Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(1):110-121. doi:10.7150/ijbs.16312 This issue Cite
Research Paper
Involvement of Rictor/mTORC2 in cardiomyocyte differentiation of mouse embryonic stem cells in vitro
Bei Zheng1, Jiadan Wang1, Leilei Tang1, Chao Tan1, Zhe Zhao2, Yi Xiao2, Renshan Ge3, 4, Danyan Zhu1 ![]()
1. Institute of Pharmacology and Toxicology, Zhejiang University, Hangzhou 310058, CHINA.
2. Undergraduate students in Research Training Project at Zhejiang University
Hangzhou 310058, CHINA.
3. The Population Council at the Rockefeller University, New York, NY 10021, USA.
4. Institute of Reproductive Biomedicine, the 2nd Affiliated Hospital of Wenzhou Medical University, Wenzhou 325027, CHINA.
Received 2016-5-28; Accepted 2016-10-21; Published 2017-1-15
Abstract

Rictor is a key regulatory/structural subunit of the mammalian target of rapamycin complex 2 (mTORC2) and is required for phosphorylation of Akt at serine 473. It plays an important role in cell survival, actin cytoskeleton organization and other processes in embryogenesis. However, the role of Rictor/mTORC2 in the embryonic cardiac differentiation has been uncovered. In the present study, we examined a possible link between Rictor expression and cardiomyocyte differentiation of the mouse embryonic stem (mES) cells. Knockdown of Rictor by shRNA significantly reduced the phosphorylation of Akt at serine 473 followed by a decrease in cardiomyocyte differentiation detected by beating embryoid bodies. The protein levels of brachyury (mesoderm protein), Nkx2.5 (cardiac progenitor cell protein) and α-Actinin (cardiomyocyte biomarker) decreased in Rictor knockdown group during cardiogenesis. Furthermore, knockdown of Rictor specifically inhibited the ventricular-like cells differentiation of mES cells with reduced level of ventricular-specific protein, MLC-2v. Meanwhile, patch-clamp analysis revealed that shRNA-Rictor significantly increased the number of cardiomyocytes with abnormal electrophysiology. In addition, the expressions and distribution patterns of cell-cell junction proteins (Cx43/Desmoplakin/N-cadherin) were also affected in shRNA-Rictor cardiomyocytes. Taken together, the results demonstrated that Rictor/mTORC2 might play an important role in the cardiomyocyte differentiation of mES cells. Knockdown of Rictor resulted in inhibiting ventricular-like myocytes differentiation and induced arrhythmias symptom, which was accompanied by interfering the expression and distribution patterns of cell-cell junction proteins. Rictor/mTORC2 might become a new target for regulating cardiomyocyte differentiation and a useful reference for application of the induced pluripotent stem cells.
Keywords: Rictor/mTORC2, embryonic stem cell, cardiomyocyte differentiation, cell-cell junction, electrophysiology.
Introduction
The mammalian target of rapamycin complex 2 (mTORC2) is a signaling protein complex that comprises mTOR, rapamycin-insensitive companion of mTOR (Rictor) and several binding partners. Rictor is important for the stability and activity of mTORC2 [1], which has been implicated in cytoskeletal organization [2]. Also, mTORC2 plays pivotal roles in embryonic development [3], tumorigenesis [4], cardioprotection in myocardial ischemia-reperfusion injury [5, 6] and cardiomyocyte functions [5]. Down-regulation of Rictor could decrease the terminal differentiation of C2C12 myoblasts by interfering the phosphorylation of Akt (Ser473), a known substrate of mTORC2 [7]. The developing embryos that lacked Rictor exhibit growth arrest at E9.5, and die at E11.5 [8]. However, the detailed functions and the mechanism by which Rictor/mTORC2 may affect embryonic cardiogenesis remains poorly understood.
Differentiation of mouse embryonic stem (mES) cells into cardiomyocytes offers an innovative approach to find the new target for regulating the cardiac development and to reveal underlying mechanisms for induced pluripotent stem cell researches. However, differentiation of mES cells can result in the different lineages, the atrial-, ventricular- and sinus node-like cardiomyocytes without selectivity. Previous studies showed that treatment of mES cells with suramin [9] or human ES cells with IWR-1 [10] could enhance cardiac specialization into sinus node-like or ventricular-like cells respectively. To direct the differentiation of mES cells into a desired cardiac subtype, the mechanisms of cardiac subtype specification must be covered. Identifying the key regulators of cardiac subtype specification is critical for reducing the heterogeneity of the mES cell-derived cardiomyocyte population. We hypothesize that Rictor/mTORC2 signaling may regulate the differentiation of mES cells into specific cardiomyocyte subtypes.
Cardiac gap junction (GJ) plays a pivotal role in maintaining the velocity and the safety of impulse propagation in cardiac tissue, and in keeping normal cardiac rhythm [11]. A major component of GJ is connexin 43 (Cx43), which affects cardiac electrophysiological characteristic and involves in action potential (AP) of cardiomyocyte spreading. The phosphorylation of Cx43 is proposed to be associated with gap junction communication [12, 13]. Meanwhile, cardiac adherens junction (AJ) formation is a prerequisite for gap junction assembly in cardiomyocytes [14]. Cardiac-specific deletion of N-cadherin or desmoplakin, the major cardiac AJ components, results in the reduced Cx43-containing gap junctions, accompanied by disassembly of the intercalated disc structure [15] and sudden arrhythmic death [16]. Cx43, N-cadherin and Desmoplakin involve in consistent cellular activity, information transmission, differentiation and development [17, 18], by stabilizing the cell-cell junctions and myocyte integrity [19]. In addition, Rictor/mTORC2 may regulate blood-testis barrier dynamics via its effects on the expression and distribution of Cx43 [20]. However, the relationships between Rictor/mTORC2 and Cx43/N-cadherin/Desmoplakin in regulating cardiogenesis and cardiomyocyte electrophysiology have not yet been reported.
In the present study, cardiomyocyte differentiation of mES cells is employed to evaluate the expression and function of Rictor/mTORC2 during cardiomyocyte differentiation. Specifically, the relationship between Rictor knockdown (shRNA-Rictor) and the specific cardiomyocyte subtype differentiation was examined. In addition, the electrophysiological characteristics of the differentiated cardiomyocytes were evaluated under shRNA-Rictor conditions by patch-clamp analysis. Finally, whether shRNA-Rictor affected the expressions and distributions of cardiac related junction proteins were confirmed in cardiomyocytes derived from shRNA-Rictor mES cells by immunofluorescence and western blot analysis. The results showed that Rictor knockdown could result in inhibiting the ventricular-like myocytes differentiation and inducing the arrhythmias symptom, which was accompanied by changes in expression and distribution patterns of cell-cell junction proteins.
Materials and Methods
Cell Culture and Cardiomyocyte differentiation
mES cells (Mouse ES cell D3, obtained from American Type Culture Collection, USA) were cultured in DMEM medium (Life Technologies, Germany) supplemented with 1% nonessential amino acids (NEAA, Life Technologies, Germany), 10% fetal bovine serum (FBS, Life Technologies, Germany), 0.1 mmol/L β-mercaptoethanol (Sigma Aldrich, USA), and 106 units/L mouse leukemia inhibitory factor (Chemicon, USA) in 5% CO2 atmosphere at 37 ºC. mES cells (about 600) were cultured in a hanging droplet of 30 µl to form EBs for 3 days in differentiation medium (DMEM with 20% FBS, 0.1 mmol/L β-mercaptoethanol and 1% NEAA). After cultured in hanging droplet for 3 days and floating in the petri dishes for another 2 days, EBs plated separately into gelatin (0.1%, Sigma Aldrich, USA)-coated 24-well plates. Medium was changed every two days. Morphology and beating behavior of EBs were monitored by light microscopy at 37ºC [21].
Rictor Targeted shRNA Infection
Lentivirus with Rictor short hairpin RNA (shRNA) or control shRNA were infected into mES cells [7]. shRNA targeting mouse Rictor mRNA as well as a validated negative control shRNA labeled with GFP were ordered from Genepharma Company (Shanghai, China).
Target shRNA-Rictor sequence: GCCAGTAAGATGGGAATCATT, shRNA-Con: TTCTCCGAACGTGTCACGTTC.
Briefly, mES cells, 1×105 per well, were seeded into six-well plates. On the second day, the cells were infected with an aliquot of lentivirus to achieve a multiplicity of infection of 50 PFU/cell. After infected for 24 hours, cells were harvested for EB formation or further measurement of Rictor protein.
Isolation of Cardiomyocyte-Rich Cell Clusters
The beating areas of EBs were dissociated by gentle pipetting with a glass pipette of 200 to 300μm internal diameter. The cardiomyocyte-rich cell clusters were then trypsinized with Collagenase II (Invitrogen, USA) for 30 min. Detached cells were used for further analyses [22].
Western Blotting
Cells were collected in RIPA buffer (containing 0.2% Triton X-100, 5 mmol/L EDTA, 1 mmol/L PMSF, 10 mg/ml leupeptin, 10 mg/ml aprotinin, 100 mmol/L NaF, and 2 mmol/L Na3VO4) and lysed 30 min on ice. Protein concentration was assayed using the Bio-Rad protein kit (Hercules, USA), and equal amount of proteins per sample were loaded on a sodium dodecyl sulphate (SDS)-polyacrylamide gel. Subsequently, proteins were transferred onto 0.45 μm pore size positively charged nylon membranes (PVDF, Millipore, USA) and blocked with 5% milk in PBS with 0.1% Tween-20 at room temperature. The membrane was incubated with one of the primary antibodies overnight at 4 ºC. The antibodies used include mouse monoclonal anti-GAPDH (Sigma Aldrich, G-8975, USA), anti-α-Actinin (Sigma Aldrich, A-7811, USA), mouse monoclonal anti-HCN4 (Abcam, ab-32675, USA), rabbit polyclonal anti-Akt1/2/3 (Ser 473)-R (Santa Cruz, sc-7985-R, USA), rabbit polyclonal anti-Akt1 (Thr 308) (Santa Cruz, sc-135650, USA), rabbit polyclonal anti-Akt (Santa Cruz, sc-135650, USA), rabbit monoclonal anti-GSK3β (Ser 9) (Cell Signaling Technology, 5558, USA), rabbit polyclonal anti-GSK3β (Abcam, ab-35842, USA), rabbit monoclonal anti-brachyury (Santa Cruz, sc-20109, USA), rabbit polyclonal anti-desmoplakin I/II (Santa Cruz, sc-33555, USA), rabbit polyclonal anti-Sox17 (Millpore, USA), rabbit polyclonal anti-Nkx2.5 (Abcam, ab-35842, USA), rabbit polyclonal anti-Connexin 43 (Abcam, ab-11370, USA), rabbit polyclonal anti-N-cadherin (Santa Cruz, sc-31030, USA), rabbit monoclonal anti-ANP (Santa Cruz, sc-20158, USA), rabbit monoclonal anti-MLC-2v (Abcam, ab-79935, USA), goat polyclonal anti-Rictor (Santa Cruz, sc-50678, USA), goat polyclonal anti-FGF5 (Santa Cruz, sc-1363, USA). The membrane was washed three times with TTBS and incubated with one of the conjugated secondary antibodies, HRP-conjugated goat anti-rabbit, rabbit anti-goat or goat anti-mouse antibodies, respectively. After washing with TTBS three times in 10 min each, the target protein bands were detected with an enhanced chemiluminescent substrate (ECL, Pierce, USA) [23].
Immunofluorescence Analysis
Immunofluorescence was performed with either EBs or ESC-CMs. Cells were fixed with methanol at -20 ºC for 15min, followed by permeabilization with 0.05% Triton-100. The cells were blocked with 10% FBS for 30 min at room temperature and incubated in PBS containing one of the primary antibodies overnight at 4ºC and followed by secondary antibodies for 2 hours at room temperature. The primary antibodies included mouse monoclonal anti-α-Actinin (Sigma Aldrich, A-7811, USA), rabbit polyclonal anti-Nkx2.5 (Santa Cruz, sc-376565, USA), rabbit polyclonal anti-desmoplakin I/II (Santa Cruz, sc-33555, USA), rabbit polyclonal anti-N-cadherin (Santa Cruz, sc-31030, USA) or rabbit polyclonal anti-Connexin 43 (Abcam, ab-11370, USA), mouse monoclonal anti-Connexin 43 (Abcam, ab-79010, USA), rabbit polyclonal anti-MLC-2v (Abcam, ab-79935, USA). The second antibodies included Alexa fluor 488-conjugated anti-mouse IgG (Invitrogen, Carlsbad, CA, USA), Alexa fluor 594-conjugated anti-mouse IgG (Invitrogen, USA), Alexa fluor 488-conjugated anti-rabbit IgG (Invitrogen, Carlsbad, CA, USA) or Alexa fluor 594-conjugated anti-rabbit IgG (Invitrogen, USA). After washing, the fluorescence images were taken by fluorescence inverted microscope or an Olympus IX81-FV1000 inverted multiphoton laser confocal microscope [24].
Flow Cytometry Analysis
EBs or ESC-CMs obtained on day 5+3 of differentiation were digested into single cells with Collagenase II (Invitrogen, USA). Cells were fixed with 4% paraformaldehyde for 1 h and then treated with 10% FBS for another 1 h to block non-specific antigens. Cells were then incubated in PBS containing one of the primary antibodies overnight at 4 ºC. The primary antibodies included mouse monoclonal anti-α-Actinin (Sigma Aldrich, A-7811, USA), rabbit polyclonal anti-MLC-2v (Abcam, ab-79935, USA), mouse monoclonal anti-Connexin 43 (Abcam, ab-79010, USA). After washing with PBS twice, cells were incubated with Alexa fluor 488-conjugated anti-mouse IgG or phycoerythrin-conjugated anti-rabbit IgG (Invitrogen, USA) for 1 h, and then suspended in 0.5 ml 1% BSA and analyzed on a FACScan flow cytometry (Becton-Dickson, Sparks, USA). The fluorochrome was detected at 530 nm in the FL-1 and 575nm in the FL-2 channel. Each plot represented 10,000 viable cells (nonviable cells were excluded from FACS analysis by appropriate gating). Untreated cells and cells lacking primary antibody were used as negative controls. In addition, isotype controls were used to assess the level of non-specific antibody binding [24]. All data analyses were carried out by using Cell Quest software.
Cell Proliferation Assay
The effect of shRNA-Rictor on cell growth was determined with the 3-(4,5-dmethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells of infected mESC were seeded into 96-well plates at an initial density of 1×104 cells/well in 100 μl of the culture medium for 2 days. At the experiment day, 100 μl DMEM containing 0.5 mg/ml MTT was added to each well and incubated for 4 h at 37°C in 5% CO2. The reaction was stopped by adding 100 μl of DMSO and the absorbance was measured at 560 nm using a microplate reader. Data on cell viability were expressed in percentage compared to the control [25].
Electrophysiological Recordings
The action potentials (APs) of spontaneously beating ESC-CMs were recorded by the whole-cell patch-clamp under current-clamp mode at physiological temperature (37 ± 0.3ºC) with a continuous superfusion of normal Tyrode's solution consisting of the following components (g/L): NaCl 8.1816, NaOH 0.092, KCl 0.40257, CaCl2 0.199782, MgCl2-6H2O 0.2033, HEPES 2.383, Glucose 1.9817 (pH adjusted to 7.2-7.4 with NaOH). Patch pipettes (2 to 5 MΩ) were filled with the internal solution consisting of the following components (g/L): KCl 0.0745, K-asparate 0.2739, EGTA 0.0744, HEPES 0.0477, MgATP(Na2) 0.0319, MgCl2 0.019 (pH adjusted to 7.2-7.4 with KOH). ESC-CMs were visualized with an infrared-sensitive CCD camera equipped with a ×40 water-immersion lens (Nikon, ECLIPSE FN1). The cells were recorded using whole-cell techniques (Multi Clamp 700B Amplifier, Digi data 1440A analog-to-digital converter) with pClamp 10.2 software (Axon Instruments/Molecular Devices). The APs were classified by using previously described criteria. The electrophysiological parameters were analyzed using Lab chart7 software. The numbers of cells analyzed for control group was 114 and for shRNA-Rictor group was 64 [9, 26].
Statistics
Data were expressed as mean values with standard deviation. At least 3 independent experiments were performed. Statistical analysis was performed with one-way analysis of variance. P < 0.05 was considered to be significant.
Results
Rictor knockdown suppresses cardiomyocyte differentiation from mES cells
To determine whether Rictor/mTORC2 required for cardiomyocyte differentiation of mES cells, shRNA was used to selectively knockdown Rictor. Treatment with shRNA against Rictor (shRNA-Rictor) in mES cells reduced Rictor protein levels significantly, which was also accompanied by a marked decrease in the expression of p-Akt (Ser473) and unchanged level of p-Akt (Thr308) (Figure 1A). We further detected the phosphorylation level of GSK3β which is the downstream target of Akt to investigate the activity of Akt. The results showed reduced levels of p-GSK3β (Ser9) after knockdown of Rictor demonstrating the inactivation of Akt (Figure 1A). shRNA-Rictor in mES cells decreased the differentiation ratio of the beating embryoid bodies (EBs) by 21±6% and 42±6% compared with shRNA-Con cells (40±3%, 70±6%) on day 5+2 and day 5+3, respectively (Figure 1B). To further investigate whether Rictor deficiency in mES cells could influence the blastodermic layer development, the protein levels of FGF5 (Epiblast protein), brachyury (mesoderm protein), Sox17 (endoderm protein) and Nkx2.5 (cardiac progenitor cell protein) were analyzed during the early cardiomyocyte differentiation of mES cells. Among the four proteins examined, Brachyury and Nkx2.5, selectively decreased in shRNA-Rictor cells during the early differentiation (day 3 and day 5), while FGF5 and Sox17 showed no significant changes at this stage (Figure 1C). The immunostaing of Nkx2.5 in EBs showed the reduced expression of Nkx2.5 by knockdown of Rictor, which demonstrated that cardiac progenitor cells differentiation from mES cells were inhibited (Figure 1D). This suggests that shRNA-Rictor treatment of mES cells may selectively inhibit the differentiation toward mesoderm, but have no effects on epiblast and endoderm differentiation. Meanwhile, the expression of α-Actinin (early cardiomyocyte specific protein) also decreased in shRNA-Rictor group, indicating a reduction in the differentiation of cardiomyocytes compared with shRNA-Con group (Figure 1E). Therefore, Rictor might play critical roles in the early mesoderm formation and cardiomyocyte differentiation of mES cells.
Effects of knockdown of Rictor expression by shRNA in mES cells on the cardiomyocyte differentiation. (A) Knockdown of Rictor (shRNA-Rictor) in mES cells during the differentiation. (B) The beating phenotype evaluated from day 5+1 to day 5+3 after EBs plating. (C) The expression levels of blastodermic layer proteins in the early cardiomyocyte differentiation. (D) The expression of Nkx2.5 in cryosections of EBs on day 5 by immunofluoresence analysis. (E) The expression level of sarcomeric protein detected by western blot. (F) Quantification of sarcomeric protein (α-Actinin) by flow cytometry on day 5+3. (G) Immunofluorescence analysis of the expression and distribution of sarcomeric α-Actinin (red) on day 5+3. Nuclei were stained with DAPI (blue). Bar=10 μm or 100 μm. (H) The proliferation rate of mES cells after infection by shRNA-Rictor. (I) The proportion of α-Actinin-positive cells after purification. Data were represented as means ± SD of three independent experiments. *P<0.05, **P<0.01 (shRNA-Rictor vs shRNA-Con).
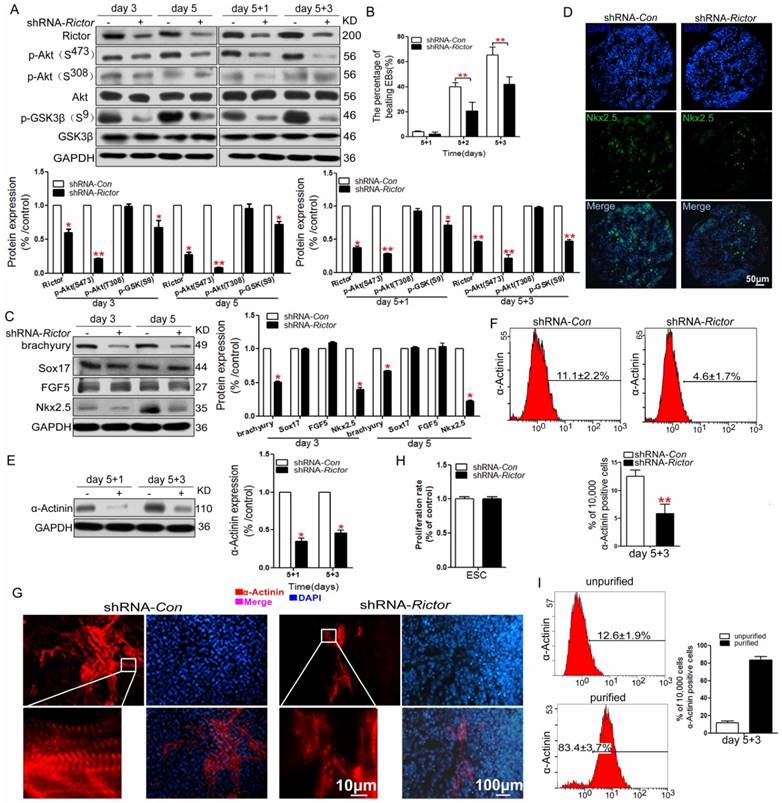
Analysis of α-Actinin expression by flow cytometry demonstrated that only 4.6±1.7% of cells were positive for the protein in shRNA-Rictor group, compared with 11.1±2.2% of the positive cells in shRNA-Con group on day 5+3 (Figure 1F). The immunofluorescence analysis was employed to reveal the expression pattern and structure of sarcomeric α-Actinin in the differentiated cardiomyocytes. The results indicated that Rictor knockdown decreased the sarcomeric protein expression associated with a disorderly sarcomeric structure (Figure 1G). These results consistently indicated that knockdown of Rictor expression by shRNA in mES cells suppressed the quantities of differentiated cardiomyocytes and impaired the normal formation of cardiac specific sarcomere.
We next sought to determine whether the suppressed effect of shRNA-Rictor on cardiomyocyte differentiation was associated with influenced proliferation of mES cells. MTT assay showed that the proliferation rate of mESC infected with shRNA-Rictor was not changed compared with the control group (Figure 1H). It indicated that the reduced cardiomyocyte differentiation by shRNA-Rictor was not due to the inhibition of mES cells proliferation.
Electrophysiological characterization of cardiomyocytes differentiated from mES cells
In order to investigate whether shRNA-Rictor treatment regulated specific cardiac cell subtype differentiation, the electrophysiological measurement of mES cells-derived cardiomyocytes (mESC-CMs) was used to distinguish the cardiomyocytes with early sinus node-, atrial-, ventricular- and purkinje-like action potentials. The proportion of cardiomyocytes after purification increased to 83.4±3.7% (Figure 1I). The action potentials (AP) of cardiomyocytes were measured by the whole cell patch-clamp technique. Based on the morphology and classification of AP properties (Table 1), four major types of AP (sinus node-like, atrial-like, ventricular-like and purkinje-like) were observed in our study (Figure 2A).
Electrophysiological properties of cardiomyocytes differentiated from mES cells on day 5+3.
| AP type | lentivirus | Vmax(V/s) | APA(mV) | APD50(ms) | APD90(ms) | n (%) |
|---|---|---|---|---|---|---|
| Sinus node-like | shRNA-Con | 3.5±1.2 | 58.8±14.2 | 173.6±22.0 | 208±14.2 | 32 |
| shRNA-Rictor | 2.93±0.9 | 17.6±14.3** | 169.3±15.7 | 226±23.3 | 45 | |
| Ventricular-like | shRNA-Con | 65.4±10.6 | 89.4±7 | 214.5±21.7 | 270±45.5 | 43 |
| shRNA-Rictor | 10.6±2.0*** | 61.6±19*** | 250.7±25.8** | 365.5±58.6** | 14 | |
| Atrial-like | shRNA-Con | 69.3±7.4 | 71.4±8.3 | 107.9±17.03 | 167.0±15.8 | 21 |
| shRNA-Rictor | 59.1±12.4 | 65.3±17.1 | 188.6±10.9** | 256.6±23.3** | 41 |
Data were means±SD. n indicated the number of cells tested. Vmax, maximum rate of AP increase. APA, AP amplitude. APD50, AP duration measured at 50% repolarisation. APD90, AP duration measured at 90% repolarisation. **P <0.01 and ***P <0.001 compared with shRNA-Con.
Electrophysiological characteristics of mESC-CMs identified by patch clamp. (A) Four major cardiac subtypes of APs discriminated in shRNA-Rictor treated mESC-CMs: sinus node-, atrial-, ventricular-, or purkinje-like APs. (B) The relative percentages of the AP types recorded from shRNA-Rictor treated cells. (C) The AP frequency recorded on shRNA-Rictor treated cells. Data were represented as means ± SD. Statistical significance was set as ***P <0.001 (shRNA-Rictor, n=64 vs shRNA-Con, n=114).
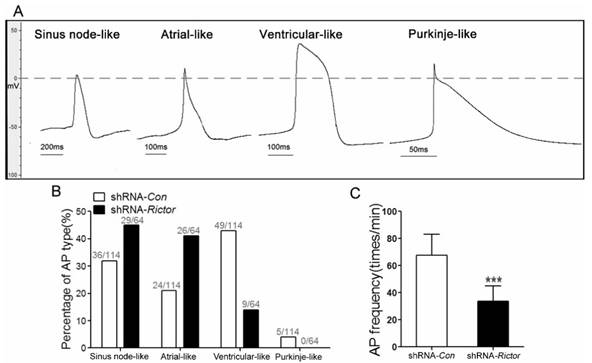
The ratios of AP for four major types were different between shRNA-Rictor group and shRNA-Con group. A significantly decreased proportion of ventricular-like cells was revealed in the shRNA-Rictor group on day 5+3, whereas the percentage of atrial- and sinus node-like cells increased compared with shRNA-Con group (Figure 2B). In the shRNA-Rictor group (n=64), 14% of myocytes possessed ventricular-like AP, significantly diminished compared with 43% in the shRNA-Con group (n=114). However, 45% and 41% of the myocytes from shRNA-Rictor group exhibited a sinus node-like and atrial-like AP, respectively. The purkinje-like cells were not identified in shRNA-Rictor group (Table 1 and Figure 2B). The AP frequency was also reduced by shRNA-Rictor treatment (Figure 2C). The patch-clamp data further suggested that shRNA-Rictor application might decrease the number of ventricular-like cells specifically and could affect the function of differentiated cardiomyocytes.
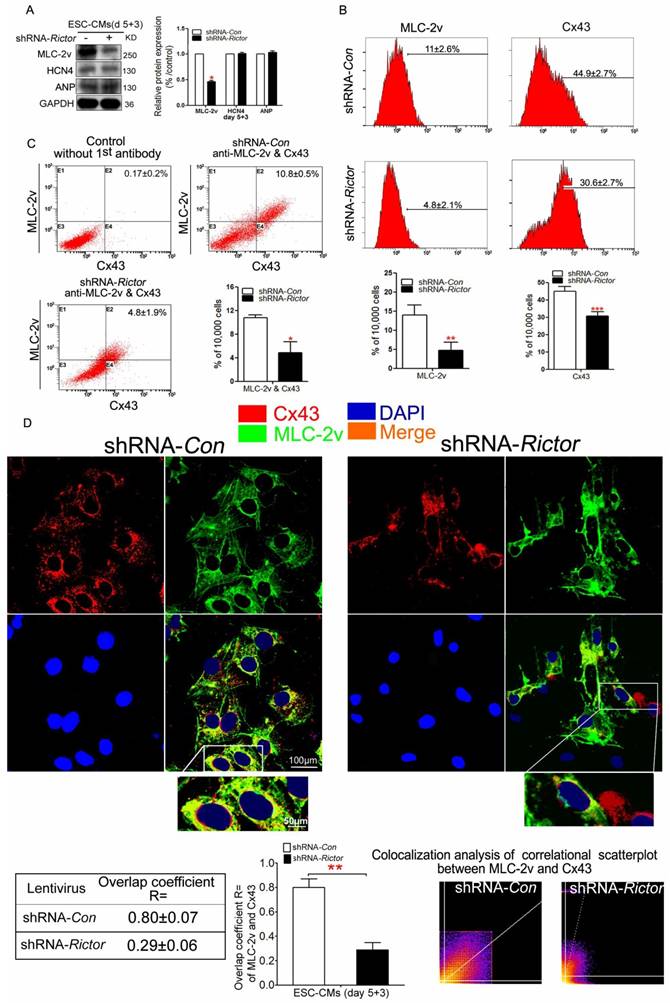
To further confirm that Rictor/mTORC2 might influence the formation of ventricular-like cells, we investigated the expressions of ventricular marker protein MLC-2v. Meanwhile, we also analyzed the expressions of ANP (the atrial marker) and HCN4 (the sinus node marker). Knockdown of Rictor indeed inhibited the expressions of MLC-2v, while the protein levels of HCN4 and ANP were not significantly affected (Figure 3A). Cx43 is a predominant gap junction protein in ventricular cells playing significant roles in normal electrophisiology. Flow cytometry analysis of shRNA-Rictor cells showed that the percentage of MLC-2v positive cells were significantly decreased to 4.8±2.1% compared with the shRNA-Con group (Figure 3B). The co-expression of Cx43 and MLC-2v was significantly reduced to 4.8±1.9% in mESC-CMs by shRNA-Rictor, compared with 10.8±0.5% of the positive-staining cells in shRNA-Con group (Figure 3C). Also, the co-expression of Cx43 and MLC-2v staining pattern was changed and significantly curtailed in shRNA-Rictor mESC-CMs by immunofluorescence analysis. Cx43 and MLC-2v staining were intense at cytoplasm in shRNA-Con mESC-CMs, while Cx43 was sparse at cytoplasm, and MLC-2v appeared less intense across the sarcomere in shRNA-Rictor mESC-CMs (Figure 3D).
Effects of Rictor/mTORC2 knockdown on the formation of ventricular-like cells and the expressions of MLC-2v and Cx43. (A) Western blot analysis indicated that shRNA-Rictor inhibited the expression of ventricular-specific MLC-2v, while the expression of HCN4 and ANP were not changed in mESC-CMs. (B) Flow cytometry analysis showed that the expressions of MLC-2v or Cx43 were reduced in shRNA-Rictor cells isolated from day 5+3 EBs. Representative histogram and the statistics analyses were shown. (C) Quantification of MLC-2v and Cx43 co-expression by flow cytometry. Representative dot blot analyses of MLC-2v and Cx43 co-expression were shown. The X-axis corresponding to fluorochrome FITC detected at 530 nm, and Y-axis corresponding to fluorochrome PE detected at 575 nm. (D) Analysis of ventricular-specific MLC-2v and Cx43 co-immunostained for MLC-2v (green) and Cx43 (red) on day 5+3. Nuclei were stained with DAPI (blue). The colocalization analysis of correlational scatterplot between MLC-2v and Cx43 were counted. Bar=100 μm or 50 μm. Similar data were obtained from at least 3 independent experiments. Data were shown as means ± SD. *P<0.05, **P <0.01, ***P<0.001 for shRNA-Rictor vs shRNA-Con.
Suppression of Rictor expression induced cardiomyocyte arrhythmia
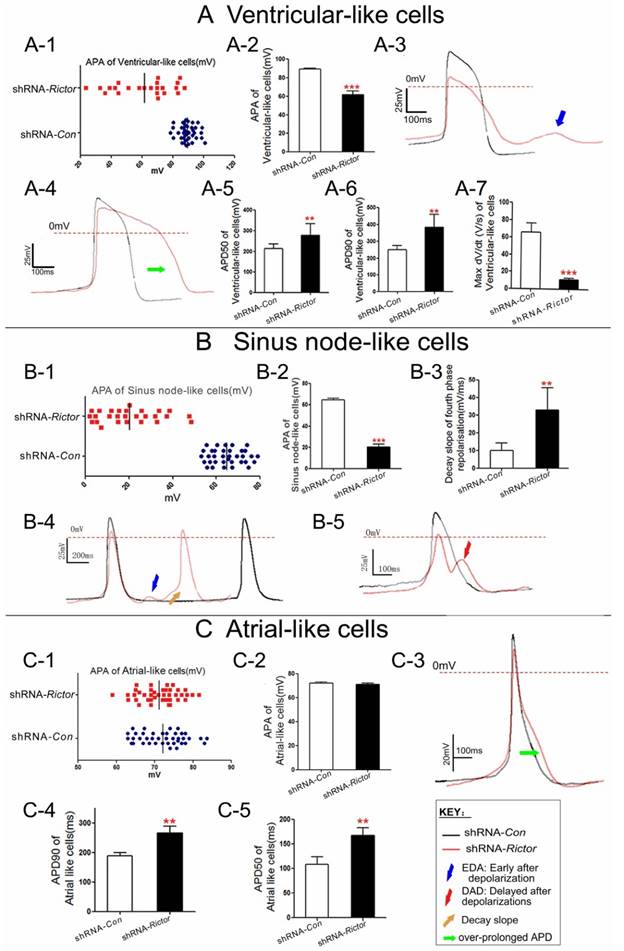
To understand the regulation of Rictor on the cardiomyocyte excitability, the action potential parameters of mESC-CMs were determined by the whole cell patch-clamp technique. The mESC-CMs displayed a variety of action potentials and activated spontaneously. The AP amplitude (APA) of ventricular and purkinje-like myocytes from shRNA-Rictor mESC-CMs were reduced, and irregularly distributed between 20 to 90 mv, while those in shRNA-Con mESC-CMs were regularly distributed between 80 to 100 mv. The results indicated that shRNA-Rictor induced the allorhythmia of mESC-CMs (Figure 4A-1/2, 4B-1/2, 4C-1/2). Action potential duration of ventricular-like and atrial-like myocytes from shRNA-Rictor mESC-CMs were prolonged compared to those of shRNA-Con (Figure 4A-4 and 4C-3). Action potential duration measured at 90% re-polarization of ventricular-like and atrial-like myocytes from shRNA-Rictor mESC-CMs were significantly prolonged (365.5±58.6 and 256.6±23.3 ms, respectively) relative to those from shRNA-Con (270±45.5 and 167.0±15.8 ms) (Table 1 and Figure 4A-6 and 4C-4). Action potential duration measured at 50% re-polarization showed the similar prolongation in ventricular-like and atrial-like myocytes with shRNA-Rictor (Table 1 and Figure 4A-5 and 4C-5). However, sinus node-like myocytes did not show any notable differences between the lines (Table 1). The maximum rate of AP increase (Vmax) was remarkably diminished in the shRNA-Rictor treated cultures, which reduced in the excitability of ventricular-like myocytes (Figure 4A-7). Meanwhile, knockdown of Rictor expression by shRNA in mES cells could decrease automatic rhythmicity of sinus node-like myocytes through the decay slope of the fourth phase re-polarization (Figure 4B-4, 4B-5). Nearly 50% ventricular-like and sinus node-like myocytes treated with shRNA-Rictor resulted in electrophysiological abnormalities, including the early after depolarizations (EADs) (Figure 4B-5) and the delayed after depolarizations (DADs) (Figure 4A-3 and 4B-4). Such events were never observed in the control myocytes. These observations, revealed by the electrophysiological characteristics of cardiomyocytes, included the over-prolonged AP duration, disordered APA, abnormal Vmax, DAD and EAD in shRNA-Rictor mESC-CMs. These suggested that the absence of Rictor might be associated with the occurrence of the arrhythmia in mESC-CMs.
Electrophysiological characteristics of cardiomyocytes differentiated from shRNA-Rictor infected mESC-CMs on day 5+3. (A) Ventricular-like APs. (B) Sinus node-like APs. (C) Atrial-like APs. Data were shown as means ± SD. Statistical significance was set as **P <0.01, ***P <0.001 (shRNA-Rictor, n=64 vs shRNA-Con, n=114).
Knockdown of Rictor inhibited cardiomyocyte functions via its effects on Cx43, N-cadherin and Desmoplakin
Cell-cell junctions play a key role in the cellular response to electrophysiological signals. The action potentials of cardiomyocytes are mediated through GJ, which involves in synchronize heart contraction. Cx43 is one of the most important component of GJ among cardiomyocytes. We hypothesized that Rictor/mTORC2 affected electrophysiological response of the cardiomyocytes by influencing the cell-cell junction proteins.
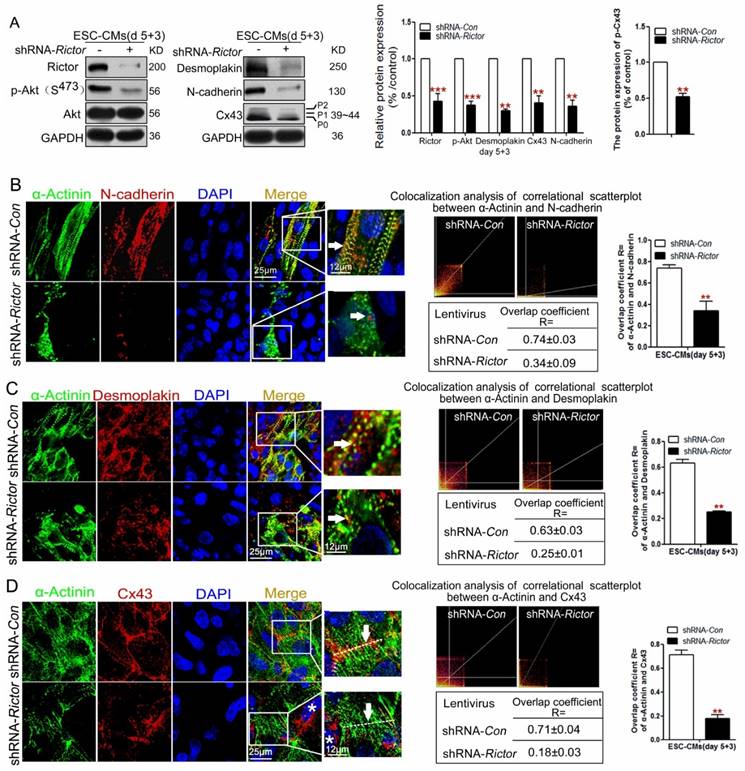
As expected, both western blot and immunofluorescence analysis consistently indicated that knockdown of Rictor in mESC-CMs resulted in the decrease in three cell junction proteins, Cx43, Desmoplakin and N-cadherin. Meanwhile, there was a marked reduction in the phosphorylation forms (P1, P2) of Cx43 (Figure 5A). Also, Rictor knockdown caused changes in the distribution of Desmoplakin and N-cadherin within the cells. Normally the two proteins localize tightly in the cell-cell interface or cytoplasm, whereas they disorderly distributed in the cytoplasm of shRNA-Rictor mESC-CMs on day 5+3 (Figure 5B, C). Besides, Desmoplakin or N-cadherin did not tightly co-localize with α-Actinin in Rictor-knockdown mESC-CMs, where only 0.34±0.09 of cells co-stained for both α-Actinin and N-cadherin (Figure 5B), and 0.25±0.01 of cells co-stained for α-Actinin and Desmoplakin, respectively (Figure 5C). Also, Cx43 normally displayed intensely at cell-to-cell contacts in shRNA-Con mESC-CMs, while it is sparse at cell-cell contacts in shRNA-Rictor group, and also scattered across the whole cells with intense staining in perinuclear location (*) (Figure 5D).
Knockdown of Rictor blocked cardiomyocyte functions via its effects on Cx43, N-cadherin and Desmoplakin protein expressions on day 5+3. (A) Western blot analysis of the expressions of Cx43, p-Cx43, N-cadherin and Desmoplakin. Densitometric quantification of the target proteins, with the values corrected by GAPDH as a loading control. (B) Immunofluorescence analysis showed co-localizations (orange, arrow) of α-Actinin (green) with N-cadherin (red) in mESC-CMs. The co-localization analysis of correlational scatterplot between α-Actinin and N-cadherin were counted. (C) Immunofluorescence analysis showed the co-localizations (orange, arrow) of α-Actinin (green) with Desmoplakin (red) in mESC-CMs. The colocalization analysis of correlational scatterplot between α-Actinin and Desmoplakin were counted. (D) Immunofluorescence analysis showed the co-localizations of α-Actinin (green) and Cx43 (red). Nuclei were stained with DAPI (blue). Bar=25 μm or 12 μm. Similar data were obtained from at least 3 independent experiments. Data were shown as means ± SD. **P <0.01, ***P <0.001 (shRNA-Rictor vs shRNA-Con).
Discussion
Cardiomyocytes differentiation of stem cells provides a promising approach that not only is useful in studying cardiac myocytes development, but also has a therapeutic potential [27]. The ES cells derived from the inner cell mass of the embryo emerged as a special tool to understand the molecular mechanisms involved in the process of cardiomyocyte differentiation [28]. Our previous studies have reported that PPARα and mGluR5 could facilitate cardiogenesis of mES cells [29, 30]. In this study, we extended our effort to examine a possible role of mTORC2 in cardiomyocyte differentiation. The mTORC2 is a multi-protein complex constituted of mTOR, Rictor plus several binding partners that are all required for its activity [1]. Rictor is a core component of the complex which is important to actin cytoskeletal organization [2, 31]. Furthermore, mTORC2 has been linked to protein synthesis and maturation [32], mitochondrial respiration [33], and embryonic development [3, 8]. Specifically, the differentiation of myoblasts was also shown to require Rictor/mTORC2 [7]. Knockdown of Rictor or disruption of the complex by a prolonged rapamycin treatment could inhibit terminal myoblast differentiation. Sciarretta and colleagues have found that Rictor/mTORC2 could preserve cardiac structure and function by restraining the activity of MST1 kinase. Suppression of Rictor/mTORC2 can increase cardiomyocyte apoptosis and tissue damages after myocardial infarction [34]. mTORC2 could promote post-natal heart growth and maintain heart function in mice by synergistically interacting with PDK1 [35]. It is known that spontaneous apoptosis occured during the process of differentiation [36]. mTORC2 was proved to be associated with apoptosis in osteosarcoma cell and breast cancer [37, 38]. It is tempting to speculate the possible relationship between mTORC2 and apoptosis involved in cardiomyocyte differentiation. In this study, we used a lentivirus encoding Rictor shRNA to test the functional involvement of Rictor/mTORC2 in cardiomyocyte differentiation. The differentiation of mES cells was characterized by the specific proteins that regulated the germ layer development. Brachyury, FGF5 and Sox17 were selected as mesoderm, ectoderm and endoderm marker, respectively [39, 40]. Nkx2.5 was indentified as cardiac progenitor cells marker [41]. The expression of α-Actinin, a cardiac-specific protein, was used to monitor the formation of cardiomyocytes from mES cells [42, 43]. Our results showed that the proteins related to cardiomyocyte differentiation were significantly decreased by the Rictor knockdown in mES cells. It suggested that ablation of Rictor inhibited the cardiomyocyte differentiation of mES cells.
Patch-clamp analysis was used to analyze the electrophysiology of cardiomyocytes and to distinguish cardiomyocytes subtypes. Previous studies reported that the differentiation of atrial- or ventricular-like cells could be accelerated by alternating retinoid signals in human embryonic stem cell [26]. Treatment with suramin induced mES cells differentiation toward to sinus node-like cells [9]. Our data showed that the proportion of ventricular-like cells was markedly reduced after knockdown of Rictor. Also, the electrophysiological characteristics of cardiac myocytes arrhythmia, including over-prolonged AP duration, disordered APA, abnormal Vmax, DAD and EAD, appeared in shRNA-Rictor cardiomyocytes. These findings support the conclusions that Rictor/mTORC2 is important for directing ventricular-like cell differentiation and forming electrophysiologically normal cardiomyocytes differentiation of mES cells.
To further investigate the mechanisms by which Rictor/mTORC2 may influence the electrophysiology of cardiomyocytes differentiated from mES cells, we have examined the expression and distribution of junctional proteins. It has been reported that the abnormal expression levels, the structural confusion and disorder distributions of Cx43, N-cadherin and Desmoplakin may all contribute to the impaired myocyte integrity, cardiac arrhythmias and cardiac remodeling [44]. As the components of three distinct junctional complexes (gap junction, adherens junction and desmosome) that constituted to interacted disk (ID), these proteins work together to mediate mechanical and electrical coupling of cardiomyocytes [45]. In the development, Cx43 is expressed both in the working myocardium of the ventricular cells and the atrial cells [44]. The interaction between Cx43 and Nav1.5, which was mediated by Desmoplakin, could be crucial for normal AP propagation [46]. Cx43 knockdown or over-expression experiments proved its association with cell coupling which ultimately influenced electrophysiological conduction of cardiomyocytes [47]. Also, the phosphorylation level of Cx43 played an important role in gap junction status and function [12, 13]. Prior studies had shown that cardiac-specific ablation of N-cadherin could result in decreased levels of gap junctions proteins Cx43 and Cx40, reduced ventricular conduction velocity [48] and impaired GJ communications [49]. Desmoplakin mutation, on other hand, lead to the occurrence of arrhythmogenic ventricular cardiomyopathy [50]. In this study, we found that knockdown of Rictor in mES cells significantly affected the expression and distribution of Cx43, Desmoplakin and N-cadherin proteins in the differentiated cardiac myocytes. These data indicated that disruption of Rictor/mTORC2 signaling not only resulted in the reduced cardiomyocyte differentiation efficacy of mES cells, but also affected the quality of the differentiated cells with the abnormal distribution and expression of the cell-cell adhesion and connexin proteins that are associated with changes in electrophysiological characteristics and conduction velocity.
In conclusion, knockdown of Rictor in mES cells resulted in reductions in cardiomyocytes of mES cell-derived mesoderm protein Brachyury, early cardiomyocyte protein Nkx2.5, and α-Actinin, followed by reduction of ventricular-like cells specific marker MLC-2v. Reduction in Rictor could also impact the cardiomyocyte electrophysiology and induced cardiomyocyte arrhythmia by affecting the expressions and distributions of cell-cell junction proteins (Cx43, N-cadherin, Desmoplakin). Overall, these results suggest that Rictor/mTORC2 might play an important role in the differentiation of cardiomyocytes. The protein would become a potential new target for regulating the cardiomyocyte differentiation. It might also provide a useful reference for the potential application of the induced pluripotent stem cells.
Acknowledgements
This work was supported by grants from the National Natural Sciences Foundation of China (No 81573426, No 30873068); Public Welfare Project of Zhejiang Provincial Science and Technology Department (No 2016C33157), Zhejiang Provincial Natural Science Foundation of China (No Y13H310001), Key Creative Team of Zhejiang Province (No 2010R50047).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Oh WJ, Jacinto E. mTOR complex 2 signaling and functions. Cell Cycle. 2011;10:2305-2316
2. Sarbassov DD, Ali SM, Kim DH. et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2014;14:1296-1302
3. Valli A, Rosner M, Fuchs C. et al. Embryoid body formation of human amniotic fluid stem cells depends on mTOR. Oncogene. 2010;29:966-977
4. Tandon M, Chen Z, Pratap J. Runx2 activates PI3K/Akt signaling via mTORC2 regulation in invasive breast cancer cells. Breast Cancer Res. 2014;16:16-28
5. Volkers M, Konstandin MH, Doroudgar S. et al. Mechanistic target of rapamycin complex 2 protects the heart from ischemic damage. Circulation. 2013;128:2132-2144
6. Gurusamy N, Lekli, Mukherjee S. et al. Cardioprotection by resveratrol: a novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc Res. 2010;86:103-112
7. Shu L, Houghton PJ. The mTORC2 complex regulates terminal differentiation of C2C12 myoblasts. Mol Cell Biol. 2009;29:4691-4700
8. Shiota C, Woo JT, Lindner J. et al. Multi-allelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell. 2006;11:538-589
9. C Wiese, T Nikolova, I Zahanich. et al. Differentiation induction of mouse embryonic stem cells into sinus node-like cells by suramin. Int J Cardiol. 2011;147:95-111
10. Karakikes GD Senyei, J Hansen et al. Small molecule-mediated directed differentiation of human embryonic stem cells toward ventricular cardiomyocytes. Stem Cells Transl Med. 2014;3:18-31
11. Kostin S, Rieger M, Dammer S. et al. Gap junction remodeling and altered connexin43 expression in the failing human heart. Mol Cell Biochem. 2003;242:135-144
12. Lampe PD, TenBroek EM, Burt JM. et al. Phosphorylation of connexin43 on serine368 by protein kinase C regulates gap junctional communication. J Cell Biol. 2000;149:1503-1512
13. Solan JL, Lampe PD. Connexin43 phosphorylation: structural changes and biological effects. Biochem J. 2009;419:261-272
14. Wei CJ, Francis R, Xu X. et al. Connexin43 associated with an N-cadherin-containing multiprotein complex is required for gap junction formation in NIH3T3 cells. J Biol Chem. 2005;280:19925-19936
15. Kostetskii I, Li J, Xiong Y. et al. Induced deletion of the N-cadherin gene in the heart leads to dissolution of the intercalated disc structure. Circ Res. 2005;96:346-354
16. Uzumcu A, Norgett EE, Dindar A. et al. Loss of desmoplakin isoform I causes early onset cardiomyopathy and heart failure in a Naxos-like syndrome. J Med Genet. 2006;43:1-8
17. Rhett JM, Gourdie RG. The perinexus: a new feature of Cx43 gap junction organization. Heart Rhythm. 2012;9:619-623
18. Reaume AG, Sousa PA, Kulkarni S. et al. Cardial malformation on neonatal mice lacking connexin 43. Science. 1995;267:1831-1834
19. Zemljic-Harpf AE, Godoy JC, Platoshyn O. et al. Vinculin directly binds zonula occludens-1 and is essential for stabilizing connexin-43-containing gap junctions in cardiac myocytes. J Cell Sci. 2014;127:1104-1116
20. Mok KW, Mruk DD, Lee W. et al. Rictor/mTORC2 regulates blood-testis barrier dynamics via its effects on gap junction communications and actin filament network. FASEB J. 2013;27:1137-1152
21. Wo YB, Zhu DY, Hu Y. et al. Reactive oxygen species involved in prenylflavonoids, icariin and icaritin, initiating cardiac differentiation of mouse embryonic stem cells. J Cell Biochem. 2008;103:1536-1550
22. Shinozawa T, Furukawa H, Sato E. et al. A novel purification method of murine embryonic stem cell- and human-induced pluripotent stem cell-derived cardiomyocytes by simple manual dissociation. J Biomol Screen. 2012;17:683-691
23. Shen G, Hu Y, Wu J. et al. A 2,6-disubstituted 4-anilinoquinazoline derivative facilitates cardiomyogenesis of embryonic stem cells. ChemMedChem. 2012;7:733-740
24. Zhu DY, Zhou LM, Zhang YY. et al. Involvement of metabotropic glutamate receptor 5 in cardiomyocyte differentiation from mouse embryonic stem cells. Stem Cells Dev. 2012;21:2130-2141
25. Zhang X, Wang X, Xu T. et al. Targeting of mTORC2 may have advantages over selective targeting of mTORC1 in the treatment of malignant pheochromocytoma. Tumour Biol. 2015;36:5273-5281
26. Zhang Q, Jiang J, Han P. et al. Direct differentiation of atrial and ventricular myocytes from human embryonic stem cells by alternating retinoid signals. Cell Res. 2011;21:579-587
27. Takahashi T, Lord B, Schulze PC. et al. Ascorbic Acid Enhances Differentiation of Embryonic Stem Cells Into Cardiac Myocytes. Circulation. 2003;107:1912-1916
28. Pal R. Embryonic stem (ES) cell-derived cardiomyocytes: A good candidate for cell therapy applications. Cell Biol Int. 2009;33:325-336
29. Zhou L, Huang Y, Zhang Y. et al. mGluR5 stimulating Homer-PIKE formation initiates icariin induced cardiomyogenesis of mouse embryonic stem cells by activating reactive oxygen species. Exp Cell Res. 2013;319:1505-1514
30. Ding L, Liang X, Zhu D. et al. Peroxisome proliferator-activated receptor alpha is involved in cardiomyocyte differentiation of murine embryonic stem cells in vitro. Cell Biol Int. 2007;31:1002-1009
31. Jacinto E, Loewith R, Schmidt A. et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122-1128
32. Oh WJ, Wu CC, Kim SJ. et al. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 2010;29:3939-3951
33. Schieke SM, Phillips D, McCoy JP Jr. et al. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem. 2006;281:27643-27652
34. Sciarretta S, Zhai P, Maejima Y. et al. mTORC2 regulates cardiac response to stress by inhibiting MST1. Cell Rep. 2015;11:125-136
35. Zhao X, Lu S, Nie J. et al. PDK1 and mTORC2 synergistically maintain post-natal heart growth and heart function in mice. Mol Cell Biol. 2014;34:1966-1975
36. Liang J, Wang YJ, Tang Y. et al. Type 3 inositol 1,4,5-trisphosphate receptor negatively regulates apoptosis during mouse embryonic stem cell differentiation. Cell Death Differ. 2010;17:1141-1154
37. Wang X, Lai P, Zhang Z. et al. Targeted inhibition of mTORC2 prevents osteosarcoma cell migration and promotes apoptosis. Oncol Rep. 2014;32:382-388
38. Li H, Lin J, Wang X. et al. Targeting of mTORC2 prevents cell migration and promotes apoptosis in breast cancer. Breast Cancer Res Treat. 2012;134:1057-1066
39. Boheler KR, Czyz J, Tweedie D. et al. Differentiation of pluripotent embryonic stem cells into cardiomyocytes. Circ Res. 2002;91:189-201
40. Xu B, Ji X, Chen X. et al. Effect of perfluorooctane sulfonate on pluripotency and differentiation factors in mouse embryoid bodies. Toxicology. 2015;328:160-167
41. Singla DK, Sobel BE. Enhancement by growth factors of cardiac myocyte differentiation from embryonic stem cells: A promising foundation for cardiac regeneration. Biochem Bioph Res Co. 2005;335:634-642
42. Ding L, Liang XG, Hu Y. et al. Involvement of p38MAPK and reactive oxygen species in icariin-induced cardiomyocyte differentiation of murine embryonic stem cells in vitro. Stem Cells Dev. 2008;17:751-760
43. Oka T, Komuro I. Differentiation of cardiomyocyte. Nihon Yakurigaku Zasshi. 2000;115:337-344
44. Miquerol L, Dupays L, Theveniau-Ruissy M. et al. Gap junctional connexins in the developing mouse cardiac conduction system. Novartis Found Symp. 2003;250:80-98
45. Vite A, Radice GL. N-cadherin/catenin complex as a master regulator of intercalated disc function. Cell Commun Adhes. 2014;21:169-179
46. Jansen JA, Noorman M, Musa H. et al. Reduced heterogeneous expression of Cx43 results in decreased Nav1.5 expression and reduced sodium current that accounts for arrhythmia vulnerability in conditional Cx43 knockout mice. Heart Rhythm. 2012;9:600-607
47. Ai X, Zhao W, Pogwizd SM. Connexin43 knockdown or overexpression modulates cell coupling in control and failing rabbit left ventricular myocytes. Cardiovasc Res. 2010;85:751-762
48. Li J, Patel VV, Kostetskii I. et al. Cardiac-specific loss of N-cadherin leads to alteration in connexins with conduction slowing and arrhythmogenesis. Circ Res. 2005;97:474-481
49. Zhu H, Wang H, Zhang X. et al. Inhibiting N-cadherin-mediated adhesion affects gap junction communication in isolated rat hearts. Mol Cells. 2010;30:193-200
50. Norman M, Simpson M, Mogensen J. et al. Novel Mutation in Desmoplakin Causes Arrhythmogenic Left Ventricular Cardiomyopathy. Circulation. 2005;112:636-642
Author contact
![]() Corresponding author: Dr. Danyan Zhu, Ph.D. Institute of Pharmacology and Toxicology, Zhejiang University, 866 Yuhangtang Road, Hangzhou 310058, China. Tel: +86 571 8820 8402, Fax: +86 571 8820 8402, E-mail address: zdyzxbedu.cn.
Corresponding author: Dr. Danyan Zhu, Ph.D. Institute of Pharmacology and Toxicology, Zhejiang University, 866 Yuhangtang Road, Hangzhou 310058, China. Tel: +86 571 8820 8402, Fax: +86 571 8820 8402, E-mail address: zdyzxbedu.cn.