ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2016; 12(11):1341-1356. doi:10.7150/ijbs.15992 This issue Cite
Research Paper
Ginsenoside Rg1 Protects against Oxidative Stress-induced Neuronal Apoptosis through Myosin IIA-actin Related Cytoskeletal Reorganization
Yan Wang1*, Qian Liu2,3*, Yingqiong Xu1, Yuanyuan Zhang1, Yanni Lv4, Yisha Tan1, Nan Jiang1, Guosheng Cao1, Xiaonan Ma5, Jingrong Wang6, Zhengyu Cao1, Boyang Yu1 ![]() , Junping Kou1
, Junping Kou1 ![]()
1. State Key Laboratory of Natural Products, Jiangsu Key Laboratory of TCM Evaluation and Translational Research, Department of Complex Prescription of TCM, China Pharmaceutical University, 639 Longmian Road, Nanjing, 211198, China;
2. Department of Neurology, Jinling Hospital, Nanjing University School of Medicine, 305 East Zhongshan Road, Nanjing, 210002, China.
3. School of Dentistry, Cardiff Institute of Tissue Engineering and Repair, Cardiff University, Heath Park, Cardiff CF14 4XY, UK.
4. Pharmacy Department, The First Affiliated Hospital of Nanchang University, 17 Yongwai Street, Nanchang, 330006, China.
5. Cellular and Molecular Biology Center, China Pharmaceutical University, 639 Longmian Road, Nanjing, 211198, China.
6. State Key Laboratory of Quality Research in Chinese Medicine, Macau Institute for Applied Research in Medicine and Health, Macau University of Science and Technology, Macau, China.
*The authors contributed equally to this work.
Abstract

Oxidative stress-induced cytoskeletal dysfunction of neurons has been implicated as a crucial cause of cell apoptosis or death in the central nervous system (CNS) diseases, such as neurodegenerative and psychiatric diseases. The application of neuroprotectants rescuing the neurons from cytoskeletal damage and apoptosis can be a potential treatment for these CNS diseases. Ginsenoside Rg1 (Rg1), one of the major active components of ginseng, has been reported possessing notable neuroprotective activities. However, there is rare report about its effect on cytoskeleton and its undergoing mechanism. The current study is to reveal the regulatory effects of Rg1 on cytoskeletal and morphological lesion in oxidative stress-induced neuronal apoptosis. The results demonstrated that pre-treatment with Rg1 (0.1-10 μM) attenuated hydrogen peroxide (H2O2)-induced neuronal apoptosis and oxidative stress through reducing the intracellular reactive oxygen species (ROS) production and methane dicarboxylic aldehyde (MDA) level. The Rg1 treatment also abolished H2O2-induced morphological changes, including cell rounding, membrane blebbing, neurite retraction and nuclei condensation, which were generated by myosin IIA-actin interaction. These effects were mediated via the down-regulation of caspase-3, ROCK1 (Rho-associated kinase1) activation and myosin light chain (MLC, Ser-19) phosphorylation. Furthermore, inhibiting myosin II activity with blebbistatin partly blocked the neuroprotective effects of Rg1. The computer-aided homology modelling revealed that Rg1 preferentially positioned in the actin binding cleft of myosin IIA and might block the binding of myosin IIA to actin filaments. Accordingly, the neuroprotective mechanism of Rg1 is related to the activity that inhibits myosin IIA-actin interaction and the caspase-3/ROCK1/MLC signaling pathway. These findings put some insights into the unique neuroprotective properties of Rg1 associated with the regulation of myosin IIA-actin cytoskeletal structure under oxidative stress and provide experimental evidence for Rg1 in CNS diseases.
Keywords: Ginsenoside Rg1, oxidative stress, neuronal apoptosis, cytoskeleton, myosin IIA-actin interaction
Introduction
Oxidative stress is a crucial hallmark in CNS diseases, such as neurodegenerative and other psychiatric diseases [1-3]. Accumulating evidence has confirmed that the extra ROS produced during neural oxidative stress could alter the arrangement of neuronal cytoskeleton [4, 5]. Since the cytoskeleton plays crucial roles in a variety of neuronal physiological processes, including organelle and vesicular trafficking, maintenance of basic architecture and polarity, neuritogenesis and migration [6-9]. It is acknowledged that the cytoskeletal dysfunction has been implicated as an underlying cause of neuronal apoptosis, and even cell death in CNS diseases [10]. Neuroprotectants preventing neuronal damage through oxidative stress-induced cytoskeletal reorganization could be extremely useful in treatment of CNS diseases.
Apoptosis leads to programmed cell death and subsequent removal of damaged, aged or superfluous cells [11]. The executive phase of apoptosis is characterized by a variety of morphological changes, including dynamic plasma membrane blebbing, cell shrinkage and chromatin condensation. The driving power of these dramatic morphological changes is generated by reorganization of actomyosin cytoskeletal structure, which is composed of non-muscle myosin II (hereafter, myosin II) and actin [12]. Myosin II is a molecular motor that binds and contracts filamentous actin (F-actin) by hydrolysis of ATP, converting chemical energy into mechanical forces [13]. The forces also contribute to numerous cellular activities in a variety of cell types, including motility [14, 15], adhesion [16], neuritogenesis [17] and cytokinesis [18]. In neuronal cells, myosin II induces synaptic dysfunction, growth cone collapse and the subsequent neurite retraction under various stimuli [19, 20]. Therefore, cytoskeletal dysfunctions associated with myosin II-actin contractility might contribute to the neuronal apoptosis and the related morphological changes.
Structurally, myosin II is a hexamer composed of two heavy chains (MHC) containing a globular motor domain and a long coiled-coil rod domain, two regulatory light chains (RLC) and two essential light chains (ELC) which bind to the neck area of MHC. The activity of myosin II is determined by the phosphorylation of RLC at Ser19, which enhances the ATPase activity of the MHC domain. RLC phosphorylation is under the monitoring of the MLC kinase (MLCK) and ROCK. As an effector of Rho family proteins, ROCK could either directly trigger the phosphorylation of MLC, or protect the phosphorylation through inhibiting MLC phosphatase [21, 22]. During the execution phase of apoptosis, ROCK1 is cleaved by activated caspase-3 at a consensus DETD1113/G sequence, with its C-terminal inhibitory domain removed. These regulations induce constitutive kinase activity which is necessary for actomyosin contractility [23, 24].
Ginseng, the root of Panax ginseng, is one of the well-known traditional Chinese medicine which has been used as both tonic and haemostatic medicine for thousands of years [25]. Ginsenoside Rg1 (Rg1, structure shown in Figure 1A), one of major active components of Ginseng, has been widely reported to possess neurotrophic and neuroprotective activities against injuries and cytotoxicity through various mechanisms, including cell proliferation, oxidative stress, apoptosis and inflammation [26-29]. It has been recently reported that Rg1 inhibits damage of actin cytoskeleton and protects podocytes from complement-mediated injury partly through reducing the production of ROS and inhibiting the activation of p38 MAPK [30]. Moreover, a combination of four active components including ginsenosides Rb1, Rg1, schizandrin and DT-13 inhibits oxidative stress-induced PC12 cell apoptosis linked with ROCK and MLC activation [31]. However, the role of Rg1 in regulating cytoskeleton organization and the related signal pathway need further investigation.
Considering the fact that the neuronal cytoskeletal disruption happens in neuronal oxidative stress, restoring cytoskeletal structure to rescue the neurons from oxidative stress could be a potential strategy for types of CNS diseases. In the present study, we aimed to investigate the regulation of Rg1 on actomyosin cytoskeleton, through which to maintain the normal morphology and promote neuronal survival following oxidative stress in CNS diseases.
Materials and methods
Materials
Rg1 was purchased from Nanjing Zelang Medical Technology (Nanjing, China. Purity > 98 %). Neurobasal medium, B-27 supplement (50x, minus antioxidants), L-glutamine, soybean trypsin inhibitor, Alexa Fluor® 488 donkey anti-rabbit antibody and Alexa Fluor® 568 phalloidin were peuchased from Thermo Fisher Scientific (San Jose, CA, USA). Penicillin, streptomycin and 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) were purchased from Amresco (Solon, OH, USA). Fetal bovine serum was obtained from ScienCell (San Diego, CA, USA). Cytosine arabinoside, DNase, poly-L-lysine, blebbistatin, N-acetyl-L-cysteine (NAC) and hydrogen peroxide (H2O2) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Y27632 and z-VAD-fmk were obtained from Selleck Chemicals (Houston, TX, USA). Anti-MAP2, anti-caspase-3, anti-MLC and anti-P-MLC (Ser-19) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-ROCK1 and anti-ROCK2 antibodies, Protein A/G PLUS-Agarose, normal rabbit IgG and normal mouse IgG were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-myosin IIA and anti-F-actin antibodies were from Abcam (Cambridge, UK). Anti-GAPDH antibody was purchased from KangCheng (Shanghai, China). HRP-conjugated secondary antibodies were from Boster (Wuhan, China). Protease inhibitor cocktail, RIPA buffer and ECL were from Vazyme Biotech (Nanjing, China). DAPI and Hoechst 33342 were purchased from Beyotime Biotech (Haimen, China). Dimethyl sulfoxide (DMSO) was obtained from SunShineBio (Nanjing, China).
Cell culture
Differentiated PC12 (rat adrenal pheochromocytoma) cells obtained from Shanghai Institute of Cell Biology, Chinese Academy of Sciences. PC12 cells were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM), supplemented with 10 % fetal bovine serum, 100 I.U./mL penicillin, and 100 μg/mL streptomycin at 37 °C in a humidified atmosphere of 5 % CO2.
Primary culture of cortical neurons
Primary cultured cortical neurons were obtained from embryonic day 16 (E16) to E18 Sprague-Dawley rats as described [32]. The pregnant rat was euthanized by CO2 asphyxiation and the neocortices of the embryos were collected within ice bath HBSS buffer. The meninges were stripped followed by trituration with a Pasteur pipette and digestion with trypsin for 25 min, at 37 °C. The suspension was then dissociated via two successive trituration and sedimentation steps in isolation buffer containing soybean trypsin inhibitor and DNase. The suspension was centrifuged, and resuspended with neuron-plating medium containing Eagles's minimal essential medium with Earle's salt (MEM), 2 mM L-glutamine, 10 % fetal bovine serum, 100 I.U./mL penicillin and 0.10 mg/mL streptomycin, pH 7.4. The cells were plated onto poly-L-lysine pre-coated plates at a cell density of 1 x 106 cells/mL and cultured at 37 °C, with 5 % CO2 and 95 % humidity. As soon as the cells attached to the culturing plate, the medium was changed to Neurobasal medium supplemented with B-27, 0.2 mM L-glutamine, 100 I.U./mL penicillin, and 0.10 mg/mL streptomycin, pH 7.4. Cytosine arabinoside (10 μM) was added to prevent the proliferation of non-neuronal cells on day 2 after plating. The culturing medium was changed every two days. The cultures were used for the following experiments between 6 and 9 days in vitro. All the protocol of the animal treatment in this study was approved by Institutional Animal Care and Use Committee of China Pharmaceutical University.
Cell treatment
PC12 cells or neurons were treated with 0.1-10 μM Rg1 (Purity > 98 %) or NAC (500 μM) for 12 h, followed by co-incubation with 100 μM H2O2 for another 12 h. Blebbistatin (1 μM, selective myosin II inhibitor), Y27632 (10 μM, ROCK-specific inhibitor), z-VAD-fmk (10 μM, caspase inhibitor) were added to the culture medium 1 h before Rg1 treatment. The control group was treated with vehicle instead of the drug. All chemicals were dissolved in DMSO and subsequently diluted in the relevant medium at final DMSO concentration of 0.1 % (V/V).
Cell viability assay
The cell viability was measured with 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT). Cells were seeded into 96-well culture plates at a density of 1 × 105 cells/mL and were cultured for 24 h before further treatment. 100 μL of MTT (5 mg/mL) was added to each well and incubated at 37 °C for 4 h. After incubation, the medium was aspirated and 150 μL of DMSO was added to each well to dissolve formazan crystals. Optical densities (OD) were determined by a microplate reader (Epoch, BioTek Instruments, Winooski, VT, USA) at 570 nm and 650 nm. Cell viability was expressed as a percentage with the control cells, which was taken as 100%.
Intracellular ROS measurement
The intracellular ROS level was detected using a non-fluorescent probe 2′,7′- dichlorofluoresein diacetate (DCFH-DA), which can penetrate into the cells where it is converted by ROS to the highly fluorescent 2′,7′-dichlorofluorescein (DCF) [33]. Cells were incubated with DCFH-DA (10 μM) for 30 min at 37 °C. The cells were washed with PBS to remove the non-specific staining. The DCF fluorescence was immediately observed by fluorescence microscopy (Olympus Corporation, Japan) at 40 × magnification. The intensity of DCF fluorescence was quantified using Image J software (National Institutes of Health, Bethesda, MD, USA).
MDA assay
The generation of malondialdehyde (MDA) was analyzed using a commercial kit (Beyotime Biotech, Haimen, China) according to the manufacturer's protocol. Briefly, treated cells were harvested by scrapping and homogenized in RIPA buffer on ice. Cell lysates were then centrifuged at 12,000 g for 10 min at 4 °C to collect the supernatant. MDA levels were detected using microplate reader at 532 nm. We used the bicinchoninic acid disodium (BCA) protein assay kit (Beyotime Biotech, Haimen, China) to quantify protein concentration. The MDA content was expressed as nmol/mg protein.
Caspase-3 activity assay
The caspase-3 activity was determined using the Caspase-3 activity kit (Beyotime Biotech, Haimen, China) as described in the manufacturer's protocol. In brief, cell lysates were prepared after treatment and then added into a 96-well microtitre plate with 10 µl caspase-3 substrate (Ac-DEVD-pNA, 2 mM) for 4 h at 37 °C. Caspase-3 hydrolyzes Ac-DEVD-pNA to produce pNA. The absorbance of pNA was measured at 405 nm using a microplate reader. The specific caspase-3 activity was normalized to total protein concentrations of cell lysates, and then expressed as the ratio of treated cells to untreated cells. Protein concentration was determined using the Bradford protein assay kit (Beyotime Biotech, Haimen, China).
Hoechst 33342 staining
PC12 cells were treated as indicated. The medium was then aspirated and Hoechst 33342 solution (1.2 µg/mL in PBS) was added to the cells. The cells were then incubated for 20 min in the dark at room temperature. Stained cells were washed with PBS and examined by fluorescent microscopy. Bright condensed and fragmented nuclei were calculated as the ratio of apoptotic nuclei to the total number of nuclei.
Analysis of cell apoptosis with flow cytometry
PC12 cells were trypsinized and resuspended into single cell solution of 1 × 106 cell/mL. Cells were then stained with the FITC-Annexin V/PI Apoptosis Detection Kit (BD Biosciences, San Diego, CA, USA) according to the manufacturer's protocol. Samples were detected with FACSCalibur flow cytometer (BD Biosciences, San Diego, CA, USA). Dot plots were analyzed using FlowJo Software (TreeStar, Inc., Ashland, OR, USA).
Membrane blebbing and neurite length of PC12 cells
After treatment as described, PC12 cells were examined for morphological changes (membrane blebbing and neurite retraction) using a phase contrast microscope (Olympus Corporation, Japan). The percent of blebbing cells was determined by the ratio of blebbing cells to the total number of cells, under blinded conditions. Neurite length was measured from the tip of the neurite to the soma of the cell by ImageJ software. Neurite length from each cell is presented as percent of control ± SD.
Transmission electron microscopy (TEM)
Cells were trypsinized, centrifuged, and washed with PBS. The cell pellet was fixed in 2.5 % glutaraldehyde in 0.1 M Phosphate buffer (pH 7.2). The cells were then post-fixed for 1 h in 1 % osmium tetroxide, dehydrated with a series of graded ethanol concentrations (30, 50, 70, 80, 90 and 100 %), embedded in Epon, and sliced to a thickness of 75 nm. TEM images were acquired on a transmission electron microscope (JEM-1010, JEOL Ltd, Japan).
Immunofluorescence
PC12 cells or neurons were seeded into 35-mm confocal dishes (Glass Bottom Dish) at a density of 1 × 104 per dish. Cells were fixed with 4 % paraformaldehyde, permeabilized with 0.1 % Triton X-100 and blocked with 5 % BSA. The cells were then washed with PBS and then incubated overnight at 4 °C with rabbit anti-myosin IIA antibody (1:200) or rabbit anti-MAP2 antibody (1:200). The cells were rinsed three times with PBS and incubated with Alexa Fluor® 488 donkey anti-rabbit antibody (1:200) for 2 h at room temperature. Filamentous actin were stained with Alexa Fluor® 568 Phalloidin (1:200). Nuclei were visualized using DAPI. Fluorescent images were observed with confocal laser scanning microscope (CLSM, LSM700, Zeiss, Germany) and processed using the ZEN imaging software. Neurite length and intensity of MAP2 fluorescence were quantified using ImageJ software.
Manders' overlap coefficients
To analyze co-localization of myosin IIA with actin, images were processed using ImageJ software. Confocal images were thresholded by background subtraction to prevent image noise from distorting the statistics. Manders' overlap coefficients were calculated using the JACoP plugin [34] according to user manual of the ImageJ software. The overlap coefficient indicates an actual overlap of the signals, with 1 representing complete correlation and 0 representing no correlation [35]. The mean ± SD were calculated by the software.
Immunoprecipitation
The protein A/G-PLUS agarose beads were washed with RIPA lysis buffer three times and then incubated with 2 μg indicated antibodies at 4 °C overnight on a rocker table. Following treatment, whole cell lysates (1 mg protein) were incubated with prepared agarose-antibody complex for 4 h at 4 °C. After centrifugation, the agarose beads were washed three times with lysis buffer to remove the unbounded protein. 30 μL of 2 x SDS loading buffer was added to the agarose and then the mixture was boiled. The immunoprecipitates were separated on 10 % SDS-PAGE and blotted with the indicated antibodies. Whole cell lysates (WCL) prior to the immunoprecipitation are shown as a reference.
Western blot analysis
Cells were lysed using RIPA buffer supplied with protease inhibitor cocktail and centrifuged at 12,000 g for 10 min at 4 °C. Cell lysates (50 μg) were separated by 10 % SDS-PAGE. The proteins then transferred onto Polyvinylidene fluoride (PVDF) membranes (Millipore Corporation, Billerica, MA, USA). The membranes were blocked with 5 % BSA and incubated overnight with primary antibodies against ROCK1 (1:300), ROCK2 (1:300), MLC (1:1000), p-MLC (Ser19), caspase-3 (1:1000), GAPDH (1:10000) followed by HRP-conjugated secondary antibodies (1:10000). After washing, detection was performed with ECL. The signal was recorded and photographed by Bio-rad gel imaging system (Bio-Rad, Hercules, CA, USA). Band intensities were quantified using ImageJ software.
Pose validation, molecular preparation and docking
The homology model of myosin IIA was constructed as previously described [36]. The atomic structure of ligand compound Rg1 was constructed using Sybyl version 6.9 (Tripos Associates, St. Louis, MO, USA). Hydrogen atoms and Gastiger-Huckel charges were added to the ligand structure using Sybyl in the tripos force field and an implicit solvent environment. Then the ligand structure was optimized with the minimization steps (1000 cycles) in steepest descent until satisfying the convergent threshold of 0.05 kcal-1•Å-1. Initial complex conformation was generated by Grid-based ligand docking energetics 4.5 (Glide 4.5, Schrödinger LLC, New York, NY, USA) program. Rg1 was then subjected to dock with myosin IIA, the binding site definement and docking method were described previously [36].
Statistical Analysis
All values are expressed as mean ± SD of three independent experiments. Statistical analysis of the data was performed with one-way analysis of variance (ANOVA) test followed by Dunnett's post-hoc test. All statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA, USA). P < 0.05 was considered statistically significant.
Results
Rg1 protects neuronal cells from H2O2-induced oxidative stress
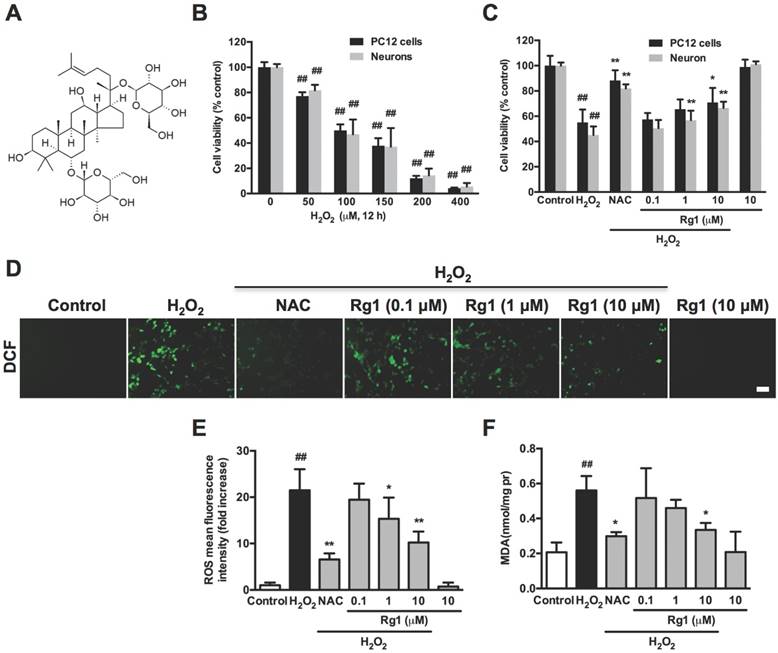
To address the regulation of Rg1 (Chemical structure shown as Figure 1A) on apoptotic cytoskeleton reorganization, we firstly detected the neuroprotective activity of Rg1 upon H2O2 exposure in PC12 cells and neurons. According to the result, 50-400 μM H2O2 induction for 12 h produced a significant decrease in the cellular viability of PC12 cells and neurons in a concentration-dependent manner. While, 50.04 ± 4.76 % of PC12 cells and 46.93 ± 11.66 % of neurons survived after 12 h treatment with 100 μM H2O2, which was adopted as the dosage for the subsequent experiments (Figure 1B). PC12 cells and neurons were pretreated with various concentrations of Rg1 (0.1-10 μM) for 12 h before the addition of 100 μM H2O2 for another 12 h. Antioxidant N-Acetyl-L-Cysteine (NAC, 500 μM) served as positive control. MTT assay showed that Rg1 protected against H2O2-induced reduction in cell viability in a concentration-dependent manner (Figure 1C). As H2O2-induced oxidative stress plays a critical role in neuronal injury [37], we evaluated the intracellular levels of ROS and MDA to determine whether Rg1 attenuates cell damage through reducing oxidative stress. In PC12 cells, H2O2 exposure increased ROS and MDA production, which were inhibited by Rg1 pretreatment (Figure 1D-F). As a positive control, antioxidant NAC (500 μM) exerted the similar effects.
Rg1 protects neuronal cells against H2O2-induced apoptosis
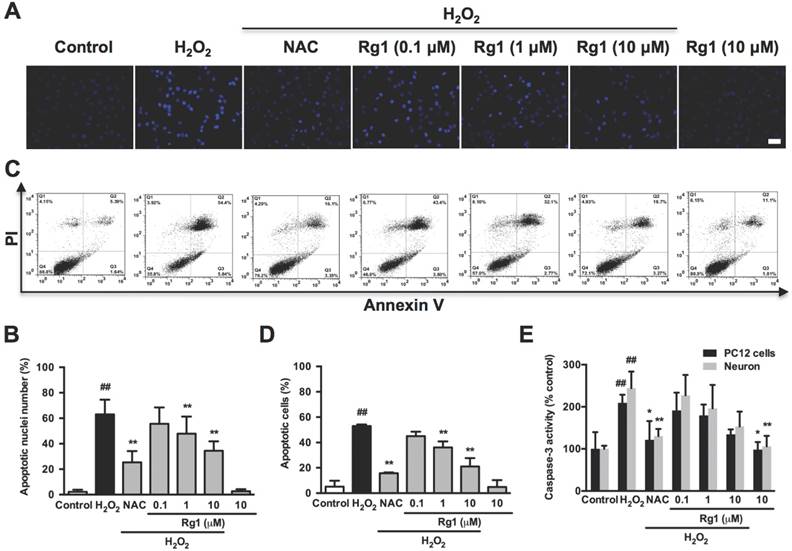
Three methods were used to assess the anti-apoptotic effects of Rg1. (i) morphology of nucleus stained by Hoechst 33342, (ii) Annexin V/PI staining analyzed by flow cytometry, (iii) active caspase-3 enzyme analyzed by caspase-3 activity assay kit. As shown in Figure 2A, we noted the typical apoptotic nuclei with H2O2 exposure, and the apoptotic ratio was 63.02 ± 11.50 %. While, either Rg1 or NAC pre-treatment effectively inhibited H2O2-induced apoptotic nuclear damages. Rg1 pretreatment caused a statistically significant reduction in apoptotic rates at 1 and 10 μM (47.89 ± 13.37 % and 34.47 ± 7.365 %, respectively) (Figure 2B). Annexin V/PI double staining assay demonstrated the similar result on neuronal apoptosis with H2O2 exposure. The oxidative damage increased late apoptosis (Annexin V+/PI+) rate to 52.97 ± 1.242 %, comparing to the corresponding rate of 5.027 ± 4.716 % in the control group. While 1 and 10 μM Rg1 pre-treatment significantly decreased the apoptosis rates to 36.17 ± 4.692 % and 21.07 ± 6.558 %, respectively (Figure 2C and 2D). As for the caspase-3 activity in the neuronal apoptosis, pre-treatment with 10 μM Rg1 significantly decreased H2O2-induced caspase-3 activity in both PC12 cells and neurons (Figure 2E). These results demonstrated the protective activity of Rg1 against oxidative stress-induced neuronal apoptosis.
Effects of Rg1 on H2O2-induced oxidative stress in PC12 cells and neurons. (A) The chemical structure of ginsenoside Rg1. (B) PC12 cells and neurons were treated with different concentration of H2O2 (50, 100, 150, 200, 400 μM) for 12 h. The cell viability was evaluated by MTT assay. (C) PC12 cells and neurons were pre-treated with different concentrations of Rg1 (0.1, 1 and 10 μM) for 12 h prior to 100 μM H2O2 exposure for another 12 h. Positive control was treated with NAC (500 µM). The cell viability of PC12 cells and neurons was evaluated by MTT assay. The data was expressed as percent of the control. (D) Endogenous ROS levels were evaluated in PC12 cells stained with DCFH-DA. Bar: 10 μm. (E) Quantitative analysis of fluorescence intensity as an indication of ROS levels in PC12 cells. (F) MDA content was assayed using the MDA assay kit. Results were obtained from three independent experiments and were expressed as mean ± SD (##P< 0.01 versus control, *P< 0.05 versus H2O2-treated cells, **P< 0.01 versus H2O2-treated cells).
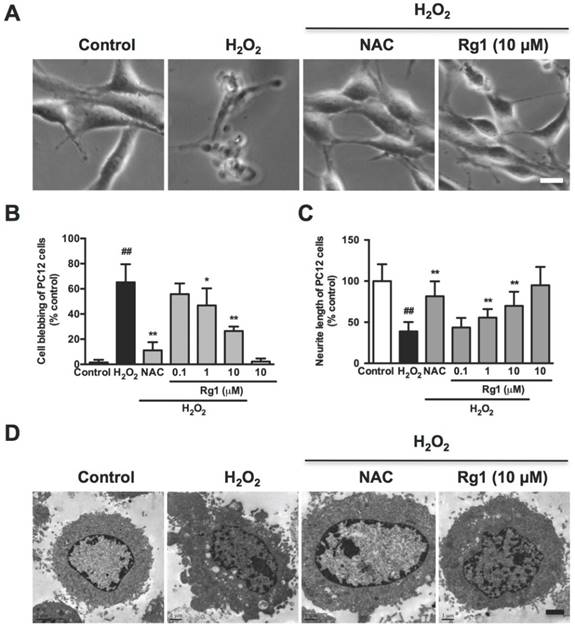
Rg1 inhibits H2O2-induced membrane blebbing and neurite retraction in neuronal cells
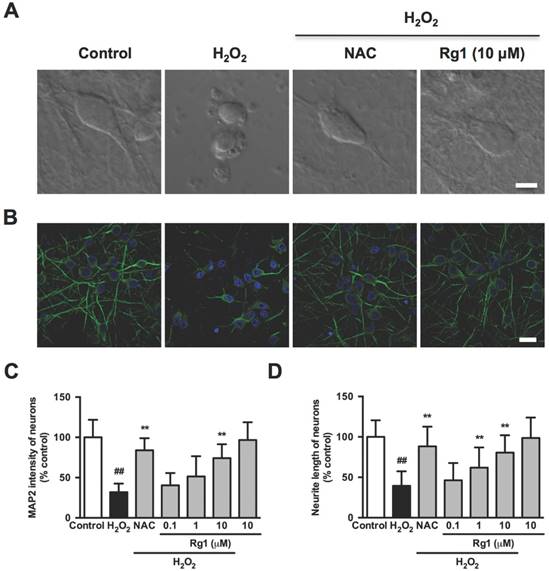
We further investigated the effect of Rg1 on morphological changes in the oxidative stress damaged neuronal cells, including cell contraction, membrane blebbing and neurite retraction, which are the characteristics of the executive phase of apoptosis [38]. Accordingly, the normal PC12 cells were with elongated and spreading morphology (Figure 3A, control). While with the H2O2 treatment, the PC12 cells lost neurite and extensions, shrunk to a rounded shape with typical membrane blebbing and detached from the culture support (Figure 3A, H2O2). Rg1 (10 μM) and NAC (500 μM) significantly attenuated H2O2 -induced membrane blebbing, cell shrinkage, detachment and neurite disruption in PC12 cells (Figure 3A, Rg1 and NAC). A statistical analysis of the aforementioned morphological changes in the PC12 cells demonstrated that pre-treatment with Rg1 (0.1-10 μM) decreased numbers of membrane blebbing cells (Figure 3B) and increased neurite progress (Figure 3C) in a concentration-dependent manner. We used TEM to observe the treated PC12 cells in high resolution. As in Figure 3D, H2O2 induced a typical apoptotic cell with significant membrane blebs and chromatin condensation, while PC12 cells pre-treated with 10 μM Rg1 displayed none bleb, keeping the nucleus intact. The protective effects of Rg1 against H2O2-induced membrane blebbing and neurite retraction were also observed in neurons (Figure 4A). As a marker protein only expressing in perikarya and dendrites of neuronal cells [39], microtubule-associated protein 2 (MAP2) is reported to degrade post ischemia and other metabolic insults, which serves as a marker of neuronal damage [40]. Under confocal microscopy, MAP2 immunoreactivity was localized predominantly to the dendrites of the normal neurons. Exposure to H2O2 caused a remarkable reduction of MAP2 fluorescence and neurite length, which was rescued by Rg1 (Figure 4B). Quantitative analysis revealed that different concentrations of Rg1 pretreatment significantly increased H2O2-induced reduction in MAP2 fluorescence and neurite length (Figure 4D and 4E). These results demonstrated that Rg1 effectively inhibited H2O2-induced membrane blebbing, cellular shrinkage, nuclear contraction and neurite retraction in PC12 cells and neurons.
Protective effects of Rg1 on H2O2-induced apoptosis in PC12 cells and neurons. PC12 cells and neurons were pre-treated with different concentrations of Rg1 (0.1, 1 and 10 μM) for 12 h prior to 100 μM H2O2 treatment for 12 h. NAC (500 µM) treated cells was tested as positive control. (A) Nuclear morphology by Hoechst 33342 staining in PC12 cells. Bar, 10 μm. (B) Quantification of condensed nuclei. (C) Flow cytometric analysis of Annexin V-FITC/PI-stained PC12 cells. Viable cells are Annexin V- and PI-, early apoptotic cells are Annexin V+ and PI- and late apoptotic cells are Annexin V+ and PI+. (D) The quantitative results were represented as the percentage of Annexin V-FITC+/PI+ cells among total cells. (E) Caspase-3 activity. Results were obtained from three independent experiments and were expressed as mean ± SD (##P< 0.01 versus control, *P< 0.05 versus H2O2-treated cells, **P< 0.01 versus H2O2-treated cells).
Rg1 regulates H2O2-induced myosin IIA-actin interaction in neuronal cells
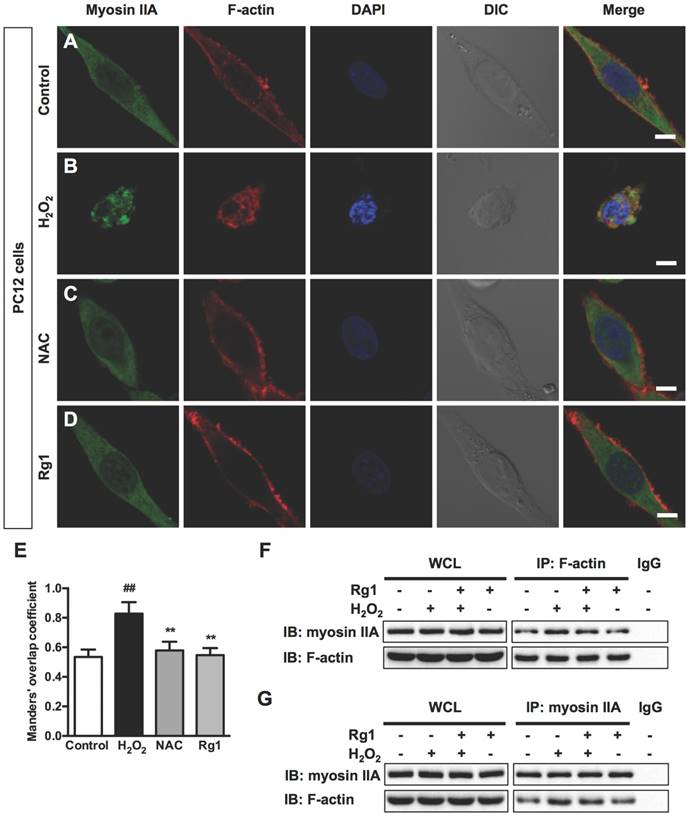
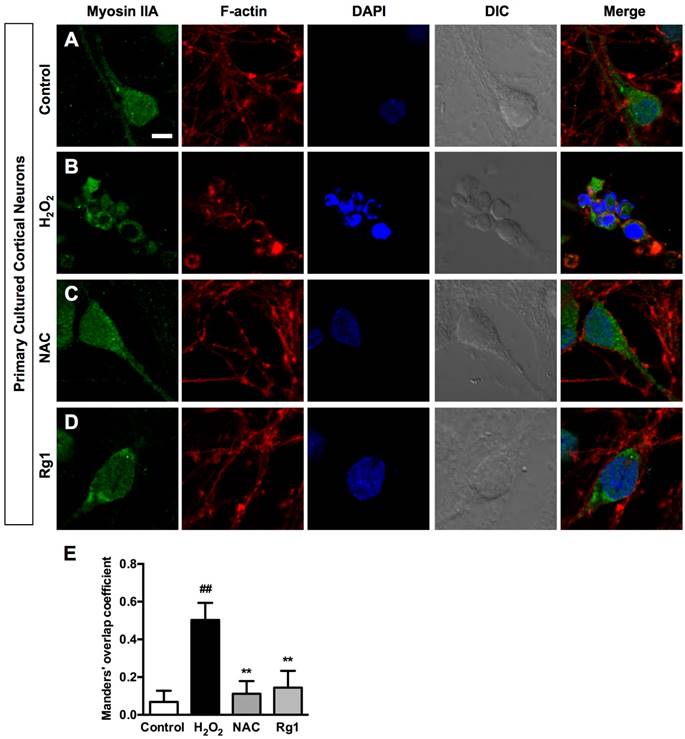
The driving power of the dramatic morphological changes is generated by contractility of myosin II and actin cytoskeletal structure [12]. Immunocytochemical analysis demonstrated a broad distribution of myosin IIA throughout the cytoplasm and relatively less expressed in the neurites of the control PC12 cells. While actin microfilaments not only located in the cell body, but also extended to the protruding areas of PC12 cells (Figure 5A). Exposure to 100 μM H2O2 for 12 h induced dramatic changes on cellular morphology and reorganization of myosin IIA and F-actin. The injured PC12 cells contracted, rounded and formed membrane blebs, which were consistent with previous observations; Myosin IIA accompanied the redistribution of F-actin, both of which accumulated around the periphery of the cell and formed a dense spherical network (Figure 5B). Both ROS scavenger NAC (Figure 5C) and 10 μM Rg1 (Figure 5D) reorganized H2O2-induced distribution of myosin IIA and F-actin, attenuated their interaction and inhibited shrinkage of cells and membrane blebbing. We applied the Manders' overlap coefficients to statistically quantify the colocalization of myosin IIA and F-actin [41]. Quantitative results revealed that the colocalization of the two proteins was more striking in H2O2-treated cells (Manders' coefficient: 0.8282 ± 0.07829) than that in the normal cells (Manders' coefficient: 0.5344 ± 0.04983), while Rg1 significantly decreased the interaction of myosin IIA and F-actin (Manders' coefficient: 0.5790 ± 0.05895) (Figure 5E). We further confirmed the interaction of myosin IIA and F-actin by co-immunoprecipitation analysis. After treatment, the cell lysates were subjected to immunoprecipitation with anti-F-actin antibody. The results of immunoblotting analysis using anti-myosin IIA antibody demonstrated that H2O2 treatment enhanced the association of myosin IIA with F-actin, which was attenuated by Rg1 pre-treatment. Rg1 alone didn't influence the interaction between myosin IIA and F-actin (Figure 5F). The consistent result was obtained when cell lysates were subjected to immunoprecipitation with anti-myosin IIA antibody (Figure 5G). Similar results were also obtained from neurons pretreated with Rg1 (Figure 6A-6D). As shown in Figure 6E, H2O2 significantly increased co-localization of myosin IIA with F-actin (Manders' coefficient: 0.5031 ± 0.09027), comparing to that of the control group (Manders' coefficient: 0.0686 ± 0.05882). While both NAC and Rg1 significantly decreased H2O2-induced co-localization of myosin IIA with F-actin (Manders' coefficients: 0.1117 ± 0.06699 and 0.1446 ± 0.08846, respectively). These findings indicated that H2O2 induced remarkable interaction of myosin IIA and F-actin, which could be inhibited by Rg1 pretreatment in PC12 cells and neurons.
Inhibition of Rg1 on H2O2-induced membrane blebbing and neurite retraction in PC12 cells. PC12 cells were pre-treated with different concentrations of Rg1 (0.1, 1 and 10 μM) for 12 h prior to 100 μM H2O2 treatment. NAC (500 µM) was used as positive control. (A) PC12 cell morphology. Bar, 10 μm. (B) PC12 cells membrane blebbing (C) The percentage of neurite length compared to control. (D) TEM of treated PC12 cells. Bar, 1 μm. Results were expressed as mean ± SD from three independent experiments (##P< 0.01 versus control, *P< 0.05 versus H2O2-treated cells, **P< 0.01 versus H2O2-treated cells).
Inhibition of Rg1 on H2O2-induced membrane blebbing and neurite retraction in neurons. Neurons were pre-treated with different concentrations of Rg1 (0.1, 1 and 10 μM) for 12 h prior to 100 μM H2O2 treatment. NAC (500 µM) was used as positive control. (A) Neurons morphology in each experimental group. Bar, 5 μm. (B) Immunofluorescence of MAP2. Nuclei staining was performed with DAPI. Bar, 20 μm. (C) Quantification of MAP2 fluorescence intensity in neurons. (D) The percentage of neurite length compared to control. Results were expressed as mean ± SD from three independent experiments (##P< 0.01 versus control, *P< 0.05 versus H2O2-treated cells, **P< 0.01 versus H2O2-treated cells).
Inhibition of Rg1 on H2O2-induced activation of caspse-3/ROCK1/MLC pathway
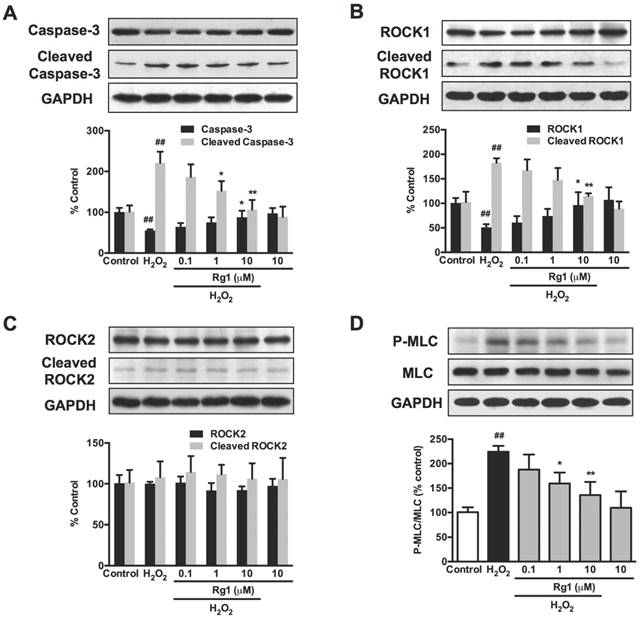
We consequently investigated the effects of Rg1 on cytoskeleton-related caspases-3/ROCK1/MLC pathway. Western blot analysis demonstrated that 100 μM H2O2 exposure for 12 h caused an increased cleaved caspase-3 comparing to the control group. A cleaved fragment (30 kDa) of ROCK1 and increased phosphorylation of MLC were also detected in PC12 cells exposed to H2O2. However, ROCK2 is not cleaved by H2O2 exposure (Figure 7C). Rg1 (0.1-10 μM) pre-treatment inhibited the up-regulated cleaved caspase-3, cleaved-ROCK1 and MLC phosphorylation induced by H2O2. These results verified the neuroprotective activity of Rg1 against oxidative stress through caspase-3/ROCK1/MLC signaling pathway in PC12 cells (Figure 7). Similar results were also observed in the neurons (data not shown).
Myosin IIA mediates the Rg1 protection against H2O2-induced neuronal apoptosis
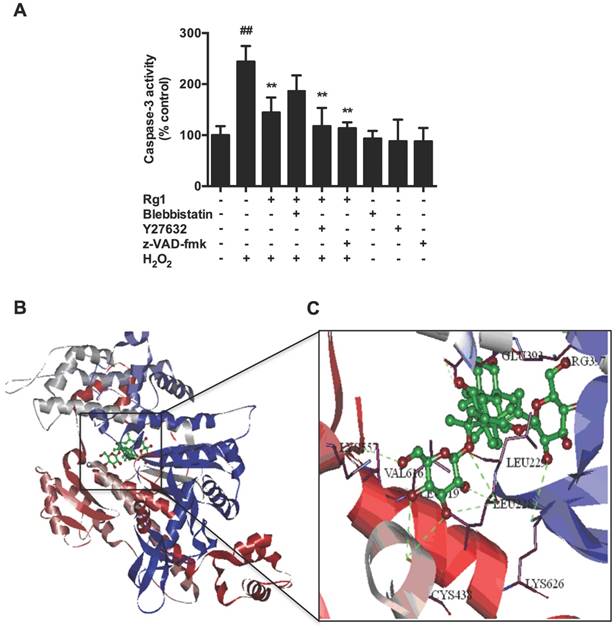
To further elucidate the mechanisms of Rg1, we combined Rg1 with inhibitors of the signaling pathway, blebbistatin, Y27632 or z-VAD-fmk. Caspase-3 activity assay revealed that the combination of Rg1 with blebbistatin treatment partly attenuated the anti-apoptotic activities of Rg1, while either Y27632 or z-VAD-fmk treatment enhanced the neuroprotective activities of Rg1. Blebbistatin, Y27632 and z-VAD-fmk treatment alone had no effects on caspase-3 activity in normal PC12 cells (Figure 8A). To further confirm the influence of blebbistatin on the effects of Rg1, computer-aided homology modeling was applied to investigate the affinity binding between Rg1 and myosin IIA. Construction of myosin IIA model was based on published structures deposited in the Protein Data Bank (PDB code: 1BR2, 1YV3) [42]. Rg1 was positioned in the cavity located within the actin binding cleft of myosin (Figure 8B). Among 5Ǻ interaction residues, LEU619, ARG397, VAL616, LEU228, GLU393, LEU229 were analyzed to get the higher frequency of occurrence of hydrogen bond, Van der waals force and hydrophobic interaction with Rg1, ARG397, LYS626, LEU228, CYS438, LYS557, LEU262 formed H-bond with Rg1 (Figure 8C). These results demonstrated that the binding site of Rg1 with myosin II was similar to that of blebbistatin, and myosin IIA might mediate the regulatory and neuroprotective effects of Rg1 on oxidative stress induced neuronal apoptosis.
Rg1 attenuates H2O2-induced myosin IIA-actin interaction in PC12 cells. PC12 cells were pre-treated with 10 μM Rg1 for 12 h prior to 100 μM H2O2 treatment for 12 h. Positive control was treated with NAC (500 µM). (A-D) Immunofluorescence of myosin IIA and F-actin under confocal microscopy. Nuclei were stained with DAPI. Differential interference contrast (DIC) images were also obtained. Bar, 10 μM. (E) The quantitative colocalization of myosin IIA with F-actin was evaluated on basis of Manders' overlap coefficients. Results were expressed as mean ± SD from three independent experiments (##P< 0.01 versus control, **P< 0.01 versus H2O2-treated cells). Co-immunoprecipitation of myosin IIA and actin was detected by Western blots with indicated antibodies. The bands of left panel are from whole cell lysates (WCL) and the bands of right panel are the same as shown on the left but immunoprecipitated with indicated antibodies. After treatment, cell lysates were subjected to immunoprecipitation with anti-F-actin antibody (F) or anti-myosin IIA antibody (G), and then the precipitates were analyzed by immunoblotting with anti-myosin IIA and anti-F-actin antibodies. Whole cell lysates (WCL) and immunoprecipitates with normal IgG were loaded as positive and negative controls, respectively. IB: immunoblotting, IP: immunoprecipitation and IgG: immunoglobulin G.
Rg1 attenuates H2O2-induced myosin IIA-actin interaction in neurons. Neurons were pre-treated with 10 μM of Rg1 for 12 h prior to 100 μM of H2O2 treatment for 12 h. Positive control was treated with 500 µM of NAC. (A-D) Immunofluorescence of myosin IIA and F-actin under confocal microscopy. Nuclei were stained with DAPI. Differential interference contrast (DIC) images were also obtained. Bar, 10 μM. (E) Quantitative colocalization of myosin IIA with F-actin was evaluated on basis of Manders' overlap coefficients. Results were expressed as mean ± SD from three independent experiments (##P< 0.01 versus control, **P< 0.01 versus H2O2-treated cells).
Discussion
Accumulating evidence has indicated that oxidative stress-induced dysfunction and disruption at the level of cytoskeleton contribute significantly to the cellular injury of CNS disorders, including neurodegenerative disorders and some psychiatric diseases [10, 43]. Any effort aimed at developing specific treatments to reduce oxidative stress, regulate cytoskeletal organization and enhance neuronal survival will be of great significance. Oxidative stress is mediated by excessive ROS, such as superoxide (O2-), hydrogen peroxide (H2O2) and singlet oxygen [44, 45]. Because of its relative stability, exogenous H2O2 has been used to mimic signaling as endogenously produced H2O2 and function as membrane-permeable second messenger [46]. In the present study, we confirmed that Rg1 prevented apoptosis and oxidative stress by reducing caspase-3 activity, nuclear condensation, intracellular ROS and MDA level in PC12 cells and neurons (Figure 1, 2). Cellular apoptosis is characterized by a variety of morphological changes. To further confirm the anti-apoptotic properties of Rg1, we then investigated the effects of Rg1 on neuronal morphology. Our results demonstrated that Rg1 inhibited H2O2-induced morphological changes of neuronal cells, such as cell rounding, membrane blebbing, neurite retraction and nuclei condensation (Figure 3, 4). Taken together, these results demonstrate that the protective effects of Rg1 are mediated by reducing oxidative stress, reorganizing morphological integrity and then attenuating apoptosis, which suggest that Rg1 may be used as a neurological cytoprotector against oxidative stress and morphological defects caused by H2O2 exposure.
Rg1 attenuates H2O2-induced caspase-3/ROCK1/MLC activation in PC12 cells. PC12 cells were pre-treated with different concentrations of Rg1 (0.1, 1 and 10 μM) for 12 h, and then co-cultured with H2O2 (100 μM) for another 12 h. Total cell lysates were subjected to western blotting. Protein level of caspase-3, cleaved caspase-3 (A), ROCK1, cleaved ROCK1 (B), ROCK2, cleaved ROCK2 (C), MLC and P-MLC (Ser-19) (D) in PC12 cells were measured using the relevant antibodies, and GAPDH as loading control. Results were obtained from three independent experiments and were expressed as mean ± SD (##P < 0.01 versus control, *P < 0.05 versus H2O2-treated cells, **P < 0.01 versus H2O2-treated cells).
It is known that actin-myosin cytoskeleton generated contractile force is a major mechanism promoting morphological changes and cellular apoptosis [47]. When under injury, F-actin is reported to polymerize at the distal end of injured neuritis and undergoes reorganization and retrograde flow generated by myosin II, the actin-binding motor protein [48, 49]. The two isoforms of myosin undertake distinct roles: myosin IIB functions as the molecular motor responsible for neurite outgrowth, while myosin IIA maintains focal contact formation and drives neurite retraction [50-52]. Furthermore, myosin IIA is reported to bind to actin more quickly and strongly than myosin IIB [53]. In our study, myosin IIA was broadly distributed throughout the cytoplasm and F-actin extended to the protruding areas under normal conditions. While under H2O2 exposure, F-actin clumped toward the cytoplasmic region of PC12 cells and neurons. The accumulation of F-actin in the myosin II-rich area might increase the interaction of myosin IIA and F-actin, which provided a basis for generating contractile forces, leading to membrane blebbing, neurite retraction and nuclei condensation. Therefore, our results provide evidences of a central role of myosin IIA in mediating force generation in H2O2-induced neuronal cells. However, further studies need to be done to explore the isoform specific roles of myosin IIB in oxidative stress-induced neuronal apoptosis. According to our results, pre-treatment with Rg1 attenuated H2O2-induced myosin IIA-actin interaction and the subsequent membrane blebbing, neurite retraction and nuclei condensation (Figure 5, 6). These results are consistent to the Manders' coefficients, which reflect the amount of myosin IIA overlapping with actin. Under physiological conditions, the Mander's overlap coefficients of PC12 cells were much higher than neuron's, with 0.5344 and 0.0686, respectively, indicating that the interaction of myosin IIA and actin varies between different cell types. The interaction of myosin IIA and actin in normal PC12 cells is significantly stronger compared to that in neurons. When with H2O2-induced damage, the Manders' coefficients increased significantly in both PC12 cells and neurons, indicating myosin IIA and acitn interaction play a critical role regardless of the cell type. The results demonstrate that the protection of Rg1 is mediated by reorganizing neuronal structure and relieving contractile forces generated by myosin IIA-actin cytoskeletal system. Our results put new insights into the neuroprotective mechanisms of Rg1 and the potential therapeutic use of Rg1 on CNS diseases associated with actomyosin cytoskeletal dysfunction.
Rg1 protects against H2O2-induced neuronal apoptosis through myosin IIA. (A) PC12 cells were pre-incubated with blebbistatin (1 μM), Y27632 (10 μM) or z-VAD-fmk (10 μM) in the presence or absence of 10 μM of Rg1 for 12 h, and then treated or untreated with 100 μM of H2O2 for another 12 h. Caspase-3 activity was assayed by caspase-3 activity assay kit. Results were expressed as mean ± SD from three independent experiments (##P < 0.01 versus vehicle-treated cells, *P < 0.05 versus H2O2 treated cells, **P < 0.01 versus H2O2 treated cells). (B) Proposed binding site of Rg1 in myosin IIA. This work used four docking programs based on different matching and shape matching algorithms to assure the accuracy of the final scores and the precision of conformers. Myosin IIA is shown as a cartoon and Rg1 is shown as green sticks in the subpanels. (C) The amplified graph showing possible interacting amino acid residues of Rg1 with myosin IIA (Range of 5 Å).
On the other hand, myosin activity is regulated by the intracellular signaling molecules, such as myosin ATPase activity, F-actin and MLC phosphorylation [38]. Our results presented that H2O2 increased phosphorylated MLC at Ser19, which is reported to enhance the ATPase activity of myosin II and automyosin contraction [54]. ROCK is reported to regulate MLC phosphorylation [22]. Abnormal activation of ROCK has been observed in the pathophysiology of CNS diseases [55, 56]. Our results revealed that ROCK1 was cleaved to form a 30-kDa band upon H2O2 exposure, while ROCK2 was not cleaved. Caspase-3 is known to cleave ROCK1 at a consensus site DETD1113/G, which is absent in the ROCK2 isoform. Cleavage of ROCK1 removes its inhibitory C-terminal domain and activates the kinase activity [23]. Therefore, ROCK1 might be the major effector for membrane blebbing. Our findings confirmed that H2O2-induced caspase-3 cleavage led to activation of ROCK1 and MLC phosphorylation. Pretreatment with Rg1 inhibited the activation of caspase-3, ROCK1 and MLC, reorganized the subsequent myosin IIA-actin cytoskeletal system, and ultimately protected against H2O2-induced neuronal apoptosis (Figure 7).
Furthermore, we used myosin IIA inhibitor blebbistatin, ROCK inhibitor Y27632 and caspase inhibitor z-VAD-fmk to verify the role of myosin II, ROCK and caspase in the protection of Rg1. It is reported that disrupting the contraction of the actomyosin contractility either by myosin II inhibitor blebbistatin or by knockdown of myosin IIA dramatically improve cell survival and cloning efficiency of individualized human embryonic stem cells (hESCs) or human-induced pluripotent stem cells (hiPSCs) [57-59]. Inhibiting ROCK with Y27632 exerted neuroprotective effects by inhibiting expression of cleaved-PARP and caspase-3 in response to proNGF or diabetes [60]. The caspase inhibitor (z-VAD-fmk) is also reported to reduce the expression and activation of caspase and effectively prevented injury-induced apoptosis [61, 62]. The co-administration of Y27632 or z-VAD-fmk with Rg1 produced greater anti-apoptotic effect, because of their different targets and effecting mechanism through the caspase-3/ROCK1/MLC/myosin IIA-actin interaction pathway. However, inhibiting myosin II activity with blebbistatin partly blocked the neuroprotective effects of Rg1 (Figure 8A). It has been established that blebbistatin binds to myosin-ADP-Pi complex at the apex of myosin IIA cleft, which is the motor domain of myosin II [36, 42]. However, the binding to blebbistatin doesn't directly disturb the binding of myosin II with actin, but slows down the phosphate release and promotes the formation of low actin-binding states [63], instead. While for Rg1, according to our result from computer-aided homology modeling, Rg1 was demonstrated preferentially bound to actin-binding cleft of myosin IIA (Figure 8B and 8C), resulting to the directly interference of the binding of myosin IIA to actin. That is believed as a potential mechanism difference between Rg1 and blebbistatin on myosin IIA activity and myosin IIA-actin interaction. On the other hand, the same binding site of Rg1 and blebbistatin might have steric hindrance effect, causing neither of them could bind to myosin IIA. Therefore, the combination treatment of blebbistatin and Rg1 showed antagonistic rather than synergistic effects on neuronal apoptosis.
Owing to the high degree of homology in amino acid sequence and structure which myosin IIA and IIB share [64], it is reasonable to assume that the binding and regulating target of Rg1 could not only be myosin IIA, but also include myosin IIB. However, our data (Figure 5 and 6) and other evidences [52, 58] have consistently demonstrated that, it is myosin IIA, but not myosin IIB who mainly contributes to actomyosin contractility in apoptotic PC12 cells and neurons. Therefore, we mainly studied the effects of Rg1 on myosin IIA, further studies are needed to clarify the regulation of Rg1 on myosin IIB. These results indicate that Rg1 might exert its neuronal protection through myosin IIA and regulate the interaction of myosin IIA and actin to release the contractile forces induced by oxidative stress. An intriguing area of future investigation will be how Rg1 regulates the activity of myosin IIA and the complex interplay between myosin IIA and actin in responses to injury.
However, if Rg1 exerted its protective effects through myosin IIA, how could it inhibit the activation of caspase-3/ROCK1/MLC pathway, which is the upstream of myosin IIA-actin interaction? It has been reported that caspase-3/ROCK1/MLC pathway might be a cyclic positive feedback to regulate apoptosis [31]. Several studies also demonstrate the involvement of myosin II in apoptotic processes in mammalian cells such as fibroblasts, suicidal erythrocyte and stem cells, in which blebbistatin inhibits various stimuli-induced caspase-3 activation [58, 65, 66]. These results suggest that myosin II might feedback to regulate the activation of caspase-3/ROCK1/MLC pathway during cellular apoptosis. In addition to the role in mediating structural rearrangements, myosin II may play a critical role in regulating the biochemical signals of neurons. Therefore, Rg1 might exert its protective effects on myosin IIA, interfere the interaction of myosin IIA with F-actin and then inhibit the related signaling pathway activation, through which Rg1 ultimately magnifies its protection against neuronal apoptosis. The cytoskeletal protein is composed of filamentous organizations of actin filaments, intermediate filaments, microtubules, and multiple regulatory binding partners such as myosin II (the actin motor protein), Tau (microtubule-stabilizing proteins) [67]. Although we focused our analysis on the actomyosin cytoskeleton, both microtubules and intermediate filaments are reported to depolymerize upon injury during the execution phase of apoptosis [68]. Whether disruption of these cytoskeletal proteins can also contribute to neuronal apoptosis and the regulation of Rg1 might be also active area of investigation.
In conclusion, we demonstrate the unique properties of Rg1 associated with reducing oxidative stress and reorganizing cytoskeletal structure to attenuate neuronal apoptosis. The potential mechanism of Rg1 is related to inhibiting myosin IIA-actin interaction and caspase-3/ROCK1/MLC signaling pathway. As normal nervous system function requires basic morphology of neuronal cells, the research that not only considers strategies to promote neuronal survival, but also incorporates ways to reorganize the cytoskeleton, may be an effective way to provide new therapeutic strategies for the CNS diseases.
Abbreviations
BCA: bicinchoninic acid disodium; CNS: central nervous system; DCFH-DA: 2',7'-dichlorofluorescein diacetate; DIC: differential interference contrast; DMSO: dimethyl sulfoxide; H2O2: hydrogen peroxide; IB: immunoblotting; IgG: immunoglobulin G; IP: immunoprecipitation; MAP2: microtubule-associated protein 2; MDA: malondialdehyde; MHC: myosin heavy chain; MLC: myosin light chain; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; myosin II: non-muscle myosin II; NAC: N-Acetyl-L-Cysteine; Rg1: ginsenoside Rg1; ROCK: Rho-associated kinase; ROS: reactive oxygen species; TEM: transmission electron microscopy.
Acknowledgements
The present research was partly supported by the Fundamental Research Funds for the Central Universities (No. 2410724), a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions. This is also an open project of State Key Laboratory of Quality Research in Chinese Medicine (Macau University of Science and Technology, MUST-SKL-2016-09) funded by the Macao Science and Technology Development Fund, Macau Special Administrative Region.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ienco EC, LoGerfo A, Carlesi C. et al. Oxidative stress treatment for clinical trials in neurodegenerative diseases. J Alzheimers Dis. 2011;24(Suppl 2):111-126
2. Agar J, Durham H. Relevance of oxidative injury in the pathogenesis of motor neuron diseases. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:232-242
3. Volpe M, Paneni F. Shooting vascular oxidative stress: new hopes for stroke patients?. Eur Heart J. 2015;36:1573-1575
4. Valdivia A, Duran C, San Martin A. The role of Nox-mediated oxidation in the regulation of cytoskeletal dynamics. Curr Pharm Des. 2015;21:6009-6022
5. Wilson C, Gonzalez-Billault C. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: implications for neuronal development and trafficking. Front Cell Neurosci. 2015;9:381
6. Kapitein LC, Hoogenraad CC. Building the Neuronal Microtubule Cytoskeleton. Neuron. 2015;87:492-506
7. Jensen CS, Watanabe S, Rasmussen HB. et al. Specific sorting and post-Golgi trafficking of dendritic potassium channels in living neurons. J Biol Chem. 2014;289:10566-10581
8. Falnikar A, Tole S, Liu M. et al. Polarity in migrating neurons is related to a mechanism analogous to cytokinesis. Curr Biol. 2013;23:1215-1220
9. Hori K, Nagai T, Shan W. et al. Cytoskeletal regulation by AUTS2 in neuronal migration and neuritogenesis. Cell Rep. 2014;9:2166-2179
10. Li H, Zhu YH, Chi C. et al. Role of cytoskeleton in axonal regeneration after neurodegenerative diseases and CNS injury. Rev Neurosci. 2014;25:527-542
11. Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189:1059-1070
12. Wickman GR, Julian L, Mardilovich K. et al. Blebs produced by actin-myosin contraction during apoptosis release damage-associated molecular pattern proteins before secondary necrosis occurs. Cell Death Differ. 2013;20:1293-1305
13. Butt T, Mufti T, Humayun A. et al. Myosin motors drive long range alignment of actin filaments. J Biol Chem. 2010;285:4964-4974
14. Devreotes P, Horwitz AR. Signaling networks that regulate cell migration. Cold Spring Harb Perspect Biol. 2015;7:a005959
15. Even-Ram S, Doyle AD, Conti MA. et al. Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat Cell Biol. 2007;9:299-309
16. Sackmann E. How actin/myosin crosstalks guide the adhesion, locomotion and polarization of cells. Biochim Biophys Acta. 2015;1853:3132-3142
17. Matsumoto Y, Inden M, Tamura A. et al. Ezrin mediates neuritogenesis via down-regulation of RhoA activity in cultured cortical neurons. PLoS One. 2014;9:e105435
18. Takaine M, Numata O, Nakano K. An actin-myosin-II interaction is involved in maintaining the contractile ring in fission yeast. J Cell Sci. 2015;128:2903-2918
19. Roland AB, Ricobaraza A, Carrel D. et al. Cannabinoid-induced actomyosin contractility shapes neuronal morphology and growth. Elife. 2014;3:e03159
20. Vallee RB, Seale GE, Tsai JW. Emerging roles for myosin II and cytoplasmic dynein in migrating neurons and growth cones. Trends Cell Biol. 2009;19:347-355
21. Kohama K, Ye LH, Hayakawa K. et al. Myosin light chain kinase: an actin-binding protein that regulates an ATP-dependent interaction with myosin. Trends Pharmacol Sci. 1996;17:284-287
22. Kimura K, Ito M, Amano M. et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science. 1996;273:245-248
23. Sebbagh M, Renvoize C, Hamelin J. et al. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001;3:346-352
24. Coleman ML, Sahai EA, Yeo M. et al. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3:339-345
25. Ong WY, Farooqui T, Koh HL. et al. Protective effects of ginseng on neurological disorders. Front Aging Neurosci. 2015;7:129
26. Liu Q, Kou JP, Yu BY. Ginsenoside Rg1 protects against hydrogen peroxide-induced cell death in PC12 cells via inhibiting NF-kappaB activation. Neurochem Int. 2011;58:119-125
27. Radad K, Moldzio R, Rausch WD. Ginsenosides and their CNS targets. CNS Neurosci Ther. 2011;17:761-768
28. Wu J, Pan Z, Wang Z. et al. Ginsenoside Rg1 protection against beta-amyloid peptide-induced neuronal apoptosis via estrogen receptor alpha and glucocorticoid receptor-dependent anti-protein nitration pathway. Neuropharmacology. 2012;63:349-361
29. Ma J, Liu J, Wang Q. et al. The beneficial effect of ginsenoside Rg1 on Schwann cells subjected to hydrogen peroxide induced oxidative injury. Int J Biol Sci. 2013;9:624-636
30. Zhang MH, Fan JM, Xie XS. et al. Ginsenoside-Rg1 protects podocytes from complement mediated injury. J Ethnopharmacol. 2011;137:99-107
31. Shen K, Wang Y, Zhang Y. et al. Cocktail of Four Active Components Derived from Sheng Mai San Inhibits Hydrogen Peroxide-Induced PC12 Cell Apoptosis Linked with the Caspase-3/ROCK1/MLC Pathway. Rejuvenation Res. 2015;18:517-527
32. Cao Z, Cui Y, Busse E. et al. Gambierol inhibition of voltage-gated potassium channels augments spontaneous Ca2+ oscillations in cerebrocortical neurons. J Pharmacol Exp Ther. 2014;350:615-623
33. Hafer K, Iwamoto KS, Schiestl RH. Refinement of the dichlorofluorescein assay for flow cytometric measurement of reactive oxygen species in irradiated and bystander cell populations. Radiat Res. 2008;169:460-468
34. Manders EM, Stap J, Brakenhoff GJ. et al. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992;103( Pt 3):857-862
35. Zinchuk V, Zinchuk O, Okada T. Quantitative colocalization analysis of multicolor confocal immunofluorescence microscopy images: pushing pixels to explore biological phenomena. Acta Histochem Cytochem. 2007;40:101-111
36. Lv Y, Lu S, Lu T. et al. Homology model of nonmuscle myosin heavy chain IIA and binding mode analysis with its inhibitor blebbistatin. J Mol Model. 2013;19:1801-1810
37. Manzanero S, Santro T, Arumugam TV. Neuronal oxidative stress in acute ischemic stroke: sources and contribution to cell injury. Neurochem Int. 2013;62:712-718
38. Ndozangue-Touriguine O, Hamelin J, Breard J. Cytoskeleton and apoptosis. Biochem Pharmacol. 2008;76:11-18
39. Dehmelt L, Halpain S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2005;6:204
40. Shelton MA, Newman JT, Gu H. et al. Loss of Microtubule-Associated Protein 2 Immunoreactivity Linked to Dendritic Spine Loss in Schizophrenia. Biol Psychiatry. 2015;78:374-385
41. Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213-232
42. Allingham JS, Smith R, Rayment I. The structural basis of blebbistatin inhibition and specificity for myosin II. Nat Struct Mol Biol. 2005;12:378-379
43. Cairns NJ, Lee VM, Trojanowski JQ. The cytoskeleton in neurodegenerative diseases. J Pathol. 2004;204:438-449
44. Radi E, Formichi P, Battisti C. et al. Apoptosis and oxidative stress in neurodegenerative diseases. J Alzheimers Dis. 2014;42(Suppl 3):S125-152
45. Kasote DM, Katyare SS, Hegde MV. et al. Significance of antioxidant potential of plants and its relevance to therapeutic applications. Int J Biol Sci. 2015;11:982-991
46. Forman HJ. Use and abuse of exogenous H2O2 in studies of signal transduction. Free Radic Biol Med. 2007;42:926-932
47. Kim T. Determinants of contractile forces generated in disorganized actomyosin bundles. Biomech Model Mechanobiol. 2015;14:345-355
48. Yu P, Santiago LY, Katagiri Y. et al. Myosin II activity regulates neurite outgrowth and guidance in response to chondroitin sulfate proteoglycans. J Neurochem. 2012;120:1117-1128
49. Medeiros NA, Burnette DT, Forscher P. Myosin II functions in actin-bundle turnover in neuronal growth cones. Nat Cell Biol. 2006;8:215-226
50. Wylie SR, Wu PJ, Patel H. et al. A conventional myosin motor drives neurite outgrowth. Proc Natl Acad Sci U S A. 1998;95:12967-12972
51. Wylie SR, Chantler PD. Myosin IIA drives neurite retraction. Mol Biol Cell. 2003;14:4654-4666
52. Cai Y, Rossier O, Gauthier NC. et al. Cytoskeletal coherence requires myosin-IIA contractility. J Cell Sci. 2010;123:413-423
53. Kolega J. The role of myosin II motor activity in distributing myosin asymmetrically and coupling protrusive activity to cell translocation. Mol Biol Cell. 2006;17:4435-4445
54. Mills JC, Stone NL, Erhardt J. et al. Apoptotic membrane blebbing is regulated by myosin light chain phosphorylation. J Cell Biol. 1998;140:627-636
55. Fujita Y, Yamashita T. Axon growth inhibition by RhoA/ROCK in the central nervous system. Front Neurosci. 2014;8:338
56. Jeon BT, Jeong EA, Park SY. et al. The Rho-kinase (ROCK) inhibitor Y-27632 protects against excitotoxicity-induced neuronal death in vivo and in vitro. Neurotox Res. 2013;23:238-248
57. Chen G, Hou Z, Gulbranson DR, Thomson JA. Actin-myosin contractility is responsible for the reduced viability of dissociated human embryonic stem cells. Cell Stem Cell. 2010;7:240-248
58. Walker A, Su H, Conti MA, Harb N, Adelstein RS, Sato N. Non-muscle myosin II regulates survival threshold of pluripotent stem cells. Nat Commun. 2010;1:71
59. Hur EM, Yang IH, Kim DH. et al. Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc Natl Acad Sci U S A. 2011;108:5057-5062
60. Al-Gayyar MM, Mysona BA, Matragoon S. et al. Diabetes and overexpression of proNGF cause retinal neurodegeneration via activation of RhoA pathway. PLoS One. 2013;8:e54692
61. Li J, Lin L, Du L. et al. Radioprotective effect of a pan-caspase inhibitor in a novel model of radiation injury to the nucleus of the abducens nerve. Mol Med Rep. 2014;10:1433-1437
62. Cao Z, Li X, Zou X. et al. Involvement of JNK and caspase activation in hoiamide A-induced neurotoxicity in neocortical neurons. Mar Drugs. 2015;13:903-919
63. Kovacs M, Toth J, Hetenyi C. et al. Mechanism of blebbistatin inhibition of myosin II. J Biol Chem. 2004;279:35557-35563
64. Ma X, Adelstein RS. The role of vertebrate nonmuscle Myosin II in development and human disease. Bioarchitecture. 2014;4:88-102
65. Iwasaki T, Katayama T, Kohama K. et al. Myosin phosphatase is inactivated by caspase-3 cleavage and phosphorylation of myosin phosphatase targeting subunit 1 during apoptosis. Mol Biol Cell. 2013;24:748-756
66. Lang E, Qadri SM, Zelenak C. et al. Inhibition of suicidal erythrocyte death by blebbistatin. Am J Physiol Cell Physiol. 2011;301:C490-498
67. Kevenaar JT, Hoogenraad CC. The axonal cytoskeleton: from organization to function. Front Mol Neurosci. 2015;8:44
68. Oropesa Avila M, Fernandez Vega A, Garrido Maraver J. et al. Emerging roles of apoptotic microtubules during the execution phase of apoptosis. Cytoskeleton (Hoboken). 2015;72:435-446
Author contact
![]() Corresponding author: Prof. Junping Kou and Prof. Boyang Yu, State Key Laboratory of Natural Products, Jiangsu Key Laboratory of TCM Evaluation and Translational Research, Department of Complex Prescription of TCM, China Pharmaceutical University, 639 Longmian Road, Nanjing, 211198, China. Tel & Fax: 86-25-86185158; E-mail: junpingkouedu.cn; boyangyu59com
Corresponding author: Prof. Junping Kou and Prof. Boyang Yu, State Key Laboratory of Natural Products, Jiangsu Key Laboratory of TCM Evaluation and Translational Research, Department of Complex Prescription of TCM, China Pharmaceutical University, 639 Longmian Road, Nanjing, 211198, China. Tel & Fax: 86-25-86185158; E-mail: junpingkouedu.cn; boyangyu59com
Received 2016-4-28
Accepted 2016-8-8
Published 2016-10-25