Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Zta and Rta synergistically...
3. Host factors contributing to...
4. The viral self-regulation of...
5. The pathogenic role of lytic...
6. Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(11):1309-1318. doi:10.7150/ijbs.16564 This issue Cite
Review
Epstein-Barr virus lytic reactivation regulation and its pathogenic role in carcinogenesis
Hongde Li1,2,3, Sufang Liu4, Jianmin Hu1,2,3, Xiangjian Luo1,2,3, Namei Li1,2,3, Ann M.Bode5, Ya Cao1,2,3 ![]()
1. Key Laboratory of Carcinogenesis and Invasion, Chinese Ministry of Education, Xiangya Hospital, Central South University, Changsha 410078, China
2. Cancer Research Institute, Xiangya School of Medicine, Central South University, Changsha 410078, China
3. Key Laboratory of Carcinogenesis, Chinese Ministry of Health, Changsha 410078, China
4. Division of Hematology, Institute of Molecular Hematology, the Second Xiangya Hospital, Central South University, Changsha, Hunan 410011, China
5. The Hormel Institute, University of Minnesota, Austin, MN 55912, USA
Received 2016-6-22; Accepted 2016-8-20; Published 2016-10-18
Abstract

Epstein-Barr virus (EBV) has been associated with several types of human cancers. In the host, EBV can establish two alternative modes of life cycle, known as latent or lytic and the switch from latency to the lytic cycle is known as EBV reactivation. Although EBV in cancer cells is found mostly in latency, a small number of lytically-infected cells promote carcinogenesis through the release of growth factors and oncogenic cytokines. This review focuses on the mechanisms by which EBV reactivation is controlled by cellular and viral factors, and discusses how EBV lytic infection contributes to human malignancies.
Keywords: Zta, Rta, Epstein-Barr virus, latency, reactivation, carcinogenesis
1. Introduction
EBV is an oncogenic virus that is linked with several malignancies, including nasopharyngeal carcinoma (NPC), Hodgkin's lymphoma (HL), Burkitt's lymphoma (BL), and gastric carcinoma [1]. In the host, EBV can establish two types of infection known as latent and lytic. During latency, only a limited number of viral genes are expressed and the viral genome exists in the nucleus as an episome. Upon reactivation, EBV briefly passes through three consecutive lytic phases, including immediate early (IE), early (E), and late (L) [2]. The viral IE genes BZLF1 and BRLF1 are first transcribed to encode the transactivators, Zta and Rta, respectively, followed by expression of the early genes required for EBV genome replication. After EBV DNA replication, late genes are expressed that encode mainly viral structural proteins, including capsid antigens and membrane proteins, followed by viral genome encapsidation and the production of mature virions. Although all EBV-associated cancers involve the latent cycle of EBV, the viral lytic cycle also contributes to the development and maintenance of malignancies through the induction of growth factors and oncogenic cytokine production [3-5].
In this review, we describe recent advances regarding the mechanisms underlying EBV reactivation, focusing on the control of the host and the virus itself, and discuss the contribution of viral lytic infection to EBV-associated malignancies.
2. Zta and Rta synergistically trigger EBV reactivation
Following various stimuli, such as 12-O-tetradecanoylphorbol-13-acetate (TPA), sodium butyrate, anti-Ig, and transforming growth factor-beta (TGF-β), EBV reactivation can be triggered by two immediate early (IE) transactivators, Zta and Rta. Together, both IE proteins turn on the entire lytic viral cascade of gene expression and EBV replication. Zta, a member of the basic-region leucine zipper (bZIP) family of transcription factors, activates the expression of lytic EBV genes by binding to the activator protein (AP)-1-like motif known as Zta response elements (ZREs) [6,7]. In addition, it also functions as a replication factor for EBV genomic DNA by binding the lytic origin of replication, oriLyt [8]. Similar to Zta, the Rta protein can transactivate lytic target promoters by direct binding to Rta response elements (RREs) [9]. Although Rta is unable to recognize oriLyt, it plays an indispensable role in the process of lytic DNA replication by activating the expression of the BHLF1 gene which encodes replication proteins [10]. This synergy is achieved because Zta and Rta activate both their own and one another's promoters, which greatly amplifies their lytic-inducing effects [11]. Zta can directly activate transcription from its own BZLF1 promoter (Zp), by binding to the ZIIIA and ZIIIB elements of Zp [12] and the BRLF1 promoter (Rp) by binding to three known ZREs (ZRE1, ZRE2 and ZRE3) within Rp [13]. However, Rta activates its own promoter through an indirect mechanism involving a direct interaction with specificity protein (Sp1) through an intermediary protein, MCAF1, to form a complex on Sp1-binding sites [14]. Rta also activates Zp indirectly through activation of the mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3-K) pathways, resulting in phosphorylation of transcription factors that bind to a ZII cyclic AMP response element, such as activating transcription factor-2 (ATF-2) or c-Jun [15,16].
3. Host factors contributing to the regulation of EBV reactivation
3.1. The role of post-translational modifications in the functional activities of Zta and Rta
The balance between EBV latent and lytic infection in host cells is initially implicated in transcriptional control of the BZLF1 and BRLF1 genes. Cellular transcription factors and their binding motifs within Zp and Rp have been well-studied [17,18]. However, activation of both IE promoters is not sufficient for induction of EBV reactivation. The ability of Zta and Rta to trigger EBV reactivation is also regulated through post-translational mechanisms. Among them, phosphorylation is the most common post-translational modification and modulates the transcriptional potential of transcription factors regardless of whether they are encoded by the host cell or the virus. Phosphorylation of serine residue 173 (Ser173), located in the DNA binding domain of Zta, promotes viral replication by enhancing Zta's affinity for DNA, but is not required for activation of early lytic genes [19]. Ser186 of Zta is phosphorylated by protein kinase C after stimulation with TPA. The phosphorylation of Ser186 is essential for the full functional activity of Zta during the lytic cycle [20]. In addition to Ser173 and Ser186, Zta was shown to be constitutively phosphorylated at multiple sites [21]. Nonetheless, the role of phosphorylation in the functional activity of Zta remains largely unknown. Unlike phosphorylation, sumoylation modification often negatively affects Zta transcriptional activity [22,23]. Recent evidence revealed that sumoylation of lysine 12 results in Zta repression of viral gene expression, promoting EBV latency and, also, that the EBV-encoded protein kinase (EBV-PK) reverses the sumoylation of Zta during EBV reactivation [22]. Subsequently, Murata et al. demonstrated that the inhibitory effect of sumoylation on Zta activity is mainly mediated by recruiting histone deacetylase (HDAC) complexes [23].
In addition, post-translational modifications have been shown to affect Zta and Rta activities through protein-protein interactions. In EBV-infected cells, the transcription factors Ikaros, Oct-1, and TAF4 and the retinoblastoma (Rb) protein directly interact with Rta, and the interactions are thought to be important for Rta-mediated disruption of viral latency [14,24-26]. Mutation analysis revealed that the interactions require the DNA-binding/dimerization domain of Rta. Transducer of regulated CREB protein 2 (TORC2) and C/EBP have been identified as co-activators for Zta to activate its own promoter, Zp [27,28]. Interestingly, both EBV IE transactivators, Zta and Rta, have been shown to interact with cAMP response element binding (CREB)-binding protein (CBP), which exhibits histone acetylase activity. The interactions enhance Zta and Rta transactivator activity and increase their ability to induce the lytic form of EBV infection in latently-infected cells [29]. Alternatively, protein-protein interactions are also related to the reduced transcriptional activity and weak affinity for DNA of IE proteins. For example, the cellular transcription factors, Oct-2, Pax-5, NF-κB, and c-Myc, inhibit the induction of EBV lytic reactivation by interacting with Zta [30-33].
3.2. Cellular signaling pathways involved in EBV reactivation
In the human host, B-cell receptor (BCR) antigen stimulation is known to reactivate EBV latency. An accumulation of data demonstrated that the PKC, MAPK, and PI3-K signaling pathways are involved in BCR induction of the EBV lytic cycle [34]. Eventually, a network with crosstalk of these signaling pathways leads to activation of several positive transcription factors on Zp or Rp, thereby stimulating the latent-lytic switch (Figure 1).
Signaling pathways of BZLF1 promoter activation. EBV reactivation can be induced by BCR-mediated signaling pathways or cellular stress (e.g., DNA damage). PKC, MAPK (ERKs, JNKs, and p38), and PI3K pathways as well as ATM-dependent mechanisms appear to be involved. A network with crosstalk of four major signaling pathways leads to activation of several positive transcription factors, followed by transcription from Zp.
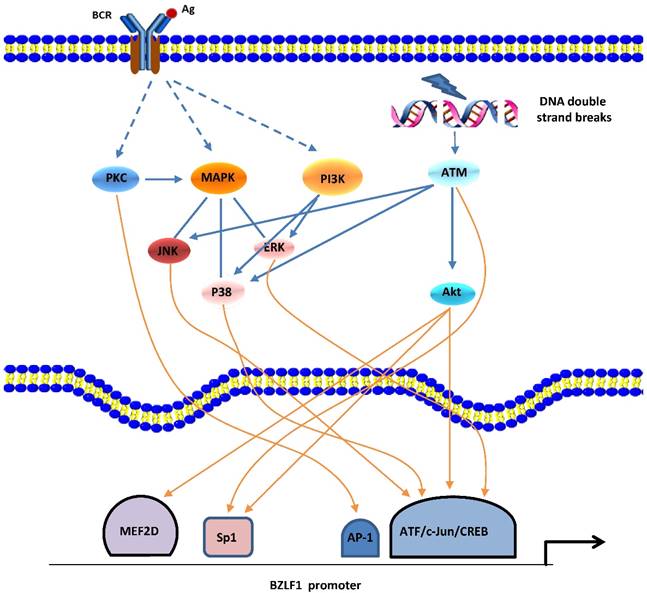
Gao et al. first reported the involvement of the PKC pathway in the latent-lytic switch [35]. The switch, marked by Zta expression, can be induced by active NF-κB and AP-1 through PKC. This report was followed by similar reports from other groups [36,37]. The MAPK family consists of 3 member cascades, extracellular signal regulated kinases (ERKs), c-Jun NH2-terminal kinases (JNKs), and p38 and is usually activated as a partner of the PKC signaling pathway. JNK signaling leads to the phosphorylation of c-Jun and c-Jun/c-Fos cooperate with Smads proteins to bind the AP-1 motif and the Smad4-binding element within Zp, followed by expression of the BZLF1 gene [38]. By inducing the phosphorylation of c-Jun/ATF2, the MAPK signaling pathways are also required for Rta-mediated activation of Zp [25]. Iwakiri and colleagues demonstrated that PI3-K/Akt signal transduction contributes to transcription from the promoters of the BZLF1 gene [39], which is consistent with our recent study showing that the PI3-K/Akt and ERKs pathways are involved in the EBV spontaneous lytic cycle cascade [40]. PI3-K/Akt signaling has been shown to activate cellular transcription factors c-Jun, ATF2, CREB, Sp1, and myocyte-specific enhancer factor 2D (MEF2D) that activate Zp and/or Rp [18]. Moreover, in cells that are not responsive to BCR-mediated EBV reactivation, active PI3-K activates signaling cascades for the ERKs and p38 pathways, resulting in initiation of the EBV lytic cycle [39].
Recently, the ataxia telangiectasia mutated (ATM) activation that occurs in response to DNA damage or oxidative stress has been shown to induce EBV reactivation through a p53-dependent mechanism [41,42]. ATM and downstream signaling pathways p38 and JNKs are responsible for phosphorylation of p53 at multiple sites, and the activated p53 protein mediates expression of the BRLF1 gene by directly binding Rp.
3.3 Epigenetic regulation of EBV reactivation
Epigenetic factors, including viral genome methylation and histone modifications, also play an important role in regulating the state of EBV infection in host cells.
DNA methylation
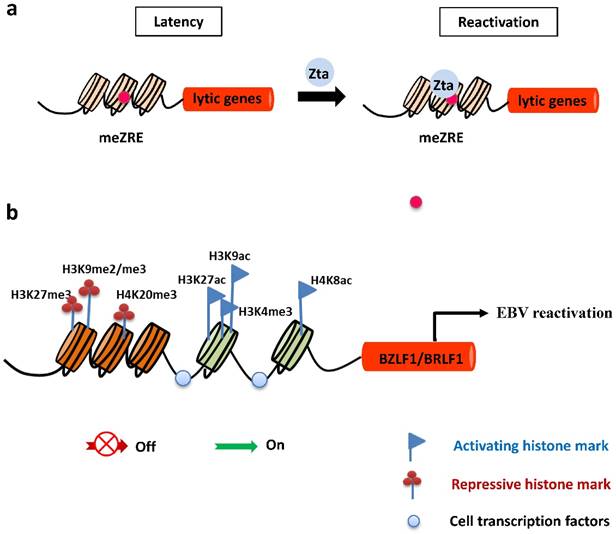
In latently-infected cells, the promoters of EBV lytic genes are intensively repressed and silenced by host-driven DNA methylation [43]. The repressive epigenetic player, CpG DNA methylation of the viral genome decreases the ability of Rta to activate most early lytic promoters [44]. Importantly, the virus has evolved a strategy to overcome the silencing of DNA methylation and withdraw into a latent state. The unique ability of Zta to bind preferentially and activate methylated CpG-containing ZREs is essential for initiating EBV reactivation in cells latently infected with a methylated viral genome (Figure 2). Of note, Ser186 is required for Zta activation of the methylated form of EBV lytic promoters [45].
Epigenetic control of EBV lytic reactivation. a. During latency, EBV lytic promoters are silenced by host-driven methylation. The IE protein Zta preferentially binds to methylated ZREs (meZREs) in lytic promoters to initiate replication from this epigenetic repressed state. b. The IE promoters Zp and Rp are controlled by several repressive histone modifications during EBV latent infection. After nucleosomes are removed locally, activating histone marks are established and allow the access of transcription factors to induce expression of both IE genes.
Histone modifications
Histone modifications have a central epigenetic role in regulating activation of BZLF1 and BRLF1 genes. The local acetylation state of histones H3 and H4 around Zp and Rp, such as histone H3 Lys27 acetylation (H3K27ac), H3K9ac, and H4K8ac, help to establish the open chromatin configuration so as to allow access of transcription factors, followed by transcription of the BZLF1 and BRLF1genes [46-48]. In contrast, the methylation modifications of histones were suggested to be marks of heterochromatin formation and transcriptional repression. Repressive histone modifications, such as H3K27me3, H3K9me2/me3, and H4K20me3 have been identified at Zp or Rp, and correlate with inactivation of both IE promoters, maintaining viral latency. Nevertheless, H3K4me3 allows the virus to express Zta [49-51] (Figure 2).
3.4 Cellular stresses contributing to EBV reactivation
Increasing evidence suggests that severe host cell stress in response to many different toxic stimuli, including chemotherapy and γ irradiation, can induce lytic EBV infection, which ensures that the virus spreads from host to host.
Oxidative stress
Oxidative stress, generally induced by chemotherapy and irradiation (IR), leads to EBV reactivation through the induction of BZLF1 gene expression [52]. Increasing evidence suggests a role for reactive oxygen species (ROS), resulting from oxidative stress, as intermediates of intracellular signal transduction pathways [53]. Huang, et al. demonstrated a novel signaling mechanism by ROS for induction of EBV reactivation [42]. They found that various signaling pathways including ATM, p38 and JNKs are activated by ROS and involved in the induction of EBV reactivation in a p53-dependent manner. Also, phosphorylation of the ATF2 transcription factor by p38 and JNKs has been reported to activate Zp [16], implying deregulated ROS signaling might similarly induce EBV reactivation from latency through modification of other redox-sensitive transcription factors that activate Zp and/or Rp, such as early growth response 1 (EGR1) [54,55], Sp1 [56], Stat3 [57], and c-Jun [58].
Hypoxia
Hypoxia-inducible factor 1 (HIF-1) is a transcription factor that consists of α and β subunits, and is responsible for hypoxia induction of EBV reactivation [59]. In hypoxic conditions, HIF-1 accumulates to a high level, consequently the α subunit up-regulates expression of the BZLF1 gene by binding to HIF-1-responsive elements (HREs) in Zp (Kraus RJ, Yu X, Sathiamoorthi S, Ruegsegger N, Nawandar DM, Kenney SC, et al. Unpublished data. n.d.).
Autophagy
Although autophagy normally serves as a defense mechanism against viral infection, recent research findings showed that EBV manipulates this mechanism to promote viral replication [60-63]. Results from various studies reveal that autophagic activation through the Rta-mediated ERKs pathway [60] and the PKCθ-p38 signaling axis [61] promotes viral lytic development in the early phase of EBV reactivation, but is soon inhibited by the early lytic products so as to prevent viral degradation in the degradative phases of autophagy [62,63].
Inflammation
Inflammatory responses against viral infection is one of the predisposing factors associated with virus-mediated tumorigenesis [64]. In the case of EBV, lytic reactivation induces expression of inflammatory cytokines, including interleukin-6 (IL-6), IL-8, IL-10, and IL-13, contributing to pathogenesis of NPC or lymphomas [65-68]. In a recent study, Gandhi, et al. elucidated the role of inflammation in EBV lytic reactivation [69]. They found that COX-2, a key mediator of inflammatory processes, induces EBV lytic reactivation through prostaglandin E2 (PGE2) by modulating the prostaglandin EP receptor-signaling pathway.
4. The viral self-regulation of EBV reactivation
4.1. Viral encoded proteins tend to maintain the temporal modes of EBV infection
In latent infection, the EBV latent membrane protein 1 (LMP1), a viral mimic of constitutively active CD40, intensifies latency in part through NF-κB activity [70]. LMP2A blocks BCR-induced EBV reactivation by inhibiting activation of tyrosine kinases by BCR [71,72]. And yet, some EBV early lytic proteins affect the activities of Zta or Rta through interactions between EBV proteins [73-75]. For example, the BRRF1 (Na)-Rta interaction enhances induction of viral lytic replication [73]. Unlike Na, the interaction of LF2 with Rta is critical for altering Rta subcellular localization and consequent functional repression [74,75]. In an earlier study, the BMRF1 protein was found to directly interact with Zta in vitro as well as in vivo, enhancing transcription from their common early BHLF1 promoter [76]. Similarly, EBV tegument protein BGLF2, encoded in the late phase of the lytic cycle, was suggested to enhance Zta expression through activation of the p38 signaling pathway [77].
EBV nuclear antigen1 (EBNA1) is expressed in both latent and lytic modes of EBV infection. Notably, two roles for EBNA1 in the EBV latent-lytic switch have been identified. First, EBNA1 is known to be required for maintenance of latency; and second, when the lytic cycle is induced, it also has a role in viral reactivation and lytic infection [78,79]. For example, EBNA1 was shown to organize the oriP regions into replication domains for lytic replication and transcription [78], and induce EBV reactivation by overcoming the PML protein- and nuclear body (NB)-suppression of lytic infection [79].
These results indicate that the viral proteins preferentially maintain the modes of EBV infection, which is most likely to facilitate viral optimal replication in host cells.
4.2. The inhibitory effects of EBV-encoded microRNAs on reactivation from latency
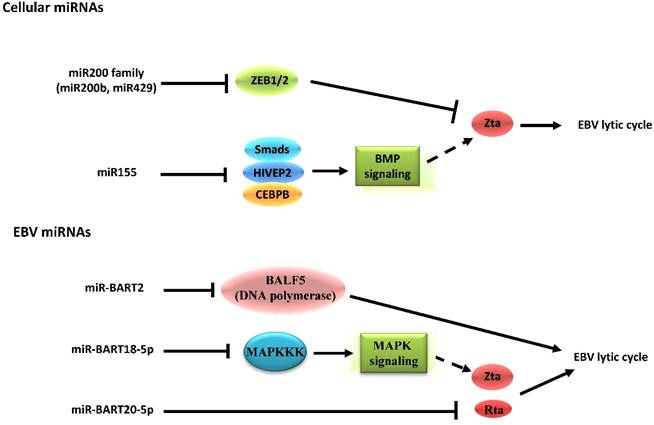
EBV is the first virus found to encode microRNAs (miRNAs) [80]. The miRNAs encoded by EBV can be divided into two clusters, including 29 miRNAs located in the introns of the viral BART gene and 3 located adjacent to the BHRF1 gene [81]. By identifying target genes, the roles of miRNAs in EBV latent-lytic switch have been established (Figure 3). For instance, the cellular miR-200b and miR-429, members of the miR200 family, are able to induce lytic replication by targeting ZEB1/2 and blocking their repressive effect on Zp [82,83]. On the other hand, miR-155 inhibits BMP-mediated lytic reactivation by targeting multiple members of the BMP signaling pathway, including SMAD1, SMAD5, and CEBPB [84]. As for EBV-encoded miRNAs, the fact that a subset of viral miRNAs is present at high copy numbers in latently-infected cells implicates them in establishing and maintaining latency [81,85]. This occurs by inhibiting expression of viral lytic genes that play essential roles in the latent-lytic switch [86,87]. Barth, et al. demonstrated that EBV-encoded miR-BART2 directly targets the transcript of the viral DNA polymerase BALF5 to inhibit the transition from latent to lytic viral replication [86]. A recent study demonstrated that the EBV-encoded miR-BART20-5p directly targets the transcripts of the lytic switch proteins, Zta and Rta [87]. The EBV-encoded miRNAs also enhance latency by targeting host transcripts [88,89]. For example, EBV-encoded miR-BART5 counteracts the pro-apoptotic function of the p53/PUMA pathway by targeting transcripts of PUMA, optimizing cellular conditions for EBV latency [88]. Additionally, EBV-encoded miR-BART18-5p directly targets the transcript of MAPKKK2 (MAP3K2), which modulates the MAPK signaling pathways that are known to be important in EBV reactivation [89].
Schematic indicating the role of cellular and EBV miRNAs in EBV latency and reactivation. Cellular miRNAs have distinct functions in EBV latency and lytic reactivation by directly targeting transcription factors on Zp or regulating the signaling pathway related to expression of Zta. Nevertheless, the EBV-encoded miRNAs inhibit the transition from latent to lytic viral replication, which occurs both through modulation of specific signaling pathways as well as through the restriction of its own gene expression. Notably, the EBV-encoded mi-BART20-5p can directly target the transcripts of the BZLF1 and BRLF1 genes.
5. The pathogenic role of lytic infection in EBV-associated malignancies
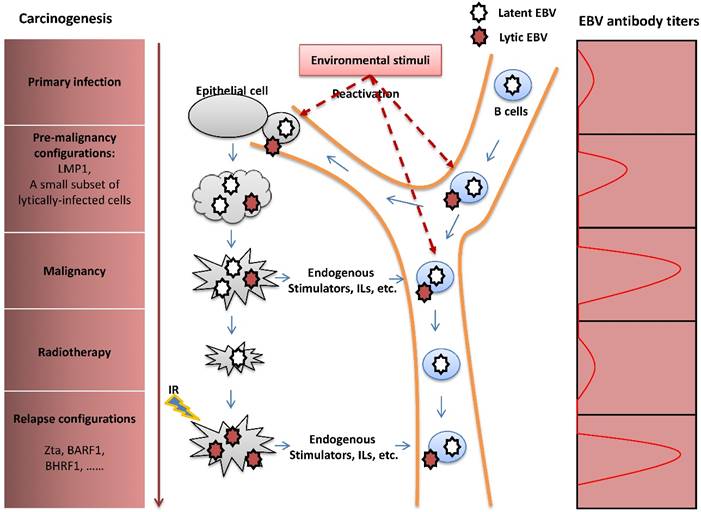
Previous studies have focused on the contributions of EBV latent infection in the pathogenesis of EBV-induced malignancies and revealed that LMP1 is an essential oncoprotein [90]. In recent years, the viral lytic cycle has been shown to play an important role in carcinogenesis through several potential mechanisms. By enhancing transmission of the virus from cell to cell, EBV lytic infection may increase the total number of latently-infected cells and thus is an essential aspect of viral pathogenesis. A small subset of lytically-infected cells is commonly detected in biopsies of EBV-associated malignancies [91-93], suggesting a potential role for viral lytic infection in promoting tumor growth in vivo. Furthermore, some studies indicated that the viral lytic cycle in a fraction of B cells promotes the transformation of B-lymphocytes in vitro [94] and growth of B cell lymphoma in vivo [7,95] through the release of paracrine growth factors and angiogenic factors. Focusing on the study of NPC, Wu, et al. found that recurrent EBV reactivation promotes genome instability, invasiveness and tumorigenesis of NPC cells, and that the contribution of the lytic cycle is more profound than latent infection [5,96,97]. Additionally, lytic replication enhances secretion of the angiogenic factor, vascular endothelial growth factor (VEGF), in NPC cells, contributing to angiogenesis and consequent metastasis or relapse of NPC after remission [98]. Clinical and epidemiological studies have revealed that individuals with elevated plasma EBV DNA load and antibody titers against the lytic viral capsid antigen (VCA) and early antigen (EA) have a high risk of NPC [99,100]. These studies also show that fluctuation of EBV antibody titers occurs prior to the onset of NPC [101,102]. These results suggest the importance of lytic infection for the initiation, progression, and metastasis or relapse of NPC (Figure 4).
The pathogenic role of EBV lytic infection in NPC carcinogenesis. After primary infection, EBV establishes latent infection in B cells. Upon reactivation, lytically-infected B cells travel through the nasopharynx tissue, followed by infection of the nasopharyngeal epithelial cells by cell-to-cell contact. Similar to the LMP1 oncoprotein, the presence of a limited number of lytically-infected epithelial cells induces genome instability and release of oncogenic cytokines, consequently promoting NPC carcinogenesis. Although NPC is sensitive to radiotherapy, extensive resistance to radiation often causes tumor metastasis or relapse after remission. IR -induced recurrent expression of lytic proteins is a potential factor that mediates the impact of EBV on NPC relapse. The fluctuation of EBV antibody titers reflects tumor progression of NPC.
In addition, understanding the function of the EBV lytic proteins in malignancies is clearly essential in determining the role of lytic infection in the carcinogenic process (Table 1). The IE protein Zta triggers paracrine secretion of the angiogenic factor VEGF [98] and several oncogenic and inflammatory cytokines, including IL-6, IL-8, IL-10, and IL-13 [3,94,95,103], and thereby, participates in the tumorigenesis of EBV-associated malignancies. Some lytic proteins, such as BGLF4 protein kinase (EBV-PK) and BGLF5 nuclease (EBV DNase), have been reported to promote genomic instability and enhance tumor progression of NPC cells [96,104]. Intriguingly, EBV encodes a series of important proteins that show homology to diverse human anti-apoptotic molecules and oncogenic cytokines. For example, early gene BCRF1, also known as viral interleukin-10 (vIL-10), encodes a homolog of IL-10 that functions as a paracrine growth factor in EBV-associated lymphomas [105]. BHRF1 exhibits homology to the human oncoprotein Bcl-2 and delays cell death during EBV lytic replication [106,107]. Chiu, et al. demonstrated that BALF3, a homologue to terminase, is not only involved in the induction of host genomic instability, but also mediates the impact of EBV on NPC relapse [108]. Another early gene BARF1 encodes a homologue to the colony-stimulating factor-1 (CSF-1) receptor, which is a product of the human oncogene, c-fms. BARF1 inhibits apoptosis by activating Bcl-2 [109], hence contributing to the tumorigenicity of NPC cells [110,111]. Thus, effective strategies that inhibit EBV lytic reactivation might be valuable in the prevention or treatment of EBV-associated malignancies and improve the clinical outcome.
EBV lytic proteins and tumorigenic functions a
| Lytic protein | Lytic phase | Human homologue | Tumorigenic function |
|---|---|---|---|
| Zta | Immediate Early | — | Induction of IL-6, IL-8, IL-10, IL-13, and VEGF secretion |
| BARF1 | Early | C-fms receptor | Anti-apoptosis |
| BHRF1 | Early | Bcl-2 | Anti-apoptosis |
| BALF3 | Early | Terminase | Induction of genomic instability |
| BCRF1 | Early | IL-10 | Anti-apoptosis |
| BGLF4 | Early | — | Induction of chromosomal abnormality and DNA damage |
| DNase | Early | — | Induction of genomic instability |
a Summarized from references 3, 94-96, 98, 103-111.
6. Conclusions
Focusing on host and viral factors, this review has covered recent advances with respect to the mechanisms underlying EBV reactivation. In host cells, in addition to transcription control, post-transcriptional modifications, signal transduction, and epigenetic regulation, can together determine whether EBV infection remains latent or becomes lytic. Simultaneously, the virus has evolved strategies to exploit cell epigenetic machinery to establish a lifelong latent infection and use a strategy to escape from the latent state. To propagate, EBV spontaneously enters into lytic replication under severe cellular stress, and this adds to our understanding of how these host-viral interactions modulate the microenvironment contributing to EBV reactivation. In addition, regulation of the latent-lytic switch in the EBV life cycle is dependent partly on gene products encoded by the virus itself. Importantly, identification of lytic proteins that result in tumorigenesis determines the pathogenic role for lytic infection in human malignancies. In summary, a better understanding of the mechanisms beneath EBV lytic reactivation and the pathogenic role of viral lytic infection in carcinogenesis helps in the design of new virus-targeted therapies aiming at lytic cycle for EBV-associated malignancies.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (nos. 81161120410 and 81101474), the Innovation Foundation of Central South University (no. 2013zzts072), and the Collaborative Innovation Center for Chemistry and Molecular Medicine of Hunan province, China.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kenney S. Theodore E. Woodward Award: development of novel, EBV-targeted therapies for EBV-positive tumors. Transactions of the American Clinical and Climatological Association. 2006;117:55-73 discussion 73-54
2. Murata T, Tsurumi T. Switching of EBV cycles between latent and lytic states. Reviews in medical virology. 2014;24:142-153
3. Hong GK, Gulley ML, Feng WH. et al. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. Journal of virology. 2005;79:13993-14003
4. Fiorini S, Ooka T. Secretion of Epstein-Barr virus-encoded BARF1 oncoprotein from latently infected B cells. Virology journal. 2008;5:70
5. Fang CY, Lee CH, Wu CC. et al. Recurrent chemical reactivations of EBV promotes genome instability and enhances tumor progression of nasopharyngeal carcinoma cells. International journal of cancer. 2009;124:2016-2025
6. Petosa C, Morand P, Baudin F. et al. Structural basis of lytic cycle activation by the Epstein-Barr virus ZEBRA protein. Molecular cell. 2006;21:565-572
7. Kalla M, Schmeinck A, Bergbauer M. et al. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:850-855
8. Rennekamp AJ, Wang P, Lieberman PM. Evidence for DNA hairpin recognition by Zta at the Epstein-Barr virus origin of lytic replication. Journal of virology. 2010;84:7073-7082
9. Heilmann AM, Calderwood MA, Portal D. et al. Genome-wide analysis of Epstein-Barr virus Rta DNA binding. Journal of virology. 2012;86:5151-5164
10. El-Guindy A, Ghiassi-Nejad M, Golden S. et al. Essential role of Rta in lytic DNA replication of Epstein-Barr virus. Journal of virology. 2013;87:208-223
11. Feederle R, Kost M, Baumann M. et al. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. The EMBO journal. 2000;19:3080-3089
12. Flemington E, Speck SH. Autoregulation of Epstein-Barr virus putative lytic switch gene BZLF1. Journal of virology. 1990;64:1227-1232
13. Packham G, Economou A, Rooney CM. et al. Structure and function of the Epstein-Barr virus BZLF1 protein. Journal of virology. 1990;64:2110-2116
14. Yang YC, Chang LK. Role of TAF4 in transcriptional activation by Rta of Epstein-Barr Virus. PloS one. 2013;8:e54075
15. Darr CD, Mauser A, Kenney S. Epstein-Barr virus immediate-early protein BRLF1 induces the lytic form of viral replication through a mechanism involving phosphatidylinositol-3 kinase activation. Journal of virology. 2001;75:6135-6142
16. Adamson AL, Darr D, Holley-Guthrie E. et al. Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 activate the ATF2 transcription factor by increasing the levels of phosphorylated p38 and c-Jun N-terminal kinases. Journal of virology. 2000;74:1224-1233
17. Murata T. Regulation of Epstein-Barr virus reactivation from latency. Microbiology and immunology. 2014;58:307-317
18. Kenney SC, Mertz JE. Regulation of the latent-lytic switch in Epstein-Barr virus. Seminars in cancer biology. 2014;26:60-68
19. El-Guindy A, Heston L, Delecluse HJ. et al. Phosphoacceptor site S173 in the regulatory domain of Epstein-Barr Virus ZEBRA protein is required for lytic DNA replication but not for activation of viral early genes. Journal of virology. 2007;81:3303-3316
20. Bhende PM, Seaman WT, Delecluse HJ. et al. BZLF1 activation of the methylated form of the BRLF1 immediate-early promoter is regulated by BZLF1 residue 186. Journal of virology. 2005;79:7338-7348
21. El-Guindy AS, Paek SY, Countryman J. et al. Identification of constitutive phosphorylation sites on the Epstein-Barr virus ZEBRA protein. The Journal of biological chemistry. 2006;281:3085-3095
22. Hagemeier SR, Dickerson SJ, Meng Q. et al. Sumoylation of the Epstein-Barr virus BZLF1 protein inhibits its transcriptional activity and is regulated by the virus-encoded protein kinase. Journal of virology. 2010;84:4383-4394
23. Murata T, Hotta N, Toyama S. et al. Transcriptional repression by sumoylation of Epstein-Barr virus BZLF1 protein correlates with association of histone deacetylase. The Journal of biological chemistry. 2010;285:23925-23935
24. Iempridee T, Reusch JA, Riching A. et al. Epstein-Barr virus utilizes Ikaros in regulating its latent-lytic switch in B cells. Journal of virology. 2014;88:4811-4827
25. Robinson AR, Kwek SS, Hagemeier SR. et al. Cellular transcription factor Oct-1 interacts with the Epstein-Barr virus BRLF1 protein to promote disruption of viral latency. Journal of virology. 2011;85:8940-8953
26. Zacny VL, Wilson J, Pagano JS. The Epstein-Barr virus immediate-early gene product, BRLF1, interacts with the retinoblastoma protein during the viral lytic cycle. Journal of virology. 1998;72:8043-8051
27. Murata T, Sato Y, Nakayama S. et al. TORC2, a coactivator of cAMP-response element-binding protein, promotes Epstein-Barr virus reactivation from latency through interaction with viral BZLF1 protein. J Biol Chem. 2009;284:8033-8041
28. Wu FY, Wang SE, Chen H. et al. CCAAT/enhancer binding protein alpha binds to the Epstein-Barr virus (EBV) ZTA protein through oligomeric interactions and contributes to cooperative transcriptional activation of the ZTA promoter through direct binding to the ZII and ZIIIB motifs during induction of the EBV lytic cycle. J Virol. 2004;78:4847-4865
29. Swenson JJ, Holley-Guthrie E, Kenney SC. Epstein-Barr virus immediate-early protein BRLF1 interacts with CBP, promoting enhanced BRLF1 transactivation. J Virol. 2001;75:6228-6234
30. Robinson AR, Kwek SS, Kenney SC. The B-cell specific transcription factor, Oct-2, promotes Epstein-Barr virus latency by inhibiting the viral immediate-early protein, BZLF1. PLoS pathogens. 2012;8:e1002516
31. Raver RM, Panfil AR, Hagemeier SR. et al. The B-cell-specific transcription factor and master regulator Pax5 promotes Epstein-Barr virus latency by negatively regulating the viral immediate early protein BZLF1. Journal of virology. 2013;87:8053-8063
32. Gutsch DE, Holley-Guthrie EA, Zhang Q. et al. The bZIP transactivator of Epstein-Barr virus, BZLF1, functionally and physically interacts with the p65 subunit of NF-kappa B. Molecular and cellular biology. 1994;14:1939-1948
33. Rodriguez A, Jung EJ, Yin Q. et al. Role of c-myc regulation in Zta-mediated induction of the cyclin-dependent kinase inhibitors p21 and p27 and cell growth arrest. Virology. 2001;284:159-169
34. Goswami R, Gershburg S, Satorius A. et al. Protein kinase inhibitors that inhibit induction of lytic program and replication of Epstein-Barr virus. Antiviral research. 2012;96:296-304
35. Gao X, Ikuta K, Tajima M. et al. 12-O-tetradecanoylphorbol-13-acetate induces Epstein-Barr virus reactivation via NF-kappaB and AP-1 as regulated by protein kinase C and mitogen-activated protein kinase. Virology. 2001;286:91-99
36. Lee HH, Chang SS, Lin SJ. et al. Essential role of PKCdelta in histone deacetylase inhibitor-induced Epstein-Barr virus reactivation in nasopharyngeal carcinoma cells. The Journal of general virology. 2008;89:878-88
37. Liu YR, Huang SY, Chen JY. et al. Microtubule depolymerization activates the Epstein-Barr virus lytic cycle through protein kinase C pathways in nasopharyngeal carcinoma cells. The Journal of general virology. 2013;94:2750-2758
38. Hagemeier SR, Barlow EA, Kleman AA. et al. The Epstein-Barr virus BRRF1 protein, Na, induces lytic infection in a TRAF2- and p53-dependent manner. Journal of virology. 2011;85:4318-4329
39. Iwakiri D, Takada K. Phosphatidylinositol 3-kinase is a determinant of responsiveness to B cell antigen receptor-mediated Epstein-Barr virus activation. Journal of immunology. 2004;172:1561-1566
40. Liu S, Li H, Chen L. et al. (-)-Epigallocatechin-3-gallate inhibition of Epstein-Barr virus spontaneous lytic infection involves ERK1/2 and PI3-K/Akt signaling in EBV-positive cells. Carcinogenesis. 2013;34:627-637
41. Hagemeier SR, Barlow EA, Meng Q. et al. The cellular ataxia telangiectasia-mutated kinase promotes epstein-barr virus lytic reactivation in response to multiple different types of lytic reactivation-inducing stimuli. Journal of virology. 2012;86:13360-13370
42. Huang SY, Fang CY, Wu CC. et al. Reactive oxygen species mediate Epstein-Barr virus reactivation by N-methyl-N'-nitro-N-nitrosoguanidine. PloS one. 2013;8:e84919
43. Flower K, Thomas D, Heather J. et al. Epigenetic control of viral life-cycle by a DNA-methylation dependent transcription factor. PloS one. 2011;6:e25922
44. Wille CK, Nawandar DM, Panfil AR. et al. Viral genome methylation differentially affects the ability of BZLF1 versus BRLF1 to activate Epstein-Barr virus lytic gene expression and viral replication. Journal of virology. 2013;87:935-950
45. Yu KP, Heston L, Park R. et al. Latency of Epstein-Barr virus is disrupted by gain-of-function mutant cellular AP-1 proteins that preferentially bind methylated DNA. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:8176-8181
46. Countryman JK, Gradoville L, Miller G. Histone hyperacetylation occurs on promoters of lytic cycle regulatory genes in Epstein-Barr virus-infected cell lines which are refractory to disruption of latency by histone deacetylase inhibitors. Journal of virology. 2008;82:4706-4719
47. Jenkins PJ, Binne UK, Farrell PJ. Histone acetylation and reactivation of Epstein-Barr virus from latency. Journal of virology. 2000;74:710-720
48. Mansouri S, Wang S, Frappier L. A role for the nucleosome assembly proteins TAF-Ibeta and NAP1 in the activation of BZLF1 expression and Epstein-Barr virus reactivation. PloS one. 2013;8:e63802
49. Imai K, Kamio N, Cueno ME. et al. Role of the histone H3 lysine 9 methyltransferase Suv39 h1 in maintaining Epsteinn-Barr virus latency in B95-8 cells. The FEBS journal. 2014;281:2148-2158
50. Murata T, Kondo Y, Sugimoto A. et al. Epigenetic histone modification of Epstein-Barr virus BZLF1 promoter during latency and reactivation in Raji cells. Journal of virology. 2012;86:4752-4761
51. Ramasubramanyan S, Osborn K, Flower K. et al. Dynamic chromatin environment of key lytic cycle regulatory regions of the Epstein-Barr virus genome. Journal of virology. 2012;86:1809-1819
52. Lassoued S, Gargouri B, El Feki Ael F. et al. Transcription of the Epstein-Barr virus lytic cycle activator BZLF-1 during oxidative stress induction. Biological trace element research. 2010;137:13-22
53. Finkel T. Reactive oxygen species and signal transduction. IUBMB life. 2001;52:3-6
54. Chang Y, Lee HH, Chen YT. et al. Induction of the early growth response 1 gene by Epstein-Barr virus lytic transactivator Zta. Journal of virology. 2006;80:7748-7755
55. Zalani S, Holley-Guthrie E, Kenney S. The Zif268 cellular transcription factor activates expression of the Epstein-Barr virus immediate-early BRLF1 promoter. Journal of virology. 1995;69:3816-3823
56. Liu S, Borras AM, Liu P. et al. Binding of the ubiquitous cellular transcription factors Sp1 and Sp3 to the ZI domains in the Epstein-Barr virus lytic switch BZLF1 gene promoter. Virology. 1997;228:11-18
57. Koganti S, Clark C, Zhi J. et al. Cellular STAT3 functions via PCBP2 to restrain Epstein-Barr Virus lytic activation in B lymphocytes. Journal of virology. 2015;89:5002-5011
58. Liang CL, Chen JL, Hsu YP. et al. Epstein-Barr virus BZLF1 gene is activated by transforming growth factor-beta through cooperativity of Smads and c-Jun/c-Fos proteins. The Journal of biological chemistry. 2002;277:23345-23357
59. Jiang JH, Wang N, Li A. et al. Hypoxia can contribute to the induction of the Epstein-Barr virus (EBV) lytic cycle. Journal of clinical virology: the official publication of the Pan American Society for Clinical Virology. 2006;37:98-103
60. Hung CH, Chen LW, Wang WH. et al. Regulation of autophagic activation by Rta of Epstein-Barr virus via the extracellular signal-regulated kinase pathway. Journal of virology. 2014;88:12133-12145
61. Gonnella R, Granato M, Farina A. et al. PKC theta and p38 MAPK activate the EBV lytic cycle through autophagy induction. Biochimica et biophysica acta. 2015;1853:1586-1595
62. De Leo A, Colavita F, Ciccosanti F. et al. Inhibition of autophagy in EBV-positive Burkitt's lymphoma cells enhances EBV lytic genes expression and replication. Cell death & disease. 2015;6:e1876
63. Granato M, Santarelli R, Farina A. et al. Epstein-barr virus blocks the autophagic flux and appropriates the autophagic machinery to enhance viral replication. Journal of virology. 2014;88:12715-12726
64. Chen CJ, Hsu WL, Yang HI. et al. Epidemiology of virus infection and human cancer. Recent results in cancer research Fortschritte der Krebsforschung Progres dans les recherches sur le cancer. 2014;193:11-32
65. Bennett JM, Glaser R, Malarkey WB. et al. Inflammation and reactivation of latent herpesviruses in older adults. Brain, behavior, and immunity. 2012;26:739-746
66. Hsu M, Wu SY, Chang SS. et al. Epstein-Barr virus lytic transactivator Zta enhances chemotactic activity through induction of interleukin-8 in nasopharyngeal carcinoma cells. Journal of virology. 2008;82:3679-3688
67. Lee CH, Yeh TH, Lai HC. et al. Epstein-Barr virus Zta-induced immunomodulators from nasopharyngeal carcinoma cells upregulate interleukin-10 production from monocytes. Journal of virology. 2011;85:7333-7342
68. Tsai SC, Lin SJ, Chen PW. et al. EBV Zta protein induces the expression of interleukin-13, promoting the proliferation of EBV-infected B cells and lymphoblastoid cell lines. Blood. 2009;114:109-118
69. Gandhi J, Gaur N, Khera L. et al. COX-2 induces lytic reactivation of EBV through PGE2 by modulating the EP receptor signaling pathway. Virology. 2015;484:1-14
70. Prince S, Keating S, Fielding C. et al. Latent membrane protein 1 inhibits Epstein-Barr virus lytic cycle induction and progress via different mechanisms. Journal of virology. 2003;77:5000-5007
71. Fukuda M, Longnecker R. Epstein-Barr virus (EBV) latent membrane protein 2A regulates B-cell receptor-induced apoptosis and EBV reactivation through tyrosine phosphorylation. Journal of virology. 2005;79:8655-8660
72. Swanson-Mungerson M, Ikeda M, Lev L. et al. Identification of latent membrane protein 2A (LMP2A) specific targets for treatment and eradication of Epstein-Barr virus (EBV)-associated diseases. J Antimicrob Chemother. 2003;52:152-154
73. Hong GK, Delecluse HJ, Gruffat H. et al. The BRRF1 early gene of Epstein-Barr virus encodes a transcription factor that enhances induction of lytic infection by BRLF1. Journal of virology. 2004;78:4983-4992
74. Heilmann AM, Calderwood MA, Johannsen E. Epstein-Barr virus LF2 protein regulates viral replication by altering Rta subcellular localization. Journal of virology. 2010;84:9920-9931
75. Calderwood MA, Holthaus AM, Johannsen E. The Epstein-Barr virus LF2 protein inhibits viral replication. Journal of virology. 2008;82:8509-8519
76. Zhang Q, Hong Y, Dorsky D. et al. Functional and physical interactions between the Epstein-Barr virus (EBV) proteins BZLF1 and BMRF1: Effects on EBV transcription and lytic replication. Journal of virology. 1996;70:5131-5142
77. Liu X, Cohen JI. Epstein-Barr Virus (EBV) Tegument Protein BGLF2 Promotes EBV Reactivation through Activation of the p38 Mitogen-Activated Protein Kinase. Journal of virology. 2015;90:1129-1138
78. Daikoku T, Kudoh A, Fujita M. et al. In vivo dynamics of EBNA1-oriP interaction during latent and lytic replication of Epstein-Barr virus. The Journal of biological chemistry. 2004;279:54817-54825
79. Sivachandran N, Wang X, Frappier L. Functions of the Epstein-Barr virus EBNA1 protein in viral reactivation and lytic infection. Journal of virology. 2012;86:6146-6158
80. Barth S, Meister G, Grasser FA. EBV-encoded miRNAs. Biochimica et biophysica acta. 2011;1809:631-640
81. Qiu J, Cosmopoulos K, Pegtel M. et al. A novel persistence associated EBV miRNA expression profile is disrupted in neoplasia. PLoS pathogens. 2011;7:e1002193
82. Ellis-Connell AL, Iempridee T, Xu I. et al. Cellular microRNAs 200b and 429 regulate the Epstein-Barr virus switch between latency and lytic replication. Journal of virology. 2010;84:10329-10343
83. Lin Z, Wang X, Fewell C. et al. Differential expression of the miR-200 family microRNAs in epithelial and B cells and regulation of Epstein-Barr virus reactivation by the miR-200 family member miR-429. Journal of virology. 2010;84:7892-7897
84. Yin Q, Wang X, Fewell C. et al. MicroRNA miR-155 inhibits bone morphogenetic protein (BMP) signaling and BMP-mediated Epstein-Barr virus reactivation. Journal of virology. 2010;84:6318-6327
85. Forte E, Luftig MA. The role of microRNAs in Epstein-Barr virus latency and lytic reactivation. Microbes and infection / Institut Pasteur. 2011;13:1156-1167
86. Barth S, Pfuhl T, Mamiani A. et al. Epstein-Barr virus-encoded microRNA miR-BART2 down-regulates the viral DNA polymerase BALF5. Nucleic acids research. 2008;36:666-675
87. Jung YJ, Choi H, Kim H. et al. MicroRNA miR-BART20-5p stabilizes Epstein-Barr virus latency by directly targeting BZLF1 and BRLF1. Journal of virology. 2014;88:9027-9037
88. Choy EY, Siu KL, Kok KH. et al. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. The Journal of experimental medicine. 2008;205:2551-2560
89. Qiu J, Thorley-Lawson DA. EBV microRNA BART 18-5p targets MAP3K2 to facilitate persistence in vivo by inhibiting viral replication in B cells. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:11157-11162
90. Young LS, Dawson CW. Epstein-Barr virus and nasopharyngeal carcinoma. Chinese journal of cancer. 2014;33:581-590
91. Xue SA, Labrecque LG, Lu QL. et al. Promiscuous expression of Epstein-Barr virus genes in Burkitt's lymphoma from the central African country Malawi. International journal of cancer. 2002;99:635-643
92. Zheng XH, Lu LX, Cui C. et al. Epstein-Barr virus mir-bart1-5p detection via nasopharyngeal brush sampling is effective for diagnosing nasopharyngeal carcinoma. Oncotarget. 2016;7:4972-4980
93. Zheng XH, Lu LX, Li XZ. et al. Quantification of Epstein-Barr virus DNA load in nasopharyngeal brushing samples in the diagnosis of nasopharyngeal carcinoma in southern China. Cancer science. 2015;106:1196-1201
94. Katsumura KR, Maruo S, Takada K. EBV lytic infection enhances transformation of B-lymphocytes infected with EBV in the presence of T-lymphocytes. Journal of medical virology. 2012;84:504-510
95. Ma SD, Hegde S, Young KH. et al. A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas. Journal of virology. 2011;85:165-177
96. Wu CC, Liu MT, Chang YT. et al. Epstein-Barr virus DNase (BGLF5) induces genomic instability in human epithelial cells. Nucleic acids research. 2010;38:1932-1949
97. Huang SY, Fang CY, Tsai CH. et al. N-methyl-N'-nitro-N-nitrosoguanidine induces and cooperates with 12-O-tetradecanoylphorbol-1,3-acetate/sodium butyrate to enhance Epstein-Barr virus reactivation and genome instability in nasopharyngeal carcinoma cells. Chemico-biological interactions. 2010;188:623-634
98. Hong GK, Kumar P, Wang L. et al. Epstein-Barr virus lytic infection is required for efficient production of the angiogenesis factor vascular endothelial growth factor in lymphoblastoid cell lines. Journal of virology. 2005;79:13984-13992
99. Chien YC, Chen JY, Liu MY. et al. Serologic markers of Epstein-Barr virus infection and nasopharyngeal carcinoma in Taiwanese men. The New England journal of medicine. 2001;345:1877-1882
100. Lo YM. Quantitative analysis of Epstein-Barr virus DNA in plasma and serum: applications to tumor detection and monitoring. Annals of the New York Academy of Sciences. 2001;945:68-72
101. Cao SM, Liu Z, Jia WH. et al. Fluctuations of epstein-barr virus serological antibodies and risk for nasopharyngeal carcinoma: a prospective screening study with a 20-year follow-up. PloS one. 2011;6:e19100
102. Ji MF, Wang DK, Yu YL. et al. Sustained elevation of Epstein-Barr virus antibody levels preceding clinical onset of nasopharyngeal carcinoma. British journal of cancer. 2007;96:623-630
103. Ma SD, Yu X, Mertz JE. et al. An Epstein-Barr Virus (EBV) mutant with enhanced BZLF1 expression causes lymphomas with abortive lytic EBV infection in a humanized mouse model. Journal of virology. 2012;86:7976-7987
104. Chang YH, Lee CP, Su MT. et al. Epstein-Barr virus BGLF4 kinase retards cellular S-phase progression and induces chromosomal abnormality. PloS one. 2012;7:e39217
105. Stuart AD, Stewart JP, Arrand JR. et al. The Epstein-Barr virus encoded cytokine viral interleukin-10 enhances transformation of human B lymphocytes. Oncogene. 1995;11:1711-1719
106. Desbien AL, Kappler JW, Marrack P. The Epstein-Barr virus Bcl-2 homolog, BHRF1, blocks apoptosis by binding to a limited amount of Bim. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5663-5668
107. Milian E, Prats E, Cairo JJ. et al. BHRF1 exerts an antiapoptotic effect and cell cycle arrest via Bcl-2 in murine hybridomas. Journal of biotechnology. 2015;209:58-67
108. Chiu SH, Wu CC, Fang CY. et al. Epstein-Barr virus BALF3 mediates genomic instability and progressive malignancy in nasopharyngeal carcinoma. Oncotarget. 2014;5:8583-8601
109. Wang Q, Tsao SW, Ooka T. et al. Anti-apoptotic role of BARF1 in gastric cancer cells. Cancer letters. 2006;238:90-103
110. Sakka E, Zur Hausen A, Houali K. et al. Cellular localization of BARF1 oncoprotein and its cell stimulating activity in human epithelial cell. Virus research. 2013;174:8-17
111. Takada K. Role of EBER and BARF1 in nasopharyngeal carcinoma (NPC) tumorigenesis. Seminars in cancer biology. 2012;22:162-165
Author contact
![]() Corresponding author: Prof. Ya Cao, Key Laboratory of Carcinogenesis and Invasion, Chinese Ministry of Education, Xiangya Hospital, Central South University, Changsha 410078, China; Telephone: +86-731-84805448, Fax: +86-731-84470589; E-mail: ycao98sina.com.
Corresponding author: Prof. Ya Cao, Key Laboratory of Carcinogenesis and Invasion, Chinese Ministry of Education, Xiangya Hospital, Central South University, Changsha 410078, China; Telephone: +86-731-84805448, Fax: +86-731-84470589; E-mail: ycao98sina.com.