Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(12):1523-1532. doi:10.7150/ijbs.16233 This issue Cite
Research Paper
Genes involved in angiogenesis and mTOR pathways are frequently mutated in Asian patients with pancreatic neuroendocrine tumors
Wen-Chi Chou1, Po-Han Lin2, Yi-Chen Yeh3,4, Yi-Ming Shyr4,5, Wen-Liang Fang4,5, Shin-E Wang4,6, Chun-Yu Liu4,5, Peter Mu-Hsin Chang4,5, Ming-Han Chen4, Yi-Ping Hung4,5, Chung-Pin Li4,7, Yee Chao4,5*, Ming-Huang Chen4,5* ![]()
1. Department of Hematology-Oncology, Chang Gung Memorial Hospital, Linkou, Taiwan
2. Department of Medical Genetics, National Taiwan University Hospital, Taiwan
3. Department of Pathology, Taipei Veterans General Hospital, Taipei, Taiwan
4. School of Medicine, National Yang-Ming University, Taipei, Taiwan;
5. Department of Oncology, Taipei Veterans General Hospital, Taipei, Taiwan
6. Division of General Surgery, Department of Surgery, Taipei Veterans General Hospital, Taipei, Taiwan
7. Division of Gastroenterology and Hepatology, Department of Medicine, Taipei Veterans General Hospital, Taipei, Taiwan
* These authors contributed equally to this work.
Received 2016-5-19; Accepted 2016-9-25; Published 2016-11-25
Abstract
Introduction: To address the issue of limited data on and inconsistent findings for genetic alterations in pancreatic neuroendocrine tumors (pNETs), we analyzed sequences of known pNET-associated genes for their impact on clinical outcomes in a Taiwanese cohort. Methods: Tissue samples from 40 patients with sporadic pNETs were sequenced using a customized sequencing panel that analyzed 43 genes with either an established or potential association with pNETs. Genetic mutations and clinical outcomes were analyzed for potential associations. Results: Thirty-three patients (82.5%) survived for a median 5.9 years (range, 0.3-18.4) of follow up. The median number of mutations per patient was 3 (range, 0-16). The most frequent mutations were in ATRX (28%), MEN1 (28%), ASCL1 (28%), TP53 (20%), mTOR (20%), ARID1A (20%), and VHL (20%). The mutation frequencies in the MEN1 (including MEN1/PSIP1/ARID1A), mTOR (including mTOR/PIK3CA/AKT1/PTEN /TS1/TSC2/ATM), DAXX/ATRX, and angiogenesis (including VHL/ANGPT1/ANGPT2 /HIF1A) pathways were 48%, 48%, 38%, and 45%, respectively. Mutations in ATRX were associated with WHO grade I pNET (vs. grade II or III, p = 0.043), and so were those in genes involved in angiogenesis (p = 0.002). Patients with mutated MEN1 and DAXX/ATRX pathways showed a trend toward better survival, compared to patients with the wild-type genes (p = 0.08 and 0.12, respectively). Conclusion: Genetic profiles of Asian patients with pNETs were distinct from Caucasian patient profiles. Asian patients with pNETs were more frequently mutated for the mTOR and angiogenesis pathways. This could partially explain the better outcome observed for targeted therapy in Asian patients with pNETs.
Keywords: neuroendocrine tumor, genetic mutation, angiogenesis, mTOR pathway
Introduction
Pancreatic neuroendocrine tumors (pNETs) arise from endocrine cells of the pancreas. This tumor type represents less than 3% of primary pancreatic neoplasms (1). A majority of pNETs are sporadic, and the rest can be associated with hereditary diseases including multiple endocrine neoplasia type I (MEN1), von Hippel Lindau (VHL) syndrome, neurofibromatosis (NF) type I, and tuberous sclerosis (TS). The prevalence rate of pNETs has increased gradually over the past 30 years in the United States as well as in Asian countries (2-4). This increase might partially be explained by improvements in diagnosis, clinical awareness, and a uniform nomenclature of pathological grading introduced by the World Health Organization (WHO) in 2010 (5). Interestingly, pNET incidence and stage distribution at initial diagnosis are distinct between Caucasian and Asian populations. Asian populations have a lower incidence of pNET and fewer patients present with metastatic disease at initial diagnosis than the Caucasian population (2-4).
pNETs are distinct from pancreatic exocrine carcinomas with respect to their indolent clinical behavior, specific tumor immunohistochemical profiles, hormone-secreting ability (some tumors), and promising treatment response to biological and novel targeted agents rather than to traditional chemotherapy in metastatic disease. The distinguishing clinical features between pNETs and exocrine carcinomas were recently described based on their marked difference in genetic alterations. Jiao et al. first determined by whole-exome sequencing that DAXX/ATRA, MEN1, and mTOR pathways are frequently mutated in pNETs in Caucasian patients, but such mutations are rarely seen in exocrine carcinomas (6). Subsequently, Yuan et al. confirmed that DAXX/ATRX and mTOR pathways were frequently mutated in Chinese patients with pNETs (7). However, the frequencies and prognostic values of these mutations differ remarkably with respect to the ethnicities of patients with pNETs. These inconsistencies in pNET-associated genetic alterations can be partially explained by disease rarity, small cohort numbers, and differences in gene sequencing methodologies.
The differences in epidemiological distribution of and genetic alterations in pNETs in Western and Eastern populations imply different pNET pathogenesis between patients of different ethnicities. Furthermore, reports on the distributions and frequencies of genetic mutations are still rare for patients with pNETs, especially from Asian populations. In this study, we developed a customized sequencing panel containing 43 genes with either a known or potential association with pNETs (Table 1), based on a detailed literature review (6-27). The selected genes were categorized into five genetic pathways: the DAXX/ATRX (DAXX and ATRX), MEN1 (MEN1, ARID1A, and PSIP1), mTOR (PIK3CA, PTEN, TSC1, TSC2, AKT1, ATM, and mTOR), angiogenesis (VHL, ANGPT1, ANGPT2, HIF1A, NF1, NF2, TEK, and THBS1), and p53 (ASCL1, CCNB1, CCND1, CCND2, CCND3, CCNE1, CCNE2, CDKN1B, RB1, and TP53) pathways; and miscellaneous genes (ALCAM, BIN2, EIF1AX, FOXA2, GERM1, IGFR1, KIT, MGMT, NEUROD1, PDGFRA, PGF, RET, and RHEB). Using this panel, we aimed to perform a comprehensive genomic analysis to assess mutation frequencies and determine correlations between genetic alterations and clinical outcome in Taiwanese patients with pNETs.
List of genes chosen for sequencing
| Gene | Genomic position | Length | Exons | Coding exons | Transcrip length | Involved pathway | Reference |
|---|---|---|---|---|---|---|---|
| AKT1 | Chromosome 14: 105,235,686-105,262,088 | 26,402 | 15 | 13 | 2,866 | mTOR | Zitzmann K, et al. 20128 |
| ALCAM | Chromosome 3: 105,085,753-105,295,744 | 2,930 | 16 | 15 | 4,701 | mesenchymal stem cell differentiation * | Tachezy M, et al. 20119 |
| ANGPT1 | Chromosome 14: 21,152,336-21,162,345 | 10,009 | 2 | 1 | 1,222 | angiogenesis | Chen MH, et al. 201310 |
| ANGPT2 | Chromosome 8: 6,499,651-6,563,409 | 63,758 | 9 | 4 | 5,416 | angiogenesis | Speisky D, et al. 201211 |
| ARID1A | Chromosome 1: 27,022,524-27,108,595 | 86,071 | 20 | 20 | 8,577 | MEN1 | Fernandez-Cuesta L, et al. 201412. |
| ASCL1 | Chromosome 12: 103,351,464-103,354,294 | 209,991 | 16 | 15 | 4,701 | p53 | Shida T, et al. 200813 |
| ATM | Chromosome 11: 108,093,559-108,239,826 | 146,267 | 63 | 62 | 13,147 | mTOR | Shin JU, et al. 201214 |
| ATRX | Chromosome X: 76,760,356-77,041,702 | 281,346 | 35 | 35 | 11,167 | DAXX/ATRX | Jiao Y, et al. 20116 |
| BIN2 | Chromosome 12: 51,674,822-51,717,948 | 43,126 | 13 | 13 | 2,221 | phagocytosis * | Sánchez-Barrena MJ, et al. 201215 |
| CCNB1 | Chromosome 5: 68,462,837-68,474,072 | 11,235 | 9 | 9 | 2,169 | p53 | Shin JU, et al. 201214 |
| CCND1 | Chromosome 11: 69,455,855-69,469,242 | 13,387 | 5 | 5 | 4,307 | p53 | Jiao Y, et al. 20116 |
| CCND2 | Chromosome 12: 4,273,772-4,305,350 | 31,578 | 5 | 5 | 6,840 | p53 | Jiao Y, et al. 20116 |
| CCND3 | Chromosome 6: 41,934,933-42,050,357 | 115,424 | 5 | 5 | 2,085 | p53 | Jiao Y, et al. 20116 |
| CCNE1 | Chromosome 19: 30,302,805-30,315,215 | 12,410 | 12 | 11 | 2,043 | p53 | Jiao Y, et al. 20116 |
| CCNE2 | Chromosome 8: 94,879,770-94,896,678 | 16,908 | 12 | 11 | 3,330 | p53 | Jiao Y, et al. 20116 |
| DAXX | Chromosome 6: 33,286,335-33,290,791 | 4,456 | 8 | 7 | 2,613 | DAXX/ATRX | Jiao Y, et al. 20116 |
| EIF1AX | Chromosome X: 20,142,636-20,159,962 | 17,326 | 7 | 7 | 4,427 | gene expression * | Fernandez-Cuesta L et al. 201412. |
| FOXA2 | Chromosome 20: 22,561,643-22,566,093 | 4,450 | 2 | 2 | 2,422 | FOXA2 and FOXA3 transcription factor networks * | Gupta A, et al. 200816 |
| GREM1 | Chromosome 15: 33,010,175-33,026,870 | 16,695 | 2 | 1 | 4,168 | signaling by BMP * | Chen MH, et al. 201310 |
| HIF1a | Chromosome 14: 62,162,258-62,214,976 | 52,718 | 15 | 15 | 3,919 | angiogenesis | Pinato DJ, et al. 201417 |
| IGFLR1 | Chromosome 15: 99,192,200-99,507,759 | 315,559 | 21 | 21 | 11,803 | mRNA splicing * | Hansel DE, et al. 200418 |
| KIT | Chromosome 4: 55,524,085-55,606,881 | 82,796 | 21 | 21 | 5,186 | signaling by GPCR * | Han X, et al. 201319 |
| MEN1 | Chromosome 11: 64,570,986-64,578,766 | 7,780 | 10 | 9 | 3,162 | MEN1 | Jiao Y, et al. 20116 |
| MGMT | Chromosome 10: 131,265,448-131,566,271 | 300,823 | 5 | 5 | 1,759 | DNA double-strand break repair * | Schmitt AM, et al. 201420 |
| mTOR | Chromosome 1: 11,166,592-11,322,564 | 155,972 | 58 | 57 | 8,677 | mTOR | Jiao Y, et al. 20116 |
| NEUROD1 | Chromosome 2: 182,541,194-182,545,603 | 4,409 | 2 | 1 | 2,852 | neural stem cell differentiation * | Shida T, et al. 200813 |
| NF-1 | Chromosome 17: 29,421,945-29,704,695 | 282,750 | 58 | 58 | 12,425 | angiogenesis | Jiao Y, et al. 20116 |
| NF-2 | Chromosome 22: 29,999,547-30,094,587 | 95,040 | 16 | 16 | 6,025 | angiogenesis | Jiao Y, et al. 20116 |
| CDKN1B | Chromosome 12: 12,870,058-12,875,305 | 5,247 | 3 | 2 | 2,657 | p53 | Guo SS, et al. 200121 |
| PDGFRA | Chromosome 4: 55,095,264-55,164,414 | 69,150 | 23 | 22 | 6,576 | signaling by GPCR * | Knösel T, 201222 |
| PGF | Chromosome 14: 75,408,537-75,422,487 | 13,950 | 7 | 7 | 1,927 | signaling by GPCR* | Speisky D, et al. 201211 |
| PIK3CA | Chromosome 3: 178,866,311-178,957,881 | 91,570 | 21 | 20 | 9,093 | mTOR | Jiao Y, et al. 20116 |
| PSIP1 | Chromosome 9: 15,464,064-15,511,017 | 46,953 | 16 | 15 | 3,391 | MEN1 | Fernandez-Cuesta L et al. 201412. |
| PTEN | Chromosome 10: 89,622,870-89,731,687 | 108,818 | 9 | 9 | 9,027 | mTOR | Jiao Y, et al. 20116 |
| RB1 | Chromosome 13: 48,877,911-49,056,122 | 178,211 | 27 | 2 | 4,840 | p53 | Yachida S, et al. 201223 |
| RET | Chromosome 10: 43,572,475-43,625,799 | 53,324 | 20 | 20 | 5,659 | signaling by GPCR* | Falchetti A, et al. 200824 |
| RHEB | Chromosome 7: 151,163,098-151,217,010 | 53,912 | 8 | 8 | 2,075 | signaling by GPCR* | Pollizzi KN, et al. 201525 |
| THBS1 | Chromosome 15: 39,873,280-39,891,667 | 18,387 | 22 | 21 | 7,775 | angiogenesis | Chan AO, et al. 200326 |
| TEK | Chromosome 9: 27,109,147-27,230,173 | 121,026 | 23 | 23 | 4,760 | angiogenesis | Detjen KM, et al. 201027 |
| TP53 | Chromosome 17: 7,565,097-7,590,856 | 25,760 | 11 | 10 | 2,579 | p53 | Yachida S, et al. 201223 |
| TSC1 | Chromosome 9: 135,766,735-135,820,008 | 53,273 | 23 | 21 | 8,604 | mTOR | Jiao Y, et al. 20116 |
| TSC2 | Chromosome 16: 2,097,466-2,138,716 | 41,250 | 42 | 41 | 6,156 | mTOR | Jiao Y, et al. 20116 |
| VHL | Chromosome 3: 10,183,319-10,195,354 | 12,035 | 3 | 3 | 3,737 | angiogenesis | Yuan F, et al. 20147 |
* There genes were categorized as “miscellaneous” of the involved pathway.
Materials and Methods
Forty patients with pNETs, treated at the Taipei Veteran General Hospital between 1998 and 2013, were enrolled in this study retrospectively. All patients had a definite pathological diagnosis of pNETs, and those who had mixed adeno-endocrine carcinomas were excluded. All tumor specimens were formalin-fixed and paraffin-embedded. Patient data on demographics, tumor stage, and pathological grade [as classified according to the World Health Organization 2010 definition (Ki67 index)] were collected. Patients were followed up with, and overall survival was analyzed. The study was approved by the local institutional review board of the Taipei Veterans General Hospital (2014-01-002A).
Constructing a target-enriched sequencing library
The cancerous portions of pancreatic neuroendocrine tumors were isolated by a pathologist and then the genomic DNA (gDNA) was extracted from formalin-fixed paraffin-embedded tissues by a QIAamp DNA FFPE Tissue Kit (Qiagen Inc., Valencia, CA USA). The purity and concentration of gDNA samples were first confirmed by electrophoresis, Nanodrop 2000 (Thermo Scientific, USA), and a Qubit 2.0 Fluorometer (Life Technologies, USA). Double-stranded DNA (dsDNA; 2 μg) that passed quality-control steps was sheared to fragments of approximately 300 bp with an M220 focused ultrasonicator (Covaris, USA). Size distribution of the fragmented DNA was confirmed using a Bioanalyzer 2100 (Agilent Technologies, USA), and shotgun genomic libraries were then generated using KAPA Hyper Prep kits (Kapa Biosystems, USA). These libraries were examined with the Agilent Bioanalyzer 2100. In order to comprehensively analyze the 43 genes of interest, the target gene library was enriched by a NimblGen Seqcap EZ system (Roche NimblGen, INC.) targeting their exons (Supplementary Table 1) according to the manufacturer's protocol. Captured libraries were further amplified by performing 12 cycles of ligation-mediated PCR with HiFi HotStart ReadyMix (Kapa Biosystems), followed by purification with AMPure XP beads for MiSeq sequencing.
MiSeq high-throughput sequencing and data processing
After library concentration was determined by real-time quantitative PCR, libraries were denatured and sequenced on a MiSeq platform with v2 reagent for paired-end sequencing (2 × 150 bp). Instrument control, cluster generation, image capture, and base calling were processed with Real Time Analysis software 1.18.54, MiSeq Control software 2.4.1.3, and MiSeq Report software 2.4.60.8. FASTQ files generated by MiSeq Report were used for further analysis. The algorithm used for analysis was described previously (28). Briefly, the FASTQ files were aligned to the human reference genome (February 2009, GRCh37/hg19) using BWA software (version 0.7.4) (29). Picard tools (version 1.90) were used to perform the necessary data conversion, sorting, and indexing (30). For single nucleotide polymorphisms (SNPs) and small insertion/deletions (indels), Genome Analysis Toolkit (GATK; version 2.5-2) was used for variant calling by using Base/indel-calibrator and HaplotypeCaller (31). Genetic variants larger than 100 bp cannot be identified by GATK, so Pindel or Breakdancer software were used to find structural variants, such as large deletions, insertions, and duplications (32). The variant calls were then annotated using ANNOVAR (2014-09-09) (33). Only the allele frequency of the mutation greater than 10% was included.
To exclude germline variants, those variants with allelic frequencies >1% in either the 1000 Genomes Project or NHLBI-ESP 6500 exome project were filtered. The definition of the pathogenic mutation was mainly based on the American College of Medical Genetics guideline (34). Briefly, the frameshift insertion or deletion (indel), nonsense, and splice-site variants with >1% allele frequencies were considered as pathogenic somatic mutations. Missense variants were evaluated by SIFT and PolyPhen2 scores (27, 28); and only missense variants with scores >0.95 in PolyPhen2 and <0.05 in SIFT scores were further analyzed.
Statistical analysis
Chi-square (χ2) and Fisher's exact tests were performed to determine significant associations between genetic variables (PIK3CA, AKT, KRAS mutations, and PTEN loss in pancreatic cancer tissues) and clinical characteristics. Kaplan-Meier survival curves were drawn and tested by Log-rank test to evaluate differences in disease-free survival between the variables. The effects of mutations on prognosis were analyzed by univariate and COX multivariate risk models. Significant differences were defined as P < 0.05.
Results
Clinical characteristics of patients and DNA sequencing on the Illumina platform
Clinical characteristics of the patients are listed in Table 2. The median age at diagnosis was 55 years of age and 58% of patients were male. Most patients (67.5%) had early-stage tumors (AJCC stage I or II). The tumors were functional in 9 out of 40 patients (22.5%). Four patients (10%) had multiple tumors within the pancreas by tissue proof at the time of NET diagnosis. The percentages of patient tumors with WHO grade I, II, and III or unknown were 77.5%, 17.5%, and 5%, respectively.
Patient clinical characteristics
| Variable | Category | No (%) |
|---|---|---|
| Median Age, years (range) | 55 (19-71) | |
| Gender | male | 23 (57.5) |
| female | 17 (42.5) | |
| AJCC stage | 1 | 12 (30) |
| 2 | 15 (37.5) | |
| 3 | 4 (10) | |
| 4 | 9 (22.5) | |
| T classification | 1 | 7 (17.5) |
| 2 | 22 (55) | |
| 3 | 3 (7.5) | |
| 4 | 8 (20) | |
| N classification | 0 | 31 (77.5) |
| 1 | 9 (22.5) | |
| M classification | 0 | 31 (77.5) |
| 1 | 9 (22.5) | |
| Functional status | yes | 9 (22.5) |
| Tumor number | single | 36 (90) |
| multiple | 4 (10) | |
| Pancreatic location | head | 16 (40) |
| other site | 24 (60) | |
| WHO grade | 1 | 31 (77.5) |
| 2 | 7 (17.5) | |
| Variable | Category | No (%) |
| WHO grade | 3 | 2 (5) |
Capture enrichment and NGS
Two independent capture experiments and MiSeq runs were performed to complete the NGS sequencing for the 40 patients. The average output was 3745.3 million per experiment and Q30 was greater than 94.0%, with ~68.0% of reads (range, 61.4-75.0%) mapping to the target region. The average mean coverage for the targeted exons was 180.0 ± 47.1, 79.9% of the exons had a coverage of ≥ 100 reads, and 90.1% of the exons had a coverage of ≥ 75 reads.
Genetic mutations
A total of 139 genetic mutations were identified in the 43 genes in the 40 patients. The mean and median number of mutations per patient were 3.5 and 3.0 (range, 0-16), respectively (Figure 1, supplementary table 1 and table 2). The order of the most frequently mutated genes (over 10%) per person was: ATRX (28%), MEN1 (28%), ASCL1 (28%), TP53 (20%), mTOR (20%), ARID1A (20%), VHL (20%), NF1 (18%), TSC2 (18%), DAXX (15%), and ANGPT2 (13%) (Table 3). Both ATRX and DAXX mutations were identified in two patients (5%). We next selected genes within the same pathway for further analysis. The mutation frequencies of the MENI-, mTOR-, DAXX/ATRX-, and angiogenesis-pathway genes per person were 48%, 48%, 38%, and 45%, respectively (Table 4).
All gene mutations identified in the 40 Taiwanese patients with pancreatic neuroendocrine tumors (pNETs). The yellow, blue, green, red, purple, and gray color represent gene involved the DAXX/ATRX, MEN1, mTOR, angiogenesis, TP53, and miscellaneous pathway, respectively.

Commonly mutated genes in pancreatic neuroendocrine tumors in Taiwanese, Chinese, and Caucasian cohorts
| Study | Current study (Taiwanese cohort) n = 40 | Chinese cohort7 n = 37 | Caucasian cohort6 n = 68 |
|---|---|---|---|
| ATRX | 11 (27.5%) | 13 (35.1) | 12 (17.6%) |
| MEN1 | 11 (27.5%) | 13 (35.1) | 30 (44.1%) |
| ASCL1 | 11 (27.5%) | n/s | 0 |
| TP53 | 8 (20%) | 5 (13.5%) | 2 (2.9%) |
| mTOR | 8 (20%) | n/s | 0 |
| ARID1A | 8 (20%) | n/s | 0 |
| VHL | 8 (20%) | 15 (40.5%) | 0 |
| NF1 | 7 (17.5) | n/s | 0 |
| TSC2 | 7 (17.5%) | 16 (43.2%) | 6 (8.8%) |
| DAXX | 6 (15%) | 11 (29.7) | 17 (25%) |
| ANGPT2 | 5 (12.5%) | n/s | 0 |
| PIK3CA | 3 (7.5%) | n/s | 1 (1.5%) |
| PTEN | 3 (7.5%) | 7 (18.9%) | 5 (7.4%) |
n/s: no sequencing
Mutation frequencies in cellular pathways in pancreatic neuroendocrine tumors in Taiwanese, Chinese, and Caucasian cohorts
| Study | Current study (Taiwanese cohort) n = 40 | Chinese cohort7 n = 37 | Caucasian cohort6 n = 68 |
|---|---|---|---|
| MEN1 pathway | 48% | 35%a | 44% |
| DAXX/ATRX | 38% | 54% | 43% |
| TP53 pathway | 20% | 14%b | 3% |
| VHL pathway | 45% | 41%c | 0% |
| mTOR pathway | 48% | 54%d | 15% |
asequencing MEN1 gene only, bsequencing TP53 only, csequencing VHL gene only,
dsequencing PTEN and TSC2 genes only.
Association between genetic mutations and clinicopathological characteristics
Only ATRX mutations were more frequently detected in WHO grade I than in grade II or III tumors (35% vs. 0%, p = 0.043) (Table 5). There were no differences in the patients' clinicopathological characteristics in terms of age, gender, tumor stage, number of tumors, tumor location within the pancreas, functional status, and WHO grade for other mutations. However, a trend toward higher MEN1 mutation rates was found in patients with multiple tumors compared to those with single tumors (75% vs. 22%, p = 0.056). Patients with metastatic disease and T2-T4 primary tumor stage of (>2 cm in largest diameter) had higher TP53 mutation rates than those without metastatic disease (44% vs 13%, p = 0.059), and T1 stage of primary tumor (≤2 cm in largest diameter) (28% vs 0%, p = 0.080).
Associations between genetic mutations and clinicopathological characteristics in pancreatic neuroendocrine tumors
| Gene | Variable | Mutation no/total patient (%) | p value |
|---|---|---|---|
| ATRX | WHO grade I | 11/31 (35%) | 0.043 |
| WHO grade II or III | 0/9 (0%) | ||
| MEN1 | Single tumor | 8/36 (22%) | 0.056 |
| Multiple tumors | 3/4 (75%) | ||
| TP53 | Metastatic disease | 4/9 (44%) | 0.059 |
| No metastatic disease | 4/31 (13%) | ||
| TP53 | Primary tumor >2 cm | 8/29 (28%) | 0.080 |
| Primary tumor ≤2 cm | 0/11 (0%) |
Only variables in Chi-square (χ2) test or Fisher's exact test with p < 0.10 are presented in the table.
When analyzing clinicopathological characteristics by rate of mutation within the same pathway (Table 6), patients with WHO grade I tumors had a higher incidence of mutations in angiogenesis-pathway genes than those with WHO grade II-III tumors (58% vs 0%, p = 0.002). Patients with multiple tumors had a higher incidence of mutations in genes in the MEN1 pathway than those with single tumors (100% vs. 42%, p = 0.042), while patients with T2-T4 stage primary tumors had a trend toward lower incidence of MEN1 pathway genes mutation than those with T1 stage (38% vs. 73%, p = 0.078).
Associations between pathway mutations and clinicopathological characteristics in pancreatic neuroendocrine tumors
| Pathway | Variables | Mutation no/total patient (%) | p value |
|---|---|---|---|
| MEN1 | Single tumor | 15/36 (42%) | 0.042 |
| Multiple tumors | 4/4 (100%) | ||
| MEN1 | Primary tumor >2 cm | 11/29 (38%) | 0.078 |
| Primary tumor ≤2 cm | 8/11 (73%) | ||
| Angiogenesis | WHO grade I | 18/31 (58%) | 0.002 |
| WHO grade II or III | 0/9 (0%) |
Only variables in Chi-square (χ2) test or Fisher's exact test with p < 0.10 are presented in the table.
Association between genetic mutations, survival outcome, and treatment response
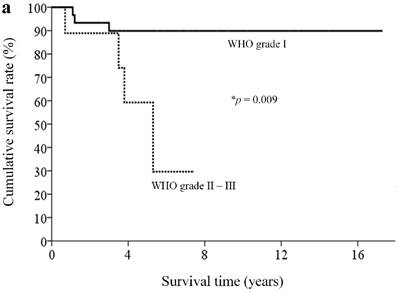
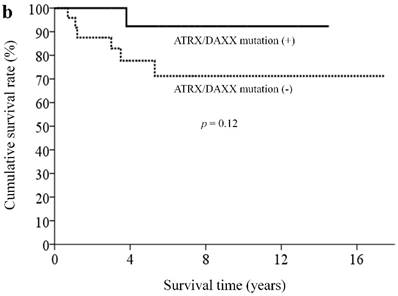
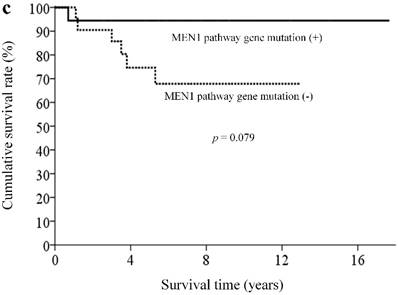
With a median follow-up of 5.9 (range, 0.3-18.4) years, 33 patients (82.5%) remained alive. By clinicopathological factors, patients with WHO grade I tumors (Ki67 less than 2%) had significantly better survival outcomes than those with either WHO grade II or III tumors (median survival time not reached vs. 5.3 years, p = 0.009) (Figure 2a). We first assessed alteration status of single genes with a mutation rates over 10% in this study for survival analysis. Owing to the small number of patients and the rarity of genetic mutations and deaths, both could limit the statistical power; we did not find statistically significant associations between the patient survival and presence or absence of certain genetic alterations. When analyzing survival outcome by pathway mutation rate, patients with genes mutated in the DAXX/ATRX pathway showed a trend toward better survival outcome than patients without mutations in this pathway (log rank, p = 0.12) (Figure 2b). Patients with mutated genes in the MEN1 pathway showed a trend toward better survival than those without mutations in this pathway (long rank, p = 0.079) (Figure 2c). Totally, 6 patients in our cohort ever received everolimus treatment. The best tumor response was partial response in 3 patients (two of these patients had mutated gene in the mTOR pathway) and stable disease in another 3 patients (two of these three patients had mutated gene in the mTOR pathway). One patient, who had no mutated gene in the angiogenesis pathway, ever received sunitinib treatment, with a best tumor response of stable disease.
Kaplan-Meier overall survival curves for patients with pNETs, stratified by the status of (a) WHO grade, (b) ATXX/DAXX mutations, and (c) MEN1-pathway gene mutations.
Discussion
Our study of a 43-gene panel for 40 Taiwanese patients with pNETs identified 139 somatic mutations. Similar to previous reports, our study showed that the majority of the mutated genes were involved in the MEN1, mTOR, or DAXX/ATRX pathways. However, there were some differences between the genetic alterations in Caucasian and Asian patients with pNETs. First, there were fewer mutated genes per patient in Asian populations (mean number of mutated genes were 3.5 in this study and 3.6 in a Chinese cohort) compared to Western population (16 from the whole exome sequencing in a Western cohort) (6, 7). However, when using the same 43 genes panel in our study to examine the Western cohort (6), only 1.1 mutated genes were identified. Second, genes involved in the mTOR and angiogenesis pathways were more frequently mutated in Asian populations. Our study indicated a discrepancy between mutation profiles of Asian and Caucasian patients with pNETs. Doctors might consider these differences in ethnicity when strategizing targeted therapy for patients with pNETs.
pNETs are characterized by their high degree of vascularization and increased expression of VEGFR2/3 within tumors (35-39). Microvessel density around the tumor tissues was shown to be greater in low-grade tumors than in high-grade ones (40), and increased expression of angiogenesis-related genes was correlated with cell differentiation and good survival outcome in patients with pNETs (36). Sunitinib (sutent, Pfizer Inc, NY, USA), a small tyrosine kinase inhibitor for angiogenesis, has demonstrated clinical efficacy in improving survival outcome in patients with well-differentiated advanced pNETs, in a phase-III trial (41). Sunitinib efficacy is higher in non-white patients than in white patients, and in low-grade (Ki67 < 5%) than in high-grade tumors. However, the relationship between specific genes involved in angiogenesis in pNETs was previously unclear. None of the mutated genes involved in angiogenesis were reported in Caucasian patients with pNETs, whereas Yuan et al. reported that 41% of Chinese patients with pNETs harbored a mutated VHL (7). Similar to the report by Yuan et al., our study identified that 45% of the angiogenesis-pathway genes were mutated in the Taiwanese patients with pNETs. Importantly, our study showed that such mutations were frequent in patients with WHO grade I tumors, which may account for the relevant clinical benefit seen in non-white, low-grade tumor patients receiving sunitinib treatment (41). The treatment response of sunitinib with regard to mutated angiogenesis pathway in patients with pNETs warrants further exploration.
The mTOR pathway is central to the control of transcription and translation, and cellular migration, metabolism, proliferation, and survival (42). Activation of the mTOR pathway is one of the common tumor-related signaling abnormalities, which are detected in various tumors including NETs and those in the breast and lung (42-44). Around 15% of Caucasian patients with pNETs harbored gene mutations in the mTOR pathway (including those in PTEN, TSC2, and PIK3CA). By sequencing mTOR pathway genes with only PTEN and TSC2, Yuan observed that 54% of Chinese patients with pNETs carried gene mutations in the mTOR pathway (7). In line with Yuan's report, our study showed that the mTOR-pathway genes were frequently mutated in the Asian patients with pNETs. In addition to the most well-known mutated genes in the mTOR pathway, our study found that other genes including mTOR, AKT1, TSC1, and ATM were also frequently mutated in the Asian patients. Therefore, detection of mutations in the mTOR pathway by sequencing only PTEN, TSC2, and PIK3CA in Asian patients with pNETs is not sufficiently comprehensive.
Everolimus (Afinitor, Norvatis Pharmaceuticals, NJ, USA), an mTOR inhibitor, has recently demonstrated efficacy in a phase-III trial in patients with low- or intermediate-grade pNETs (RADIANT-3 trial) (45). Everolimus is more efficacious in Japanese patients than in the overall population, with 5.2- and 2.4-fold improvements in progression-free survival, respectively, compared with that by placebo (46). Similar improved progression-free survival has been noted in Asian vs. Western patients treated with everolimus for other gastrointestinal and pulmonary NETs, from another phase-III (RADIANT-4) trial (47) and one multi-institution observational study in Taiwan (48). In patients with advanced breast cancer, everolimus efficacy was positively correlated with the PI3K/AKT/mTOR-pathway mutation status (49). The relative abundance of mutations in the mTOR pathway in Asian patients with pNETs might partially explain their improved response to treatment with mTOR inhibitors (50). However, the correlation of mTOR-pathway mutation status with everolimus efficacy in pNETs needs to be validated in further prospective studies.
Mutations in DAXX and ATRX, both of which encode proteins involved in chromatin remodeling, were detected in 40% of pNETs (6). Initially, mutations in these genes were found to be mutually exclusive (6). Our study found that 2 out of 11 patients with mutated ATRX also have mutations in DAXX. A similar finding was also reported by Yuan et al., who identified 4 out of 13 patients with mutations in both ATRX and DAXX (7). Tumors with wild-type DAXX and ATRX show intact nuclear expression of these genes, whereas this nuclear localization is typically lost in tumors with mutations in DAXX or ATRX. Loss of DAXX or ATRX, which had been found only in well-, but not poorly differentiated pNETs (50), indicated that alterations in DAXX/ATRX genes represent unique biological pNET entities. Jiao et al. reported that mutation in DAXX/ATRX in pNETs were associated with prolong survival relative to those patients who lacked these mutations (6). In line with report from Jiao et al., our findings also confirmed that metastatic pNETs with mutations in either DAXX or ATRX has favorable outcomes. However, Yuan et al. reported the contradiction report about the prognostic value of DAXX/ATRX in patients with pNETs. Furthermore, mutations in both DAXX and ATRX were previously associated with shortened survival and chromosomal instability in pNETs (51) and leiomyosarcomas (52). Collectively, the biological role and prognostic value of DAXX or ATRX mutations in patients with pNETs are discordant and this question needs further exploration.
There are some limitations to our study. Given the study's retrospective nature and long recruitment period, clinical practice patterns might have changed over time. Second, our study included various tumor stages, WHO grades, and antitumor strategies, and the effectiveness of each antitumor strategy on a particular tumor stage or grade may potentially affect survival. Third, because small number of patients and deaths (7 of 40 patients, 17.5%) after the median follow-up of 5.9 years, the impact of genetic alternation on survival outcome was difficult to analyze statistically. Fourth, the discrepancy of tissue sequencing among different studies may bias the results. Jiao et al. sequenced both tumor and matched normal tissue DNA, while our study and Yuan et al. sequenced tumor DNA only (6, 7). In addition, different genome analysis kit used between our study and Yuan et al. report (7) for variant calling to confirm the SNPs and small indels in Asian population may turn out different results as the ethnic variations of the known SNPs. All of these divergences may partially explain there were fewer genes mutated per patient in Asian population than in Caucasian cohort. Finally, our study showed a trend toward better survival outcome in patients with mutated genes in the MEN1 pathway or DAXX/ATRX pathway than patients without those mutations, however, the difference of survival outcomes were statistically insignificance, which mainly due to the limitation of small patient numbers. Because of the rarity of the disease and diversity of treatment strategies, a well-designed multisite prospective study is necessary to address these limitations.
In conclusion, our study showed that genetic profiles of Asian patients with pNETs were distinct from that of Caucasian patients. Asian patients with pNETs were more frequently mutated for genes of the mTOR and angiogenesis pathway. This could partially explain the improved outcome observed for targeted therapy among Asian patients with pNETs.
Supplementary Material
Additional File 1Supplementary table 1.
Supplementary table 2.
Acknowledgements
This work was supported by the Szu-Yuan Research Foundation of International Medicine, the Taiwan Cancer Clinic Foundation, and the Chong Hin Loon Memorial Cancer and Biotherapy Research Center
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fraenkel M, Kim M, Faggiano A. et al. Incidence of gastroenteropancreatic neuroendocrine tumours: a systematic review of the literature. Endocr Relat Cancer. 2014;21:R153-63
2. Yao JC. One hundred years after ”carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063-72
3. Tsai HJ, Wu CC, Tsai CR. et al. The epidemiology of neuroendocrine tumors in Taiwan: A nation-wide cancer registry-based study. PLoS One. 2013;8:e62487
4. Ito T. Epidemiological study of gastroenteropancreatic neuroendocrine tumors in Japan. J Gastroenterol. 2010;45:234-43
5. Bioulac-Sage P, Balabaud C, Wanless I. et al. WHO Classification of Tumours of the Digestive System. IARC Press: Lyon, FR. 2010
6. Jiao Y, Shi C, Edil BH. et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199-203
7. Yuan F, Shi M, Ji J. et al. KRAS and DAXX/ATRX gene mutations are correlated with the clinicopathological features, advanced diseases, and poor prognosis in Chinese patients with pancreatic neuroendocrine tumors. Int J Biol Sci. 2014;10:957-65
8. Zitzmann K, Vlotides G, Brand S. et al. Perifosine-mediated Akt inhibition in neuroendocrine tumor cells: role of specific Akt isoforms. Endocr Relat Cancer. 2012;19:423-34
9. Tachezy M, Zander H, Marx AH. et al. ALCAM (CD166) expression as novel prognostic biomarker for pancreatic neuroendocrine tumor patients. J Surg Res. 2011;170:226-32
10. Chen MH, Yeh YC, Shyr YM. et al. Expression of gremlin 1 correlates with increased angiogenesis and progression-free survival in patients with pancreatic neuroendocrine tumors. J Gastroenterol. 2013;48:101-8
11. Speisky D, Duces A, Bièche I. et al. Molecular profiling of pancreatic neuroendocrine tumors in sporadic and Von Hippel-Lindau patients. Clin Cancer Res. 2012;18:2838-49
12. Fernandez-Cuesta L, Peifer M, Lu X. et al. Frequent mutations in chromatin-remodeling genes in pulmonary carcinoids. Nat Commun. 2014;5:3518
13. Shida T, Furuya M, Kishimoto T. et al. The expression of NeuroD and mASH1 in the gastroenteropancreatic neuroendocrine tumors. Mod Pathol. 2008;21:1363-70
14. Shin JU, Lee CH, Lee KT. et al. Prognostic significance of ATM and cyclin B1 in pancreatic neuroendocrine tumor. Tumour Biol. 2012;33:1645-51
15. Sánchez-Barrena MJ, Vallis Y, Clatworthy MR. et al. Bin2 is a membrane sculpting N-BAR protein that influences leucocyte podosomes, motility and phagocytosis. PLoS One. 2012;7:e52401
16. Gupta A, Wang Y, Browne C. et al. Neuroendocrine differentiation in the 12T-10 transgenic prostate mouse model mimics endocrine differentiation of pancreatic beta cells. Prostate. 2008;68:50-60
17. Pinato DJ, Tan TM, Toussi ST. et al. An expression signature of the angiogenic response in gastrointestinal neuroendocrine tumours: correlation with tumour phenotype and survival outcomes. Br J Cancer. 2014;110:115-22
18. Hansel DE, Rahman A, House M. et al. Met proto-oncogene and insulin-like growth factor binding protein 3 overexpression correlates with metastatic ability in well-differentiated pancreatic endocrine neoplasms. Clin Cancer Res. 2004;10:6152-8
19. Han X, Zhao J, Ji Y. et al. Expression of CK19 and KIT in resectable pancreatic neuroendocrine tumors. Tumour Biol. 2013;34:2881-9
20. Schmitt AM, Pavel M, Rudolph T. et al. Prognostic and predictive roles of MGMT protein expression and promoter methylation in sporadic pancreatic neuroendocrine neoplasms. Neuroendocrinology. 2014;100:35-44
21. Guo SS, Wu X, Shimoide AT. et al. Anomalous overexpression of p27 (Kip1) in sporadic pancreatic endocrine tumors. J Surg Res. 2001;96:4-288
22. Knösel T, Chen Y, Altendorf-Hofmann A. et al. High KIT and PDGFRA are associated with shorter patients survival in gastroenteropancreatic neuroendocrine tumors, but mutations are a rare event. J Cancer Res Clin Oncol. 2012;138:397-403
23. Falchetti A, Marini F, Luzi E. et al. Multiple endocrine neoplasms. Best Pract Res Clin Rheumatol. 2008;22:149-63
24. Pollizzi KN, Patel CH, Sun IH. et al. mTORC1 and mTORC2 selectively regulate CD8⁺ T cell differentiation. J Clin Invest. 2015;125:2090-08
25. Chan AO, Kim SG, Bedeir A. et al. CpG island methylation in carcinoid and pancreatic endocrine tumors. Oncogene. 2003;22:924-34
26. Detjen KM, Rieke S, Deters A. et al. Angiopoietin-2 promotes disease progression of neuroendocrine tumors. Clin Cancer Res. 2010;16:420-9
27. Yachida S, Vakiani E, White CM. et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol. 2012;36:173-84
28. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754-60
29. Li H, Handsaker B, Wysoker A. et al. The Sequence Alignment/Map format and SAM tools. Bioinformatics. 2009;25:2078-9
30. Lin PH, Kuo WH, Huang AC. et al. Multiple gene sequencing for risk assessment in patients with early-onset or familial breast cancer. Oncotarget. 2016;7(7):8310-20
31. McKenna A, Hanna M, Banks E. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297-303
32. Mimori T, Nariai N, Kojima K. et al. iSVP: an integrated structural variant calling pipeline from high-throughput sequencing data. BMC systems biology. 2013;7(Suppl 6):S8
33. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164
34. Richards S, Aziz N, Bale S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-24
35. Terris B, Scoazec JY, Rubbia L. et al. Expression of vascular endothelial growth factor in digestive neuroendocrine tumours. Histopathology. 1998;32:133-8
36. Couvelard A, O'Toole D, Turley H. et al. Microvascular density and hypoxia- inducible factor pathway in pancreatic endocrine tumours: negative correlation of microvascular density and VEGF expression with tumour progression. Br J Cancer. 2005;92:94-101
37. Poncet G, Villaume K, Walter T. et al. Angiogenesis and tumor progression in neuroendocrine digestive tumors. J Surg Res. 2009;154:68-77
38. Missiaglia E, Dalai I, Barbi S. et al. Pancreatic endocrine tumors: expression profiling evidences a role for AKT-mTOR pathway. J Clin Oncol. 2010;28:245-55
39. Fjallskog ML, Lejonklou MH, Oberg KE. et al. Expression of molecular targets for tyrosine kinase receptor antagonists in malignant endocrine pancreatic tumors. Clin Cancer Res. 2003;9:1469-73
40. Marion-Audibert AM, Barel C, Gouysse G. et al. Low microvessel density is an unfavorable histoprognostic factor in pancreatic endocrine tumors. Gastroenterology. 2003;125:1094-104
41. Raymond E. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:501-13
42. Liu P, Cheng H, Roberts TM. et al. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627-44
43. Wu G, Xing M, Mambo E. et al. Somatic mutation and gain of copy number of PIK3CA in human breast cancer. Breast Cancer Res. 2005;7:R609-16
44. Kawano O, Sasaki H, Okuda K. et al. PIK3CA gene amplification in Japanese non-small cell lung cancer. Lung Cancer. 2007;58:159-610
45. Yao JC, Shah MH, Ito T. et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514-23
46. Ito T, Okusaka T, Ikeda M. et al. Everolimus for advanced pancreatic neuroendocrine tumours: a subgroup analysis evaluating Japanese patients in the RADIANT-3 trial. Jpn J Clin Oncol. 2012;42:903-11
47. Yao JC, Fazio N, Singh S. et al. RAD001 in Advanced Neuroendocrine Tumours, Fourth Trial (RADIANT-4) Study Group. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (RADIANT-4): a randomised, placebo-controlled, phase 3 study. Lancet. 2015 doi: 10.1016/S0140-6736(15)00817-X
48. Liu CT, Chen MH, JS Chen. et al. The efficacy and safety of everolimus for the treatment of progressive gastroenteropancreatic neuroendocrine tumors: a multi-institution observational study in Taiwan. Asia Pac J Clin Oncol. 2016 doi: 10.1111/ajco.12571
49. Deng L, Chen J, Zhong XR. et al. Correlation between activation of PI3K/AKT/mTOR pathway and prognosis of breast cancer in Chinese women. PLoS One. 2015;10:e0120511
50. Yachida S, Vakiani E, White CM. et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol. 2012;36:173-84
51. Marinoni I, Kurrer AS, Vassella E. et al. Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology. 2014;146:453-60
52. Liau JY, Tsai JH, Jeng YM. et al. Leiomyosarcoma with alternative lengthening of telomeres is associated with aggressive histologic features, loss of ATRX expression, and poor clinical outcome. Am J Surg Pathol. 2015;39:236-44
Author contact
![]() Corresponding author: Ming-Huang Chen, M.D; PhD, The Department of Oncology, Taipei Veterans General Hospital, No. 201, Sec 2, Shih-Pai Road, Taipei 112, Taiwan; Email: mhchen9com; Tel: 886-2-28757529 Fax: 886-2-28757762
Corresponding author: Ming-Huang Chen, M.D; PhD, The Department of Oncology, Taipei Veterans General Hospital, No. 201, Sec 2, Shih-Pai Road, Taipei 112, Taiwan; Email: mhchen9com; Tel: 886-2-28757529 Fax: 886-2-28757762