikuchi K. Epigenetic regulation of alkaline phosphatase in human cells of the osteoblastic lineage. Bone. 2011;49:830-8
9. Cui J, Wang W, Li Z, Zhang Z, Bei W, Li Z. Epigenetic changes in osteosarcoma. Bulletin Du Cancer. 2011;98:E62-E8
10. Li B, Ye Z. Epigenetic alterations in osteosarcoma: promising targets. Molecular Biology Reports. 2014;41:3303-15
11. Jenuwein T, Allis C D. Translating the histone code. Science. 2001;293:1074-80
12. Hassig C A, Schreiber S L. Nuclear histone acetylases and deacetylases and transcriptional regulation: HATs off to HDACs. Current Opinion in Chemical Biology. 1997;1:300-8
13. Kuo M H, Allis C D. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays News & Reviews in Molecular Cellular & Developmental Biology. 1998;20:615-26
14. Grozinger C M, Schreiber S L. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chemistry & Biology. 2002;9:3-16
15. Kruh J. Effects of sodium butyrate, a new pharmacological agent, on cells in culture. Molecular & Cellular Biochemistry. 1982;42:65-82
16. Mariadason J M, Corner G A, Augenlicht L H. Genetic reprogramming in pathways of colonic cell maturation induced by short chain fatty acids: comparison with trichostatin A, sulindac, and curcumin and implications for chemoprevention of colon cancer. Cancer Research. 2000;60:4561-72
17. Chambers A E, Banerjee S, Chaplin T, Dunne J, Debernardi S, Joel S P. et al. Histone acetylation-mediated regulation of genes in leukaemic cells. European Journal of Cancer. 2003;39:1165-75
18. George R, Rapha?L M, Chin-Yo L, Stefanie D, David I, Tomi I. et al. Multiple mechanisms induce transcriptional silencing of a subset of genes, including oestrogen receptor alpha, in response to deacetylase inhibition by valproic acid and trichostatin A. Oncogene. 2005;24:4894-907
19. Shuang C, Pikaard C S. Transcript profiling in Arabidopsis reveals complex responses to global inhibition of DNA methylation and histone deacetylation. Journal of Biophysical. 2005;280:796-804
20. Wolffe A P, Guschin D. Review: Chromatin Structural Features and Targets That Regulate Transcription. Journal of Structural Biology. 2000;129:102-22
21. Marks P A, Richon V M, Breslow R, Rifkind R A. Histone deacetylase inhibitors as new cancer drugs. Current Opinion in Oncology. 2001;13:477-83
22. Fei P, El-Deiry W S. P53 and radiation responses. Oncogene. 2003;22:5774-83
23. Wu Y, Xing D, Liu L, Gao B. Regulation of Bax activation and apoptotic response to UV irradiation by p53 transcription-dependent and -independent pathways. Cancer Letters. 2008;271:231-9
24. Luo J, Nikolaev A Y, Imai S, Chen D, Su F, Shiloh A. et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137-48
25. Chipuk J E, Kuwana T, Bouchier-Hayes L, Droin N M, Newmeyer D D, Schuler M. et al. Direct Activation of Bax by p53 Mediates Mitochondrial Membrane Permeabilization and Apoptosis. Science. 2004;303:1010-4
26. Pang W J, Xiong Y, Wang Y, Tong Q, Yang G S. Sirt1 attenuates camptothecin-induced apoptosis through caspase-3 pathway in porcine preadipocytes. Experimental Cell Research. 2013;319:670-83
27. Vigushin D M, Ali S, Pace P E, Mirsaidi N, Ito K, Adcock I. et al. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clinical Cancer Research An Official Journal of the American Association for Cancer Research. 2001;7:971-6
28. Donadelli M, Costanzo C, Faggioli L, Scupoli M T, Moore P S, Bassi C. et al. Trichostatin A, an inhibitor of histone deacetylases, strongly suppresses growth of pancreatic adenocarcinoma cells. Molecular Carcinogenesis. 2003;38:59-69
29. Kim Y, Park H Y, Han I, Kwon H J, Woods A, Oh E S. Decreased syndecan-2 expression correlates with trichostatin-A induced-morphological changes and reduced tumorigenic activity in colon carcinoma cells. Oncogene. 2003;22:826-30
30. Chiba T, Yokosuka O, Fukai K, Kojima H, Tada M, Arai M. et al. Cell growth inhibition and gene expression induced by the histone deacetylase inhibitor, trichostatin A, on human hepatoma cells. Oncology. 2004;66:481-91
31. Yoshida M, Horinouchi S, Beppu T. Trichostatin A and trapoxin: Novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays. 1995;17:423-30
32. Finnin M S, Donigian J R, Cohen A, Richon V M, Rifkind R A, Marks P A. et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature International Weekly Journal of Science. 1999;401:188-93
33. Dong-Dong C, Qing-Cheng Y, Zhi-Chang Z, Cui-Xia Y, Yi-Wen L. Antitumor activity of histone deacetylase inhibitor trichostatin A in osteosarcoma cells. Asian Pacific Journal of Cancer Prevention Apjcp. 2012;13:1395-9
34. Marks P, Rifkind R A, Richon V M, Breslow R, Miller T, Kelly W K. Histone deacetylases and cancer: causes and therapies. Rev.latinoam.bioet. 2001;1:54-61
35. Lane A A, Chabner B A. Histone deacetylase inhibitors in cancer therapy. Journal of Clinical Oncology Official Journal of the American Society of Clinical Oncology. 2009;27:5459-68
36. Glozak M, Seto E. Histone deacetylases and cancer. Oncogene. 2012;6:579-89
37. Downward J. PI 3-kinase, Akt and cell survival. Seminars in Cell & Developmental Biology. 2004;15:177-82
38. Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A. et al. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat. 2007;10:13-29
39. Ghizzoni M, Haisma H J, Maarsingh H, Dekker F J. Histone acetyltransferases are crucial regulators in NF-κB mediated inflammation. Drug Discovery Today. 2011;16:504-11
40. Loidl P, Gr?Bner P. Postsynthetic acetylation of histones during the cell cycle: a general function for the displacement of histones during chromatin rearrangements. Nucleic Acids Research. 1987;15:8351-66
41. Williams R J. Trichostatin A, an inhibitor of histone deacetylase, inhibits hypoxia-induced angiogenesis. Expert Opinion on Investigational Drugs. 2001;10:1571-3
42. Kim M S, Kwon H J, Lee Y M, Baek J H, Jang J E, Lee S W. et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nature Medicine. 2001;7:437-43
43. Schroeder T M, Westendorf J J. Histone Deacetylase Inhibitors Promote Osteoblast Maturation †. Journal of Bone & Mineral Research the Official Journal of the American Society for Bone & Mineral Research. 2005;20:2254-63
44. Schroeder T M, Nair A K, Staggs R, Lamblin A F, Westendorf J J. Gene profile analysis of osteoblast genes differentially regulated by histone deacetylase inhibitors. Bmc Genomics. 2007;8:1993-6
45. Dudakovic A, Evans J M, Li Y, Middha S, Mcgeelawrence M E, van Wijnen A J. et al. Histone deacetylase inhibition promotes osteoblast maturation by altering the histone H4 epigenome and reduces Akt phosphorylation. Journal of Biological Chemistry. 2013;288:28783-91
46. Hu X, Zhang X, Dai L, Zhu J, Jia Z, Wang W. et al. Histone deacetylase inhibitor trichostatin A promotes the osteogenic differentiation of rat adipose-derived stem cells by altering the epigenetic modifications on Runx2 promoter in a BMP signaling-dependent manner. Stem Cells & Development. 2013;22:248-55
47. Park D S, Kim H K, Park K S, Yoon T R. Trichostatin A enhances osteogenic differentiation through activation of ERK pathways in mouse bone marrow multipotent stromal cells. Fetal ovine model for in-situ esophagus tissue engineering. 2014;11:131-6
48. Azechi T, Kanehira D, Kobayashi T, Sudo R, Nishimura A, Sato F. et al. Trichostatin A, an HDAC class I/II inhibitor, promotes Pi-induced vascular calcification via up-regulation of the expression of alkaline phosphatase. Journal of Atherosclerosis & Thrombosis. 2013;20:538-47
49. Jones K B, Zaidoun S, Sara D M, Marco G, Eugenio G, Nuovo G J. et al. miRNA signatures associate with pathogenesis and progression of osteosarcoma. Cancer Research. 2012;72:1865-77
50. Ulaner G A, Vu T H, Li T, Hu J F, Yao X M, Yang Y. et al. Loss of imprinting of IGF2 and H19 in osteosarcoma is accompanied by reciprocal methylation changes of a CTCF-binding site. Human Molecular Genetics. 2003;12:535-49
51. Bekim S, Maisa Y, Khaldoun A R, Georges M, Maria Z, Squire J A. In vitro analysis of integrated global high-resolution DNA methylation profiling with genomic imbalance and gene expression in osteosarcoma. Plos One. 2008;3:e2834
52. Kobayashi E, Hornicek F J, Duan Z. MicroRNA Involvement in Osteosarcoma. Sarcoma. 2012. 2012
53. Strait K A, Dabbas B, Hammond E H, Warnick C T, Iistrup S J, Ford C D. Cell cycle blockade and differentiation of ovarian cancer cells by the histone deacetylase inhibitor trichostatin A are associated with changes in p21, Rb, and Id proteins. Molecular Cancer Therapeutics. 2002;1:1181-90
54. Hirose T, Sowa Y S, Saito S, Yasuda C, Shindo N, Furuichi K. et al. p53-independent induction of Gadd45 by histone deacetylase inhibitor: coordinate regulation by transcription factors Oct-1 and NF-Y. Oncogene. 2003;22:7762-73
55. Sowa Y, Orita T, Minamikawa S, Nakano K, Mizuno T, Nomura H. et al. Histone Deacetylase Inhibitor Activates the WAF1/Cip1 Gene Promoter through the Sp1 Sites ☆. Biochemical & Biophysical Research Communications. 1997;241:142-50
56. Gibney E R, Nolan C M. Epigenetics and gene expression. Heredity. 2010;105:4-13
57. Peserico A, Simone C. Physical and functional HAT/HDAC interplay regulates protein acetylation balance. Biomed Research International. 2011;2011:20-34
58. Peter V N, Janja M, Barbara O. Epigenetic mechanisms in bone. Clinical Chemistry & Laboratory Medicine. 2014;52:589-608
59. Shi Y, Huang J, Zhou J, Liu Y, Fu X, Li Y. et al. MicroRNA-204 inhibits proliferation, migration, invasion and epithelial-mesenchymal transition in osteosarcoma cells via targeting Sirtuin 1. Oncology Reports. 2015;34:399-406
60. Cheng Y, Cai L, Peng J, Wang J, Chao G, Feng H. et al. SIRT1 inhibition by melatonin exerts antitumor activity in human osteosarcoma cells. European Journal of Pharmacology. 2013;715:219-29
61. Qu W, Wang Y, Wu Q, Liu J, Hao D. Emodin inhibits HMGB1-induced tumor angiogenesis in human osteosarcoma by regulating SIRT1. International Journal of Clinical & Experimental Medicine. 2015;8:15054-64
62. Yan L, Bäckesjö C M, Haldosén L A, Lindgren U. Resveratrol inhibits proliferation and promotes apoptosis of osteosarcoma cells. European Journal of Pharmacology. 2009;609:13-8
63. Manish V, Nataly S, Pastorino J G. Sirtuin-3 modulates Bak- and Bax-dependent apoptosis. Journal of Cell Science. 2013;126:274-88
64. Hawtree S, Muthana M, Wilkinson J M, Wilson A G. FRI0360 Histone Deacetylase One Contributes to the Auto-Aggressive Phenotype of Rheumatoid Arthritis Synovial Fibroblasts. Annals of the Rheumatic Diseases. 2014;73:517 -
65. Li-Teh L, Hui-Chiu C, Lien-Chai C, Wen-Chun H. Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2 activation and cancer cell invasion. Cancer Research. 2003;63:3069-72
66. Ouaïssi M, Silvy F, Loncle C, Ferraz d S D, Martins A C, Martinez E. et al. Further characterization of HDAC and SIRT gene expression patterns in pancreatic cancer and their relation to disease outcome. Plos One. 2014;9:e108520-e
67. Bjørnland K, Flatmark K, Pettersen S, Aaasen A O, Fodstad Ø, Mælandsmo G M. Matrix Metalloproteinases Participate in Osteosarcoma Invasion. Journal of Surgical Research. 2005;127:151-6
68. Purcell W T, Rudek M A, Hidalgo M. Development of matrix metalloproteinase inhibitors in cancer therapy. Journal of the National Cancer Institute. 2001;93:178-93
69. Liu L T, Chang H C, Chiang L C, Hung W C. Histone deacetylase inhibitor up-regulates RECK to inhibit MMP-2 activation and cancer cell invasion. Cancer Research. 2003;63:3069-72
70. Li P T, Tsai Y J, Lee M J, Chen C T. Increased Histone Deacetylase Activity Involved in the Suppressed Invasion of Cancer Cells Survived from ALA-Mediated Photodynamic Treatment. International Journal of Molecular Sciences. 2015;16:23994-4010
71. Ying M, Liu B, Wang Q, Zhang N, Cao J, Ye Z. Use of silent mating type information regulation 2 homolog 1 (Sirt-1) protein, as molecular marker in diagnosis and detection of osteosarcoma metastasis. UNIV THIRD MILITARY MEDICAL PLA SECOND (UYTH-Non-standard).
Author contact
![]() Corresponding authors: Dr. Yong Wang, Jiangsu Key Laboratory for Molecular Medicine, School of Medicine, Nanjing University, No. 22, Hankou Road, Nanjing 210002, Jiangsu, China; Dr. Jianning Zhao, Department of Orthopedics, Jinling Hospital, School of Medicine, Nanjing University, No. 305, Zhongshan East Road, Nanjing 210002, Jiangsu, China. E-mail: yongwangedu.cn; zhaojianning.0207com
Corresponding authors: Dr. Yong Wang, Jiangsu Key Laboratory for Molecular Medicine, School of Medicine, Nanjing University, No. 22, Hankou Road, Nanjing 210002, Jiangsu, China; Dr. Jianning Zhao, Department of Orthopedics, Jinling Hospital, School of Medicine, Nanjing University, No. 305, Zhongshan East Road, Nanjing 210002, Jiangsu, China. E-mail: yongwangedu.cn; zhaojianning.0207com
Citation styles
APA
Deng, Z., Liu, X., Jin, J., Xu, H., Gao, Q., Wang, Y., Zhao, J. (2016). Histone Deacetylase Inhibitor Trichostatin a Promotes the Apoptosis of Osteosarcoma Cells through p53 Signaling Pathway Activation. International Journal of Biological Sciences, 12(11), 1298-1308. https://doi.org/10.7150/ijbs.16569.
ACS
Deng, Z.; Liu, X.; Jin, J.; Xu, H.; Gao, Q.; Wang, Y.; Zhao, J. Histone Deacetylase Inhibitor Trichostatin a Promotes the Apoptosis of Osteosarcoma Cells through p53 Signaling Pathway Activation. Int. J. Biol. Sci. 2016, 12 (11), 1298-1308. DOI: 10.7150/ijbs.16569.
NLM
Deng Z, Liu X, Jin J, Xu H, Gao Q, Wang Y, Zhao J. Histone Deacetylase Inhibitor Trichostatin a Promotes the Apoptosis of Osteosarcoma Cells through p53 Signaling Pathway Activation. Int J Biol Sci 2016; 12(11):1298-1308. doi:10.7150/ijbs.16569. https://www.ijbs.com/v12p1298.htm
CSE
Deng Z, Liu X, Jin J, Xu H, Gao Q, Wang Y, Zhao J. 2016. Histone Deacetylase Inhibitor Trichostatin a Promotes the Apoptosis of Osteosarcoma Cells through p53 Signaling Pathway Activation. Int J Biol Sci. 12(11):1298-1308.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.