International Journal of Biological Sciences 10
Impact Factor
ISSN: 1449-2288
Impact Factor
ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(1):22-31. doi:10.7150/ijbs.16298 This issue Cite
Research Paper
Let-7a Is an Antihypertrophic Regulator in the Heart via Targeting Calmodulin
Xin Zhou1, 2*, Fei Sun1, 3*, Shenjian Luo1, Wei Zhao1, Ti Yang1, Guiye Zhang1, Ming Gao1, Renzhong Lu1, You Shu1, Wei Mu1, Yanan Zhuang1, Fengzhi Ding1, Chaoqian Xu1 ![]() , Yanjie Lu1, 3
, Yanjie Lu1, 3 ![]()
1. Department of Pharmacology (State-Province Key Laboratories of Biomedicine-Pharmaceutics of China, Key Laboratory of Cardiovascular Research, Ministry of Education), College of Pharmacy, Harbin Medical University, Harbin, Heilongjiang 150081, P. R. China.
2. Department of Cardiology (Key Laboratory of Myocardial Ischemia, Ministry of Education), The 2nd Affiliated Hospital of Harbin Medical University, Harbin, Heilongjiang 150081, P. R. China
3. Northern Translational Medicine Research and Cooperation Center, Heilongjiang Academy of Medical Sciences, Harbin Medical University, Harbin, Heilongjiang 150081, P. R. China.
* These authors contributed equally to this work.
Received 2016-5-27; Accepted 2016-10-10; Published 2017-1-1
Citation:
Zhou X, Sun F, Luo S, Zhao W, Yang T, Zhang G, Gao M, Lu R, Shu Y, Mu W, Zhuang Y, Ding F, Xu C, Lu Y. Let-7a Is an Antihypertrophic Regulator in the Heart via Targeting Calmodulin. Int J Biol Sci 2017; 13(1):22-31. doi:10.7150/ijbs.16298. https://www.ijbs.com/v13p0022.htm
Other stylesAbstract

Background: MicroRNAs (miRNAs) have been emerged as important regulator in a multiple of cardiovascular disease, including arrhythmia, cardiac hypertrophy and fibrosis, and myocardial infarction. The aim of this study was to investigate whether miRNA let-7a has antihypertrophic effects in angiotensin II (AngII)-induced cardiac hypertrophy.
Methods: Neonatal rat ventricular myocytes (NRVMs) were exposed to AngII for 36 h as a cellular model of hypertrophy; subcutaneous injection of AngII for 2 weeks was used to establish a mouse model of cardiac hypertrophy in vivo study. Cell surface area (CSA) was measured by immunofluorescence cytochemistry; expression of hypertrophy-related genes ANP, BNP, β-MHC was detected by Real-time PCR; luciferase activity assay was performed to confirm the miRNA's binding site in the calmodulin (CaM) gene; CaM protein was detected by Western blot; the hypertrophy parameters were measured by echocardiographic assessment.
Results: The expression of let-7a was decreased in AngII-induced cardiac hypertrophy in vitro and in vivo. Overexpression of let-7a attenuated AngII-induced increase of cell surface area and repressed the increased mRNA levels of ANP, BNP and β-MHC. Dual-luciferase reporter assay showed that let-7a could bind to the 3'UTR of CaM 1 gene. Let-7a downregulated the expression of CaM protein. In vivo, let-7a produced inhibitory effects on cardiac hypertrophy, including the downregulation of cross-sectional area of cardiomyocytes in mouse heart, the reduction of IVSD and LVPWD, the suppression of hypertrophy marker genes ANP, BNP, β-MHC mRNA level, and the downregulation of CaM protein level.
Conclusions: let-7a possesses a prominent anti-hypertrophic property by targeting CaM genes. The findings provide new insight into molecular mechanism of cardiac hypertrophy.
Keywords: Cardiac hypertrophy, miRNA, let-7a, angiotensin Ⅱ, calmodulin.
Introduction
Cardiac hypertrophy is a physiological or adaptive response that is often associated with increased cell size, protein synthesis and fibroblast proliferation [1-3]. Pathological cardiac hypertrophy can be caused by a number of pathological stimuli, such as hypertension, myocardial infarction, and hypertrophic agents including AngⅡ, isoproterenol and endothelin1 [4]. However, prolonged cardiac hypertrophy may lead to heart failure and sudden death [5, 6]. It has been known that multiple signaling pathways or molecules participate in cardiac hypertrophy [7-9]. A role for CaM has been reported in several physiological and pathological processes in the cardiac hypertrophy [10, 11].
MicroRNAs (miRNAs) are small non-coding RNAs that modulate gene expression by inhibiting translation or promoting degradation of mRNA [12]. Recent studies demonstrated that miRNAs play a critical role in regulation of cardiac developmental and cardiovascular diseases [13-15]. It has been reported that let-7 plays a crucial role in multiple biologic functions, including tumor suppression [16, 17], axon regeneration [18], arrhythmia inhibition [19], and cardiac fibrosis and apoptosis reduction [20]. A study by Gray et al showed that upregulation of let-7 may be involved in regulation of cardiac hypertrophy process in rats [21]. Furthermore, Yang et al reported that upregulation of let-7 induced by thioredoxin 1 attenuates cardiac hypertrophy by targeting cyclin D2 [22].
One of biological functions of miRNA is that single miRNA may have multiple mRNA targets by binding to their 3'untranslated region. By analyzing miRNA target, we found that let-7a and the 3'UTR of the CaM 1 gene have complementary binding sites, indicating CaM gene may be another target of let-7 in regulation of cardiac hypertrophy process. We therefore hypothesized that let-7a might play a role in regulating cardiac hypertrophy by targeting CaM. Thereby, the aim of this study was to examine this hypothesis.
Methods
Cardiomyocytes culture and treatment
Briefly, the isolated hearts from 1 to 3-day-old Sprague-Dawley rats were cut into l.0 mm pieces and digested in 0.25% trypsin at 37 °C. Subsequently, cells in the supernatant were isolated by centrifugation (1500 rpm, 5 min) at room temperature. The cells were then cultured in DMEM (Hyclone Laboratories, UT, USA) supplemented with 10% fetal bovine serum (Gibco, Invitrogen, CA, USA) for 2 h. After fibroblast adherence, the cells in the supernatant were seeded onto cell culture dishes. After 48 h culture, NRVMs were treated with AngII (Sigma-Aldrich Co, St. Louis, MO, USA) 100 nM for 36 h to induce cell hypertrophy, which has been proven to induce cardiomyocyte hypertrophy [23].
Animal experiments
Healthy male C57BL/6 mice (20 ± 2 g) used in the current study were housed under standard animal room conditions (temperature at 21 ± 1°C, humidity of 55 ± 5%). Cardiac hypertrophy model was established by AngII administration. The mice were subcutaneously injected with AngII for 2 weeks at a concentration of 1.5 mg/kg/day. Saline-injected mice served as controls. All animal experiments were approved by the ethics committee of Harbin Medical University.
Synthesis of let-7a mimics and let-7a mimics transfection
Let-7a mimics (5'UGAGGUAGUAGGUUGUAUAGUU-3'), its anti-miRNA oligonucleotides (AMO-let-7a, 5'-AACUAUACAACCUACUACCUCA-3') and scrambled RNA (5'-UUCUCCGAACGUGUCACGU-3') as a negative control (NC) were synthesized by Ribo Biotechnology Co, Ltd (Guangzhou, China). miRNA transfection was performed using X-tremeGene siRNA Transfection Reagent (Roche, USA). In briefly, 100 nM of let-7a mimics/NC or 200 nM AMO-let-7a were added to the medium at room temperature to form transfection complexes. The cells were incubated in serum-free DMEM with the transfection complexes for 6 h, and then the medium was replaced with DMEM containing 10% FBS.
Construction and infection of lentivirus
Lentivirus vectors expressing let-7a, AMO-let-7a, or NC sequence were constructed by Invitrogen Co. (Shanghai, China). Virus-containing solution (20 µL, 1×108 TU) including len-negative control (len-NC), precursor let-7a (len-pre-let-7a), AMO-let-7a (len-AMO-let-7a) were injected into left ventricular chamber of mouse heart by blocking aorta of short duration.
Measurement of cell surface area
Cells with various treatments were washed with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. After that, the cells were washed and treated with 0.4% Triton X-100. After 90 min, the cells were incubated with goat serum at room temperature for 60 min. Then the samples were incubated with anti-sarcomeric actin antibody (Sigma-Aldrich, St. Louis, MO, USA) at 4°C overnight. Next day, cells were stained with FITC-conjugate goat anti-mouse antibody for 1 h. Images were obtained under a fluorescence microscope (Nikon 80i, Japan). Cell surface area was determined using Image-Pro Plus Data Analysis Software.
Quantitative Real-time PCR
Total RNA was extracted from cells or heart tissues using TRIZOL reagent (Invitrogen, USA). The expression levels of ANP, BNP and β-MHC mRNAs were performed on ABI 7500 fast Real Time PCR system (Applied Biosystems, USA) by quantitative real-time PCR. The sequences of ANP, BNP, β-MHC primers were listed in Table 1 and synthesized by Guangzhou Ribo Biotechnology Co. Ltd (Guangzhou, China). GAPDH served as an internal control. Reaction conditions were set as: 95°C for 10 min, 40 cycles of 95°C for 15 sec, 60°C for 30 sec, and 72°C for 30 sec. The relative quantitative expression was determined using method 2-^^Ct.
Table 1

The sequence of PCR primers.
Dual-luciferase reporter assay
To confirm that CaM was targeted by let-7a, we constructed a chimeric luciferase reporter system (pMIR-REPORT, Ambion, CA, USA) tagged with the 3'UTR region of rat CaM1 containing the seed match sequence with or without two nucleotide mutations. HEK293 cells were transfected with 200 ng of pMIR-REPORT luciferase vector and 8 ng of Prl-SV40 renilla luciferase control vector in the presence of the let-7a mimics or scrambled-oligonucleotide (as negative control) using Lipofectamine 2000 (Invitrogen, USA). Luciferase activity was assayed after 48 h using the Dual-luciferase reporter assay system (Promega, USA) normalized to Renilla luciferase activity.
Western blot analysis
Total protein was extracted from the cardiomyocytes and left ventricular tissues of heart with RIPA lysis buffer (Beyotime Institute of Biotechnology, China). Then, the homogenates were centrifuged at 13,500 rpm for 15 min at 4°C. The supernatant was collected and protein concentrations were determined using BCA Protein Assay Kit (Beyotime Institute of Biotechnology, China). The protein samples (100 μg) were separated to SDS-PAGE on 10% polyacrylamide gels, and subsequently transferred to nitrocellulose membrane (Millipore, Bedford, MA, USA), and then blocked with a solution of skim milk powder for 2 h at room temperature. The membranes were incubated overnight with the primary antibody for CaM (Abcam, MA, USA) at 4°C. The next day, the membranes were washed 3 times with PBS-T and conjugated with a secondary rabbit or mouse polyclonal antibody at room temperature for 1 h. Western blot bands were quantified using Odyssey v1.2 software and normalized to GAPDH.
Cardiac hypertrophy measurements
After 2 weeks of AngII administration, all mice were sacrificed. The mouse hearts were immediately dissected out and weighed. The heart weight (HW), body weight (BW), and left ventricular weight (LVW) were recorded. The HW/BW and LVW/BW were calculated to estimate the extent of cardiac hypertrophy.
Histological examination
The mouse hearts were washed with saline and fixed with 10% formalin. Then the hearts were cut into 7 μm slices, and stained with hematoxylin and eosin (H&E). Cross-sectional images of cardiomyocytes were scanned at 400× magnification to estimate the degree of cardiomyocyte hypertrophy. Cross-section area of cells was measured with Image Pro-Plus 4.0 system.
Echocardiographic assessment of cardiac function
The male C57BL/6 mice were lightly anesthetized and cardiac function was evaluated by using Vevo2100 High-Resolution Imaging system (Visualsonics, Toronto, Canada) equipped with a 30 MHz microscan transducer. M-mode traces were obtained in both parasternal long and short axis views. Ventricular parameters including interventricular septum depth (IVSD), diastolic left ventricular posterior wall thicknesses (LVPWD) and left ventricular internal diastolic diameter (LVIDD) were measured. Fractional shortening (FS) and ejection fraction (EF) were calculated based on the Vevo2100 High-Resolution Imaging system.
Statistical analysis
All experimental data are expressed as mean ± SD. Statistical significance was estimated by ANOVA or student's t-test for multiple group or two group comparisons, using SPSS 13.0 software. Values of P < 0.05 were considered to indicate statistically significant differences.
Results
Downregulation of let-7a in angiotensin II-induced cardiac hypertrophy
We first established in vitro and in vivo mouse models of cardiac hypertrophy. Neonatal rat ventricular myocytes (NRVMs) were treated with angiotensin II (AngII) 100 nM for 36 h to induce myocardial hypertrophy characterized by increased cell surface area (CSA) (P < 0.01, Fig. 1A and 1B). AngII 1.5 mg/kg/day was subcutaneously injected in mice for 2 weeks to establish cardiac hypertrophy characterized by increased cross-sectional area of cardiomyocytes (P < 0.01, Fig. 1C and 1D).
In order to investigate whether let-7 is involved in AngII-induced cardiac hypertrophy, we performed Real-time PCR to quantify the expression of let-7 family miRNAs in cardiac hypertrophic models. The expression of let-7a, let-7d, and let-7e was significantly downregulated in AngII-induced cellular model (Fig. 2A) and mouse model of hypertrophy (Fig. 2B), expression of let-7b and let-7c had no obviously changed. Among them, let-7a showed the most downregulation in expression after AngII treatment both in vivo and in vitro. We therefore chose let-7a as a candidate miRNA in the following experiments.
Figure 1
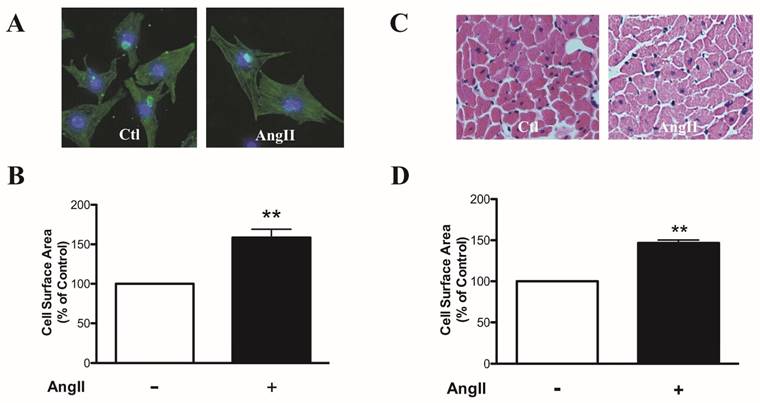
Cell surface area of cardiomyocytes in vitro and in vivo. (A) Representative fluorescence microscopy images of sarcomere organization (200×magnification). (B) Summarized data of cell surface area of NRVMs. (C) Representative fluorescence microscopy images of hematoxylin and eosin (H&E) staining. (D) Summarized data of cross-sectional area of cardiomyocytes. n = 6 experiment for each group, **P < 0.01vs.control (Ctl).
Figure 2
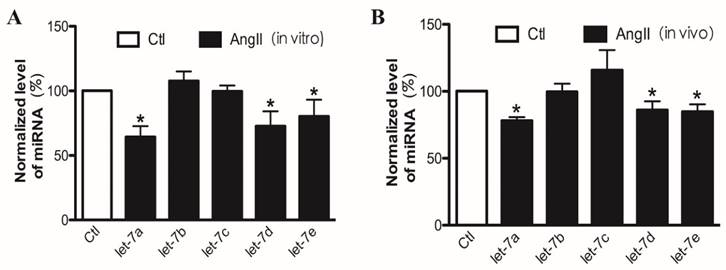
The expression of let-7a/b/c/d/e in cardiomyocytes treated with AngII in vitro and in vivo. Real-time PCR analysis of let-7a/b/c/d/e levels in AngII-treated cardiomyocytes (A) and AngII-treated mouse hearts (B). n = 6 experiment for each group; *P < 0.05 vs. Ctl.
Figure 3
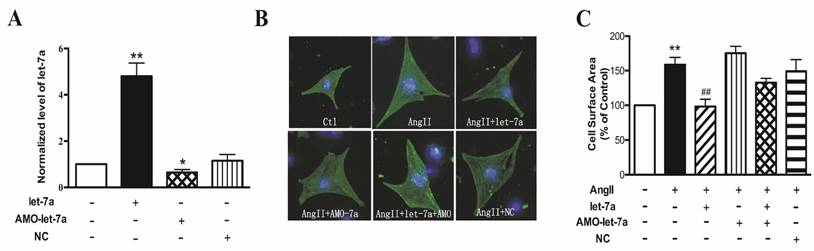
Effects of let-7a on cell surface area of cardiomyocytes. (A) Let-7a level in NRVMs transfected with let-7a, AMO-let-7a or NC. (B) Representative fluorescence microscopy images of sarcomere organization (200× magnification). (C) Summarized data from different treatment groups, n = 60 cells from four experiments. n = 4 experiment for each group; *P < 0.05 & **P < 0.01 vs. Ctl; ##P < 0.01vs. AngII.
Figure 4
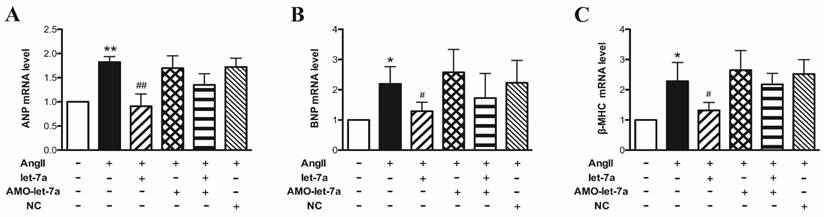
Effects of let-7a on expressions of hypertrophy marker genes in cardiomyocytes. The mRNA expressions of ANP (A), BNP (B), and β-MHC (C) in different groups were detected by Real-time PCR. Data are expressed as mean ± SD, n = 4 experiments for each group; *P < 0.05 & **P < 0.01 vs. Ctl; #P < 0.05 & ##P < 0.01vs. AngII.
Overexpression of let-7a inhibited cardiomyocyte hypertrophy
Based on the above results, we then investigated whether let-7a is involved in AngII-induced cardiomyocyte hypertrophy. We first detected the transfection efficiency of let-7a in NRVMs. Compared with control group, the level of let-7a was significantly elevated by about 5 folds in NRVMs transfected with let-7a mimics. The let-7a level was decreased by 35.5 ± 1.3% in NRVMs treated with AMO-let-7a (anti-miRNA let-7 oligonuoleotides). The NC (negative control) had no effect on let-7a expression (Fig. 3A). We used CSA of cardiomyocytes to evaluate cardiac hypertrophy. Compared with the control group, CSA was significantly increased by 158.5 ± 10.6% in AngII group (P < 0.01). Let-7a transfection could significantly decrease CSA induced by AngII; AMO-let-7a antagonized the antihypertrophic effects of let-7a; negative control (NC) of let-7a failed to do so. AMO-let-7a alone had no effect on the CSA (Fig. 3B-3C).
To further examine the effects of let-7a on cardiomyocyte hypertrophy, we tested the cardiac markers of pathological hypertrophy, atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and β-myosin heavy chain (β-MHC). Our results showed that AngII significantly increased the expression of ANP (Fig. 4A), BNP (Fig. 4B), and β-MHC (Fig. 4C) at the mRNA level (P < 0.05). And as expected, overexpression of let-7a markedly inhibited the upregulation of these hypertrophic markers in AngII-treated cells (P < 0.05). AMO-let-7a abolished the effects of let-7a, and AMO-let-7a or NC alone had no effects on expression of these markers. These results indicate that let-7a produced anti-hypertrophic effects in cardiomyocytes.
Camodulin1 gene was a target of let-7a
To elucidate the molecular mechanism of antihypertrophic effect of let-7a, we first performed Targetscan analysis to predict the possible target gene of let-7a. We found that Camodulin1 (CaM1) gene contains the “seed site” of let-7a in its 3'UTR, which is highly conserved among the species of human, rat, and mouse (Fig. 5A).
To confirm that let-7a directly targets the 3'UTR of CaM1, we constructed a chimeric luciferase reporter gene vector containing CaM1 3'UTR with the binding sites for let-7a. As shown in Fig. 5B, overexpression of let-7a resulted in significant reduction of luciferase activity. AMO-let-7a reversed inhibitory effect by let-7a. Neither NC nor AMO-let-7a alone affected the luciferase activity. However, co-transfection of let-7a and mutant CaM1 3'UTR did not influence the luciferase activity. These results indicated that CaM 1 gene is a direct target site of let-7a.
Western blot results showed that transfection of let-7a reduced the CaM protein level in cardiomyocyte. AMO-let-7a abolished the effects of let-7a. NC or AMO-let-7a alone had no effect on CaM expression (Fig. 6A and 6B). In addition, we demonstrated that let-7a also decreased the expression of CaM in cardiomyocytes treated with AngII. AMO-let-7a reversed the effect of let-7a. However, AMO-let-7a or NC did not influence CaM level (Fig.6C and 6D).
Let-7a inhibited cardiac hypertrophy in vivo
Lentivirus vectors containing pre-let-7a, AMO-let-7a, or NC were injected into the left ventricular chamber by blocking aorta of short duration. After 2 weeks, we measured the ratio of HW or LVW to BW and cross-sectional area of cardiomyocytes, which are indicators of cardiac hypertrophy. As shown in Fig. 7, the ratios of HW/BW and LVW/BW were significantly increased in AngII-treated mice, compared with the control group. Cross-sectional area of cardiomyocytes was also significantly increased in AngII-treated mice. Len-let-7a-infected mice exhibited significant inhibitory effects on the AngII-induced cardiac hypertrophy, including decrease in the HW/BW and LVW/BW ratios (Fig. 7A and 7B) and in the cross-sectional area (Fig. 7C and 7D), which were abolished by co-infection with len-AMO-let-7a. Len-AMO-let-7a or NC alone did not affect the indicators in hypertrophic hearts.
Figure 5
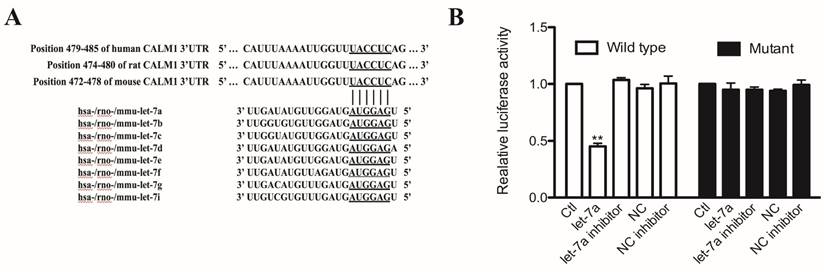
The regulatory effects of let-7 on Camodulin1 gene. (A) Predicted seed-binding sites of let-7 in CaM1 3'UTR. (B) Activities of pMIR-REPORTTM luciferase vector carrying luciferase gene and fragment of CaM1 3'UTR containing the binding sites of let-7a. Data are expressed as mean ± SD, n = 3 experiments for each group; **P < 0.01vs.Ctl.
Figure 6
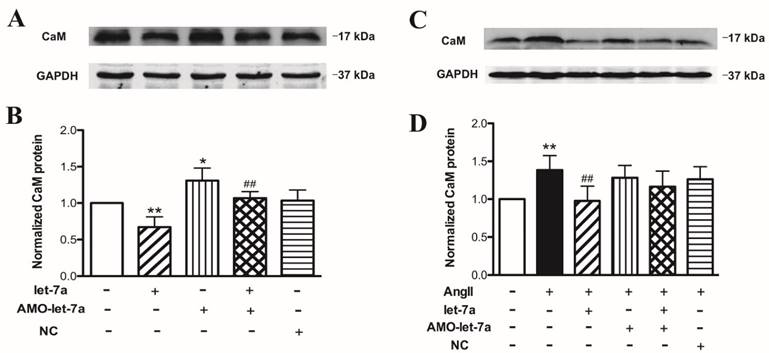
Effects of let-7a on calmodulin expression. Representative examples of Western blot bands from different treatments (A and C). Effects of let-7a on CaM at protein level in cardiomyocytes treated without (B) or with AngII (D). Data are expressed as mean ± SD, n = 3 experiments for each group; *P < 0.05 & **P < 0.01 vs.Ctl, ##P < 0.01 vs. AngII.
Figure 7
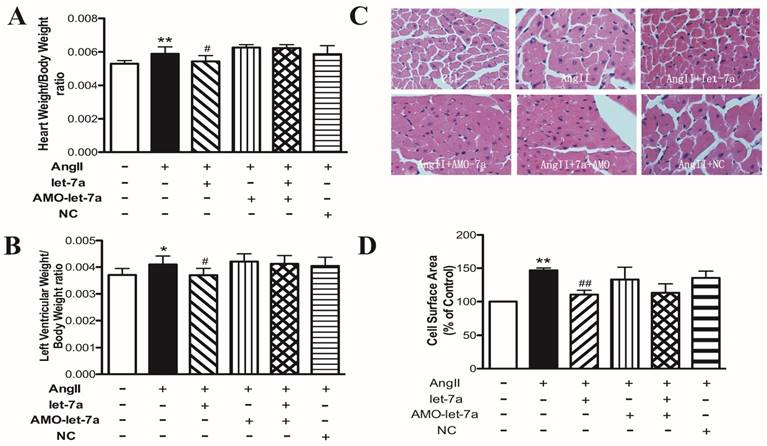
Anti-hypertrophic effects of let-7a in mouse model of cardiac hypertrophy induced by AngII. Ratios of HW/BW (A) and LVW/BW (B) in different groups of mice. (C) Representative fluorescence microscopic images (400× magnification) of hematoxylin and eosin (H&E) staining. (D) Summarized data of cross-sectional area in different groups. n = 6-8 mice in each group; *P < 0.05 & **P < 0.01 vs. Ctl; #P < 0.05 & ##P< 0.01 vs. AngII.
Figure 8
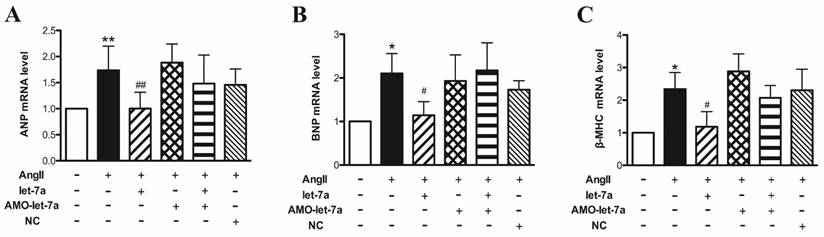
Effects of let-7a on hypertrophic markers in mouse model of cardiac hypertrophy induced by AngII. Expressions of ANP (A), BNP (B), and β-MHC (C) at mRNA level in mice with different treatments. n = 6 mice for each group, *P < 0.05 & **P < 0.01 vs. Ctl; #P < 0.05 & ##P < 0.01vs. AngII.
Then we detected the cardiac hypertrophy markers ANP, BNP, and β-MHC mRNA levels by Real-time PCR. We found that three hypertrophy markers were upregulated in left ventricular tissues in AngII-treated mice (P < 0.05). Len-let-7a suppressed the AngII-induced upregulation of ANP, BNP, and β-MHC expression. However, len-AMO-let-7a prevented the effects of len-let-7a. Len-AMO-let-7a or len-NC alone had no effects in AngII-treated mice (Fig. 8).
Western blot results showed that CaM was remarkably increased at protein level in AngII group, and len-let-7a effectively inhibited this increase. Len-AMO-let-7a prevented the action of len-let-7a. Len-AMO-let-7a or len-NC alone had no effects in AngII-treated mice (Fig. 9). These results indicate that let-7a has anti-hypertrophic effects in vivo by targeting the CaM.
Echocardiogram was performed on mice 14 days after treatment with len-pre-let-7a, len-pre-let-7a + len-AMO-let-7a, len-AMO-let-7a or len-NC. As shown in Table 2, interventricular septum depth (IVSD) and diastolic left ventricular posterior wall thickness (LVPWD) were increased in AngII-treated mice (P < 0.05), compared with the control group. Len-let-7a significantly reversed the AngII-induced increase of IVSD and LVPWD. EF and FS values were significantly reduced in AngII-treated mice (P < 0.05). The EF and FS values were slightly increased in len-let-7a, but these values did not reach statistical significance. There were no significant changes in groups of len-pre-let-7a + len-AMO-let-7a, len-AMO-let-7a or len-NC.
In addition, we applied len-pre-let-7a, len-AMO-let-7a or len-NC to the normal mice (without AngII treatment) and found no significant changes in IVSD, LVPWD, EF, and FS. These data indicate that let-7a had no effects on the cardiac structure and function in normal condition (Table 3).
Figure 9
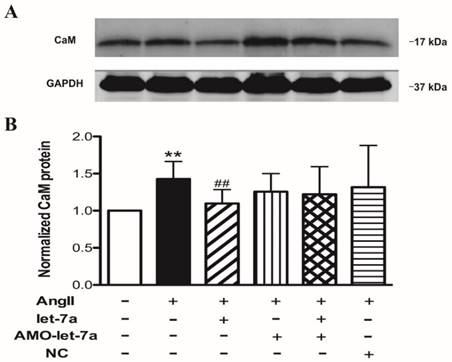
Effects of let-7a on the expression of CaM in mouse model of cardiac hypertrophy induced by AngII. Representative examples of Western blot bands from different treatments (upper). Mean data of band density of CaM in different groups (lower). n = 6 mice in each group, **P < 0.01 vs. Ctl; ##P < 0.01 vs. AngII.
Table 2
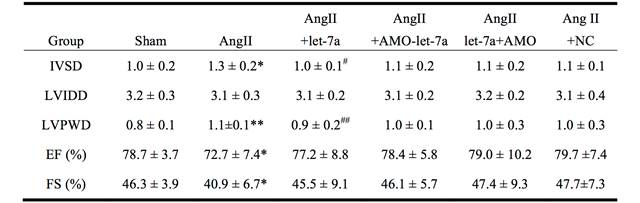
Effects of let-7a on cardiac structure and function in AngII-treated mice.
Echocardiographic parameters in different groups of mice. IVSD, interventricular septum depth; LVIDD, left ventricular internal diastolic diameter; LVPWD, diastolic left ventricular posterior wall thickness; EF, ejection fraction; FS, fractional shortening. n = 8 mice in each group. Data are expressed by mean ± SD. *P < 0.05 & **P < 0.01 vs. Ctl; #P < 0.05 & ##P< 0.01 vs. AngII.
Table 3
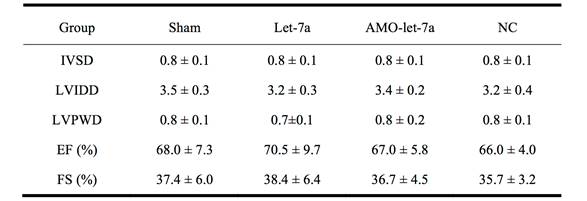
Effects of let-7a on cardiac structure and function in normal mice.
Echocardiographic parameters of mice treated with len-pre-let-7a, len-AMO-let-7a or len-NC. IVSD, interventricular septum depth; LVIDD, left ventricular internal diastolic diameter; LVPWD, diastolic left ventricular posterior wall thickness; EF, ejection fraction; FS, fractional shortening. n = 8 mice in each group. Data are expressed by mean ± SD.
Discussion
Cardiac hypertrophy is a complex event, which is closely related to multiple cardiac dysfunctions. It has been reported that a number of miRNAs, such as miR-1 [24], miR-133 [25], miR-21 [26] and miR-208 [27] are involved in the process of cardiac hypertrophy. In the present study, we found that let-7a expression was downregulated in AngII induced cardiac hypertrophy. Overexpression of let-7a mimics inhibited the AngII induced hypertrophic responses and suppressed expression of hypertrophic marker genes, ANP, BNP and β-MHC. Similar results were obtained from in vivo study in a mouse model of cardiac hypertrophy. We demonstrated that let-7a exerted its antihypertrophic effects through targeting 3'UTR of CaM gene.
Let-7 was first discovered in Caenorhabditis elegans [28]. Previous studies have demonstrated that let-7 is reduced in a number of the tumors, such as lung cancer and colon cancer [29, 30]. It participates in cellular proliferation through downregulating the RAS/c-MYC at the translational level [17]. A recent study proved that let-7 has important function in the cardiac disease. Let-7 has been demonstrated enriched in the mouse heart, and upregulated in the heart failure [31]. Li et al has demonstrated that let-7 is downregulated in the acute myocardial infarction and contributes to the anti-arrhythmic efficacy [19]. Sayed's result showed that let-7b and let-7c are upregulated, but let-7d downregulated in the TAC mice [32]. This may be due to the let-7 family members' transcript from different gene clusters, showing different function in the cardiac hypertrophy.
In present study, we found that let-7a, let-7d, let-7e were downregulated in the hypertrophic model. Our study focused on let-7a, because it demonstrated the most downregulation in hypertrophic responses both in vivo and in vitro models. Other members of the let-7 family such as let-7d and let-7e were also decreased in AngII induced hypertrophic response, indicating they may exert same function in the cardiac hypertrophy process. Notably, rest member of let-7 family let-7b, let-7c did not show significant alteration. This discrepancy of let-7 family expression may be attributed to the family members' transcript from different gene clusters playing different functions in the cardiac hypertrophy. It may be due to that members of let-7 family are located on different chromosomes (genes). Previous studies have reported that upregulation of let-7a is involved in cardiac hypertrophy [20] and miR-98/let-7 mediates the antihypertrophic effect through Trx1 [22]. These studies demonstrated the anti-hypertrophic property of let-7a and miR-98/let-7, which is consistent with our results. Taken together, let-7 family members possess antihypertrophic effect through acting on different targets, indicating let-7 family is promising target for intervening cardiac hypertrophy.
The mechanism for cardiac hypertrophy is related to changes in the organization of the sarcomeric structure that induced by altered protein and gene, such as protein C, CaM [11, 33]. CaM, in conjunction with Ca2+, activates the phosphatase protein, such as calcineurin and CAMKII, which in turn stimulate hypertrophic genes transcription [34]. We used bioinformatics approach to identify CaM as a predicted target of let-7a. Then, we proved that transfection with let-7a mimics obviously shut down the CaM protein expression and aborted hypertrophy effect of CaM. Luciferase report assay also confirmed the direct targeting of CaM by let-7a. Our experimental data suggest that let-7a has its anti-hypertrophy effect by targeting CaM.
The study by Ikeda et al [24] showed that miR-1 was downregulated in MHCα-CN mouse hearts. Although expression of let-7 family miRNAs was decreased, but they did not reach statistical significance in MHCα-CN mice. Our results showed that let-7a, let-7d, and let-7e were significantly downregulated in AngII-induced cardiac hypertrophy. These indicate that difference in miRNA expression profile is related to the various pathological conditions. They demonstrated that miR-1 negatively modulates endothelin 1-induced cardiomyocyte hypertrophy by directly targeting genes CaM and Mef2 (myocyte enhancer factor 2); the current study showed that let-7a inhibited AngII-induced cardiac hypertrophy by targeting CaM gene, indicating that a single miRNA can regulate more hypertrophic genes and one hypertrophic gene can be targeted by multiple miRNAs. Both studies showed that miR-1 or let-7a did not produce significant effects on cardiac structure and function in the normal hearts.
In summary, we show that let-7a, as negative regulator to AngII-induced hypertrophy, let-7a expression is downregulated in AngII-induced cardiomyocyte hypertrophy both in vivo and in vitro models of mouse. Overexpression of let-7a can suppress AngII induced hypertrophy, and knockdown of let-7a by its AMO prevents antihypertrophic effects of let-7a. CaM, a key regulator of cardiac hypertrophy, is a direct target of let-7a. Therefore, let-7a appears to be an interesting target for the clinical intervention of cardiac hypertrophy.
Acknowledgements
This work was supported in part by Key Program of National Natural Science Foundation of China (No 81530010), the Funds for Creative Research Groups of The National Natural Science Foundation of China (81121003), and the National Nature Science Foundation of China (No 81370245 and 81600192).
Conflicts of interest
None.
References
1. Bernardo BC, Weeks KL, Pretorius L, McMullen JR. Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacology & therapeutics. 2010;128:191-227
2. Hill JA, Olson EN. Cardiac plasticity. The New England journal of medicine. 2008;358:1370-80
3. Oka T, Komuro I. Molecular mechanisms underlying the transition of cardiac hypertrophy to heart failure. Circulation journal: official journal of the Japanese Circulation Society. 2008;72(Suppl A):A13-6
4. Ahmad F, Seidman JG, Seidman CE. The genetic basis for cardiac remodeling. Annual review of genomics and human genetics. 2005;6:185-216
5. Barry SP, Davidson SM, Townsend PA. Molecular regulation of cardiac hypertrophy. The international journal of biochemistry & cell biology. 2008;40:2023-39
6. Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation. 2000;102:470-9
7. Frey N, McKinsey TA, Olson EN. Decoding calcium signals involved in cardiac growth and function. Nature medicine. 2000;6:1221-7
8. Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J. et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215-28
9. Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochemical and biophysical research communications. 2004;322:1178-91
10. Obata K, Nagata K, Iwase M, Odashima M, Nagasaka T, Izawa H. et al. Overexpression of calmodulin induces cardiac hypertrophy by a calcineurin-dependent pathway. Biochemical and biophysical research communications. 2005;338:1299-305
11. Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA. et al. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. The Journal of clinical investigation. 2000;105:1395-406
12. Ono K, Kuwabara Y, Han J. MicroRNAs and cardiovascular diseases. The FEBS journal. 2011;278:1619-33
13. Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang F. et al. MicroRNA-328 contributes to adverse electrical remodeling in atrial fibrillation. Circulation. 2010;122:2378-87
14. Pan Z, Sun X, Shan H, Wang N, Wang J, Ren J. et al. MicroRNA-101 inhibited postinfarct cardiac fibrosis and improved left ventricular compliance via the FBJ osteosarcoma oncogene/transforming growth factor-beta1 pathway. Circulation. 2012;126:840-50
15. Yang B, Lin H, Xiao J, Lu Y, Luo X, Li B. et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nature medicine. 2007;13:486-91
16. Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A. et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120:635-47
17. Lee YS, Dutta A. The tumor suppressor microRNA let-7 represses the HMGA2 oncogene. Genes & development. 2007;21:1025-30
18. Zou Y, Chiu H, Zinovyeva A, Ambros V, Chuang CF, Chang C. Developmental decline in neuronal regeneration by the progressive change of two intrinsic timers. Science. 2013;340:372-6
19. Li X, Wang B, Cui H, Du Y, Song Y, Yang L. et al. let-7e replacement yields potent anti-arrhythmic efficacy via targeting beta 1-adrenergic receptor in rat heart. Journal of cellular and molecular medicine. 2014;18:1334-43
20. Tolonen AM, Magga J, Szabo Z, Viitala P, Gao E, Moilanen AM. et al. Inhibition of Let-7 microRNA attenuates myocardial remodeling and improves cardiac function postinfarction in mice. Pharmacology research & perspectives. 2014;2:e00056
21. Gray C, Li M, Patel R, Reynolds CM, Vickers MH. Let-7 miRNA profiles are associated with the reversal of left ventricular hypertrophy and hypertension in adult male offspring from mothers undernourished during pregnancy after preweaning growth hormone treatment. Endocrinology. 2014;155:4808-17
22. Yang Y, Ago T, Zhai P, Abdellatif M, Sadoshima J. Thioredoxin 1 negatively regulates angiotensin II-induced cardiac hypertrophy through upregulation of miR-98/let-7. Circulation research. 2011;108:305-13
23. Yu SS, Cai Y, Ye JT, Pi RB, Chen SR, Liu PQ. et al. Sirtuin 6 protects cardiomyocytes from hypertrophy in vitro via inhibition of NF-kappaB-dependent transcriptional activity. British journal of pharmacology. 2013;168:117-28
24. Ikeda S, He A, Kong SW, Lu J, Bejar R, Bodyak N. et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Molecular and cellular biology. 2009;29:2193-204
25. Dong DL, Chen C, Huo R, Wang N, Li Z, Tu YJ. et al. Reciprocal repression between microRNA-133 and calcineurin regulates cardiac hypertrophy: a novel mechanism for progressive cardiac hypertrophy. Hypertension. 2010;55:946-52
26. Yan M, Chen C, Gong W, Yin Z, Zhou L, Chaugai S. et al. miR-21-3p regulates cardiac hypertrophic response by targeting histone deacetylase-8. Cardiovascular research. 2015;105:340-52
27. van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575-9
28. Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE. et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901-6
29. Akao Y, Nakagawa Y, Naoe T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biological & pharmaceutical bulletin. 2006;29:903-6
30. Esquela-Kerscher A, Trang P, Wiggins JF, Patrawala L, Cheng A, Ford L. et al. The let-7 microRNA reduces tumor growth in mouse models of lung cancer. Cell cycle. 2008;7:759-64
31. Thum T, Catalucci D, Bauersachs J. MicroRNAs: novel regulators in cardiac development and disease. Cardiovascular research. 2008;79:562-70
32. Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circulation research. 2007;100:416-24
33. Hunter JJ, Chien KR. Signaling pathways for cardiac hypertrophy and failure. The New England journal of medicine. 1999;341:1276-83
34. Pan ZW, Zhang Y, Mei DH, Zhang R, Wang JH, Zhang XY. et al. Scutellarin exerts its anti-hypertrophic effects via suppressing the Ca2+-mediated calcineurin and CaMKII signaling pathways. Naunyn-Schmiedeberg's archives of pharmacology. 2010;381:137-45
Author contact
![]() Corresponding authors: Yanjie Lu (E-mail: yjluedu.cn) or Chaoqian Xu (E-mail: xuchaoqianhrbmu.edu.cn), Department of Pharmacology (the State-Province Key Laboratories of Biomedicine-Pharmaceutics of China), Harbin Medical University, Harbin, Heilongjiang 150081, P.R. China (Fax. +86 451 8667-1354, Tel. +86 451 8667-1354).
Corresponding authors: Yanjie Lu (E-mail: yjluedu.cn) or Chaoqian Xu (E-mail: xuchaoqianhrbmu.edu.cn), Department of Pharmacology (the State-Province Key Laboratories of Biomedicine-Pharmaceutics of China), Harbin Medical University, Harbin, Heilongjiang 150081, P.R. China (Fax. +86 451 8667-1354, Tel. +86 451 8667-1354).
Citation styles
APA
Zhou, X., Sun, F., Luo, S., Zhao, W., Yang, T., Zhang, G., Gao, M., Lu, R., Shu, Y., Mu, W., Zhuang, Y., Ding, F., Xu, C., Lu, Y. (2017). Let-7a Is an Antihypertrophic Regulator in the Heart via Targeting Calmodulin. International Journal of Biological Sciences, 13(1), 22-31. https://doi.org/10.7150/ijbs.16298.
ACS
Zhou, X.; Sun, F.; Luo, S.; Zhao, W.; Yang, T.; Zhang, G.; Gao, M.; Lu, R.; Shu, Y.; Mu, W.; Zhuang, Y.; Ding, F.; Xu, C.; Lu, Y. Let-7a Is an Antihypertrophic Regulator in the Heart via Targeting Calmodulin. Int. J. Biol. Sci. 2017, 13 (1), 22-31. DOI: 10.7150/ijbs.16298.
NLM
Zhou X, Sun F, Luo S, Zhao W, Yang T, Zhang G, Gao M, Lu R, Shu Y, Mu W, Zhuang Y, Ding F, Xu C, Lu Y. Let-7a Is an Antihypertrophic Regulator in the Heart via Targeting Calmodulin. Int J Biol Sci 2017; 13(1):22-31. doi:10.7150/ijbs.16298. https://www.ijbs.com/v13p0022.htm
CSE
Zhou X, Sun F, Luo S, Zhao W, Yang T, Zhang G, Gao M, Lu R, Shu Y, Mu W, Zhuang Y, Ding F, Xu C, Lu Y. 2017. Let-7a Is an Antihypertrophic Regulator in the Heart via Targeting Calmodulin. Int J Biol Sci. 13(1):22-31.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.