ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2017; 13(2):135-144. doi:10.7150/ijbs.17758 This issue Cite
Research Paper
Lipocalin-2 Promotes Endoplasmic Reticulum Stress and Proliferation by Augmenting Intracellular Iron in Human Pulmonary Arterial Smooth Muscle Cells
Guoliang Wang1,2, Shenghua Liu1, Li Wang1, Liukun Meng1, Chuanjue Cui1, Hao Zhang1, Shengshou Hu1, Ning Ma2 ![]() , Yingjie Wei1
, Yingjie Wei1 ![]()
1. State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China.
2. Beijing Children's Hospital, Capital Medical University, Beijing, China.
Abstract

Endoplasmic reticulum (ER) stress, a feature of many conditions associated with pulmonary hypertension (PH), is increasingly recognized as a common response to promote proliferation in the walls of pulmonary arteries. Increased expression of Lipocalin-2 in PH led us to test the hypothesis that Lipocalin-2, a protein known to sequester iron and regulate it intracellularly, might facilitate the ER stress and proliferation in pulmonary arterial smooth muscle cells (PASMCs). In this study, we observed greatly increased Lcn2 expression accompanied with increased ATF6 cleavage in a standard rat model of pulmonary hypertension induced by monocrotaline. In cultured human PASMCs, Lcn2 significantly promoted ER stress (determined by augmented cleavage and nuclear localization of ATF6, up-regulated transcription of GRP78 and NOGO, increased expression of SOD2, and mild augmented mitochondrial membrane potential) and proliferation (assessed by Ki67 staining and BrdU incorporation). Lcn2 promoted ER stress accompanied with augmented intracellular iron levels in human PASMCs. Treatment human PASMCs with FeSO4 induced the similar ER stress and proliferation response and iron chelator (deferoxamine) abrogated the ER stress and proliferation induced by Lcn2 in cultured human PASMCs. In conclusion, Lcn2 significantly promoted human PASMC ER stress and proliferation by augmenting intracellular iron. The up-regulation of Lcn2 probably involved in the pathogenesis and progression of PH.
Keywords: Lipocalin-2 (Lcn2), proliferation, endoplasmic reticulum stress, Iron, pulmonary hypertension.
Introduction
Pulmonary hypertension (PH) is a severe disease of the pulmonary circulation. A key feature of vascular pathology in PH is abnormally reconstructed in small pulmonary arteries, which results in a persistent elevation of pulmonary arterial pressure (PAP) and eventually right ventricular failure [1]. Proliferation and anti-apoptosis of pulmonary arterial smooth muscle cells (PASMCs) are believed to be the essence of vascular remodeling in PH [2,3]. In animal experiments, the inhibition of proliferation of proliferated PASMCs in pulmonary vessels prevents the progression of medial hypertrophy [4-6].
Endoplasmic reticulum (ER) stress, which is caused by accumulation of unfolded proteins in ER lumen [7], has been reported recently being a common cellular response in many known PH-triggering processes, including hypoxia, viral infections, BMPRII mutation, inflammation, and Notch induction [8-13]. The activated ER stress sensor activating transcription factor 6 (ATF6) in the small pulmonary arteries in PH leading to the up-regulation of GRP78 (an important ER chaperone) and neurite outgrowth inhibitor (NOGO), and the later triggers a switch of cellular energy metabolism to a glycolytic phenotype that promotes proliferation and suppresses apoptosis [14,15]. Attenuation of ER stress prevents the PH progression by suppressing proliferation and inducing apoptosis in PASMCs in vitro and in vivo [6], suggesting that ER stress may be a novel therapeutic target in PH.
It was reported that iron could modulate ER stress-associated pathways [16-19], and its chelators have been shown to inhibit the development of pulmonary vascular remodeling induced by hypoxia [20]. Lipocalin-2 (Lcn2), a 25-KDa secretary glycoprotein, was shown initially to combat bacterial infection through impeding bacterial iron sequestration [21]. In a recent study, Lcn2 was found to mediate the proliferation and growth of thyroid carcinoma cell line by its ability to bind and transport iron inside the cells [22]. In our previous study [23], we observed that the expression levels of Lcn2 were significantly elevated in a rat PH model induced with monocrotaline and in patients with congenital heart disease-associated PH (CHD-PH), suggest that Lcn2 probably involves in the pathological process of PH. We also reported that Lcn2 could promote human PASMC proliferation by activating phosphatidylinositol-3-kinase (PI3K)/Akt pathway [24]. While inhibition of Akt phosphorylation only partly abrogated the Lcn2-promoted proliferation [24], indicating that there may be other mechanisms in Lcn2 promoted HPASMC proliferation. Therefore, the ability of Lcn2 controlling iron uptake attracts our attention to study the involvement of iron homeostasis in Lcn2-induced ER stress and proliferation in PH. The aims of this study are to investigate whether Lcn2 causes ER stress and proliferation, whether Lcn2 controls iron uptake, and whether iron involves in the molecular mechanism of ER stress and proliferation induced by Lcn2 in human PASMCs.
Methods
Animal model of pulmonary hypertension
The local animal care committee approved all protocols and surgical procedures. The animal model of pulmonary hypertension were procedure as previously described [23,25]. Briefly, Adult male Sprague-Dawley rats (6 to 7 weeks of age; weight 250 to 300 g; n=16) were subcutaneously injected with monocrotaline (MCT, 60 mg/kg) or saline. 21 days after the subcutaneous injection, the body weight was recorded, and anaesthesia was induced using 3% isofluorane in 3 L/min of oxygen and maintained using 2.5% isofluorane per liter O2. The pulmonary arterial pressure was measured using right-heart catheterization. The rats were exsanguinated immediately after hemodynamic assessment, and the lungs and hearts were removed for further analysis. The RV-free wall was dissected from the left ventricle plus septum (LV+S) and weighed separately. The degree of right ventricular hypertrophy (RVHI) was determined from the ratio RV/(LV+S).
Cell culture
Human pulmonary arterial SMCs were purchased from ScienCell (USA) and used according to the manufacturer's instructions or maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 1% penicillin / streptomycin at 5% CO2 and 37oC as previously described [23,24]. The cells were starved for 24 hours in 0.4% FBS-containing medium prior to treatment with 10 ng/ml recombinant human Lcn2 [23,24] (RayBiotech, USA), 10 μmol/L deferoxamine (Sigma), or 10 μmol/L FeSO4 (Sigma).
Immunocytochemistry
Human PASMCs were fixed in 4% paraformaldehyde for 15 min and permeabilized with 0.1% Triton X-100 in PBS for 10 min at room temperature. The cells were blocked with 5% BSA (Sigma) for 1 h at room temperature, washed with PBS, labeled with anti-Ki67 antibody (Sigma) or anti-ATF6 antibody (Santa Cruz Biotechnology) for 2 h at room temperature, rinsed in PBS, and incubated with the AlexaFluor-488 antibody (ZSGB-BIO) for 60 min at room temperature. The nuclei were stained with DAPI and fluorescent staining was visualized using a BX-61 microscope (Olympus) or confocal microscope (Leica TCS SP8), as previously described [23].
Cell proliferation assay
To determine cell growth, the immunofluorescence staining of Ki67 was performed as described above. A colorimetric immunoassay (CA 11647229001, Roche) for the quantification of the cell proliferation was also performed based on the measurement of BrdU incorporation during DNA synthesis according to the manufacturer's instructions. The cells cultured in 96-well plates were grown to 50% confluence and then growth-arrested prior to indicated treatment. The BrdU labeling reagent was added into the medium for 2 h incubation at 37oC. Absorbance values were measured at 450 nm (reference 690 nm) using an Infinite M200pro microplate reader (TECAN, Switzerland).
Quantitative real-time RT-PCR
Cells grown in 25-cm2 flasks or 6-well plates were washed with PBS and lysed directly in the culture dish with the Trizol reagent (Invitrogen). The total RNA was extracted and reverse-transcribed using Oligo (dT) and Transcriptor Reverse Transcriptase (Roche, GA) according to the manufacturer's instructions. The gene transcripts were quantified by real-time quantitative PCR (qPCR) using an Applied Biosystems 7300 Fast Real-Time PCR System (ABI, USA) with SYBR Green PCR Master Mix, as previously described [23]. The expression of gene transcripts in the test samples was normalized against the internal standard β-actin. The primers used were as follows: human GRP78, 5'- GAA CGT CTG ATT GGC GAT GC -3' and 5'- ACC ACC TTG AAC GGC AAG AA -3'; human NOGO, 5'- GAG GGT GAG TCA CGC CAA A -3' and 5'- TCC CGG AAC AAT GAG ACT GC -3'; human β-actin, 5'- TGT TTG AGA CCT TCA ACA CC -3' and 5'- TGT TTG AGA CCT TCA ACA CC -3'.
Western Blotting analyses
The western blotting analyses of lung tissue cells were performed according to our previous reports [23,24]. The proteins were detected using the following antibodies: rabbit anti-Lcn2, rabbit anti-ATF6, rabbit anti-CHOP, rabbit anti-SOD2 (from Santa Cruz Biotechnology, USA), mouse anti-β-actin, goat anti-rabbit IgG, and goat anti-mouse IgG (from ZSGB-BIO, CHN).
Intracellular Iron measurement
The intracellular iron levels were measured using the fluorescent probe Phen Green SK (PG-SK; Invitrogen) [26]. For saturating the intracellular iron pool as a positive control, the cells were treated with 100 μM FeSO4 (Sigma) for 10 min. As a negative control, cells were incubated with a 5mM well characterized membrane-permeable iron chelator 2,2'-dipyridyl (2'-DPD, Sigma) for 20 min. After washing with PBS, the cells were incubated with 10 μM PG-SK in PBS at 37 oC for 10 min. After quickly washing with PBS, the cells were fixed in 4% paraformaldehyde for 15-30min. The nuclei were stained with DAPI and fluorescent staining was visualized using a BX-61 microscope (Olympus).
A cell-permeable calcein-AM green acetoxymethyl ester was also used for the quantification of the intracellular free iron pool [27]. After the indicated treatment, the cells were washed and loaded with 1 mM calcein-AM green acetoxymethyl ester for 30 min at 37 oC. The cellular esterases cleave the acetoxymethyl ester group of calcein-AM to form the cell-impermeable compound calcein-AM green, whose fluorescence is stoichiometrically quenched upon binding to cellular labile iron pools. The decrease in fluorescence was used to calculate the increase in free iron pool. The cell monolayers were trypsinized and resuspended in Ca2+/Mg2+ PBS (0.5 mg/ml each of CaCl2 and MgCl2) at a density of 0.5×106 cells/ml. The fluorescence of the cells was monitored with a flow cytometry (ACCURY C6, BD, USA) using standard set of filters for green (FL1) fluorescence. A total of 20,000 events were collected and analyzed. Fluorescence intensity was showed as fold of control.
Determination of mitochondrial membrane potential (ΔΨm) [27]
The JC-1 staining assay was used to examine the change in ΔΨm. The cells were washed and loaded with 5 μg/ml JC-1 at 37oC for 10 min prior to treatment. After being washed twice with prewarmed PBS, the cells were trypsinized and resuspended at ~3×105 cells/ml in JC-1 staining buffer at 25oC. Measurements were immediately made using a flow cytometry (ACCURY C6, BD, USA). JC-1 dye changes its fluorescence emission from red to green when a loss of ΔΨm occurs. The green and red fluorescence of JC-1 was detected through the FL-1 and FL-2 channels, respectively. Proper compensation was established to correct the fluorescence spillover. The cell population under study was previously selected by electronic gating, measuring forward versus side light scatter. A total of 20,000 events were collected for the analysis of each sample. Cells with high and low ΔΨm are in regions R1 and R2, respectively. Data are depicted as the percentage of cells in the R1 region compared with the total selected cells. Mitochondrial depolarization (positive control) was achieved by treating cells with 5 mM CCCP for 30 min at 37oC and was indicated by a decrease in the red/green fluorescence intensity ratio.
Statistical analyses
Each independent experiment was repeated at least 3 times, and at least 4 independent experiments were performed. The data are shown as the means ± SE (standard error). Data from PH rats and controls were tested by using the unpaired student's t-test or one sample t-test. Data from BrdU incorporation assays were tested by using the one-way analysis of variance (ANOVA). The other data were tested by using the Mann-Whitney U test. A value of p<0.05 was considered significant.
Results
Up-regulated Lcn2 expression and increased ATF6 cleavage in a rat PH model
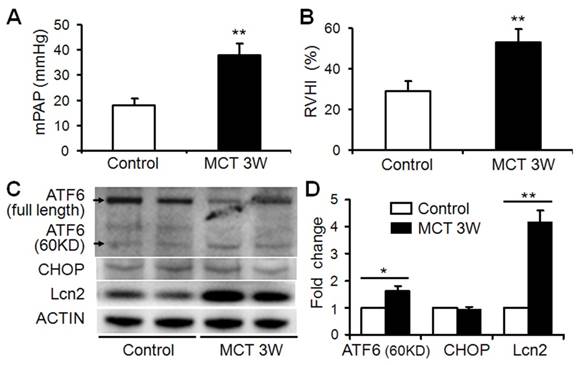
The expression level of Lcn2 is significantly elevated in a rat PH model induced with monocrotaline compared with control [23]. Because Lcn2 was reported could bind and transport iron inside the cells [21,22] and iron could modulate ER stress-associated pathways [16-19], we examined the expression levels of ER stress markers as well as Lcn2 in rat PH model. Three weeks after the MCT injection, the hemodynamic assessment showed that MCT significantly enhanced the pulmonary arterial pressure (Fig. 1A) and resulted in significant right ventricular hypertrophy (Fig. 1B). Western blotting analyses (Fig. 1C, D) revealed that the levels of Lcn2 and cleaved ATF6 were significantly increased after MCT injection. The expression levels of full length ATF6 and C/EBP homologous protein (CHOP) had no significant changes after MCT injection.
The level of Lcn2 and ER stress markers in rat PH model induced with MCT. Adult male Sprague-Dawley rats (6 to 7 weeks; 250 to 300 g) were administered a single subcutaneous injection of 60 mg/kg monocrotaline (MCT; n=8) or saline (n=8). 3 weeks after subcutaneous injection, right-heart catheterization was performed to measure the pulmonary arterial pressure, and mean pulmonary arterial pressure (mPAP) was calculated (A). The rats were exsanguinated and the hearts and lungs were removed. The RV-free wall was dissected from the left ventricle plus septum (LV+S) and weighed separately; the degree of right ventricular hypertrophy (RVHI) was determined from the ratio RV/(LV+S) (B). Total protein of lung samples was extracted and subjected to Western blotting analyses. Representative western blots (C) and quantitative analysis (D) are shown. (*, p < 0.05; **, p < 0.01).
Lcn2 promotes human PASMC proliferation
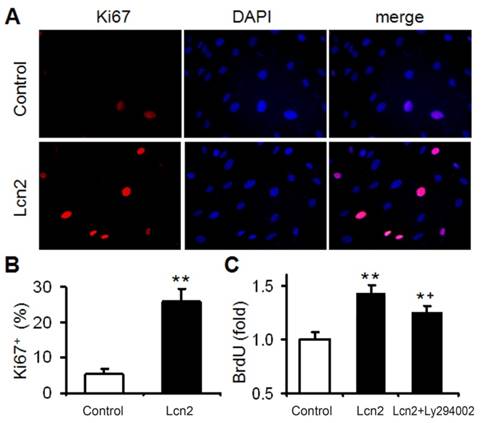
We had demonstrated that Lcn2 could promote human PASMC proliferation by activating PI3K/Akt pathway [24]. In this study, Lcn2 promoted human PASMC proliferation was also demonstrated using Ki67 immunofluorescence staining (Fig. 2A and B). We next detected human PASMC proliferation quantitatively using an ELISA Kit based on BrdU incorporation during DNA synthesis. A significant increase in human PASMC proliferation was also observed in Lcn2-treated groups compared with control (Fig. 2C). And interestingly, inhibition of Lcn2 promoted Akt phosphorylation by Ly294002 only partly abrogated the Lcn2-promoted proliferation. A significant increase in human PASMC proliferation was still observed in Lcn2+Ly294002-treated groups compared with control (Fig. 2C), indicating that there being other mechanisms in Lcn2 promoted HPASMC proliferation.
Lcn2 promotes endoplasmic reticulum stress
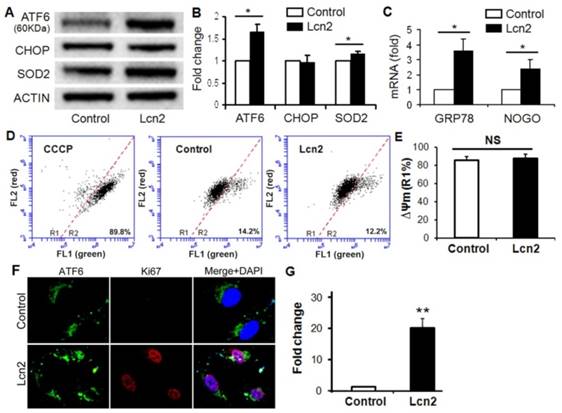
ER stress in the pulmonary circulation results in ATF6 activation and up-regulation of GRP78 and Nogo [14]. The significantly increased ATF6 cleavage (Fig. 3, A and B) and nuclear localization (Fig. 3, F and G) were observed in human PASMCs exposed to Lcn2. The mRNA levels of GRP78 and Nogo were also significantly up-regulated in the Lcn2-treated groups compared with respective control groups (Fig. 3 C). The expression level of CHOP, an indicator of proapoptotic ER stress, had no significant changes (Fig. 3, A and B). The mitochondrial membrane potential (ΔΨm), which can be influenced by ER stress [6], is an indicator of mitochondrial function. We examined the ΔΨm using JC-1 staining. When a loss of ΔΨm occurs, JC-1 changes its fluorescence emission from red to green and more cells locates in the R2 region in the images of flow cytometry. We observed a mildly increased ΔΨm in Lcn2-treated group, which suggests that the mitochondrial function was slightly inhibited, although the change was not significant (Fig. 3, D and E). The level of SOD2, another mitochondrial function indicator and important mitochondrial ROS eliminator [16], was significantly increased in Lcn2-treated group (Fig. 3, A and B), which indicates that the mitochondria-associated apoptosis was inhibited.
Lcn2 promotes human PASMC proliferation. (A) Growth-arrested human PASMCs were treated with 10 ng/ml Lcn2 or control medium for 24 h, cell growth was monitored by Ki67 immunofluorescent staining (red), and the nuclei were stained with DAPI in blue. (B) Quantitative analysis of Ki67+ cells (% of Ki67+ cells versus total, at least 50 cells per group per experiment) from five randomly selected fields in each individual experiment (n=4; *, p < 0.05). (C) Growth-arrested human PASMCs in 96-well plates were treated with 10 ng/ml Lcn2, 10 ng/ml Lcn2 + 20uM LY294002 or control medium for 48 h, cell growth was monitored using BrdU incorporation assay, data are normalized to the control group (at least 6 wells per group per experiment, n=4 experiments; *, p < 0.05 compared with control; +, p < 0.05 compared with Lcn2 group).
Lcn2 promotes ER stress in human PASMCs. Growth-arrested human PASMCs were treated with 10 ng/ml Lcn2 or control medium for 24 h. Total protein was extracted and subjected to Western blotting analyses, a representative blot (A) and quantitative analysis (B) are shown (n=4; *, p < 0.05). (C) Total RNA was extracted and reverse transcribed to cDNA, quantitative determination of GRP78 and NOGO expression using real-time PCR, corrected for the internal standard β-actin, and normalized to respective controls (n=4; *, p < 0.05). Mitochondrial membrane potential (ΔΨm) was monitored with JC-1 staining and flow cytometry, the green and red fluorescence of JC-1 was detected through the FL-1 and FL-2 channels, respectively. After appropriate electronic gating (at least 10000 gated events), cells with high and low ΔΨm are in region R1 and R2, respectively. The CCCP (5 μM, 30 min) is used as a loss of ΔΨm control. Representative dot plots (D) and quantitative analysis of ΔΨm (E) are shown (n=4; NS, not significant). (F) Immunofluorescent staining and microscope for ATF6 (green) and Ki67 (red), and the nuclei were stained with DAPI in blue. (G) Quantitative analysis of nuclear ATF6 localization (green colocalizing with blue nuclei) with Image-Pro Plus 5.0 soft ware. At least 50 cells per group per experiment from five randomly selected fields in each individual experiment are analyzed (n=4; **, p < 0.01).
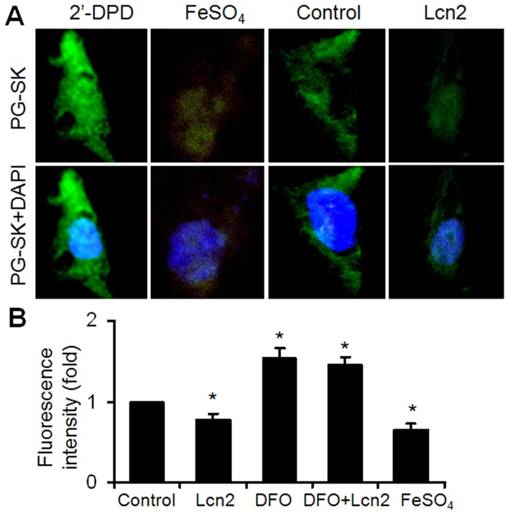
Lcn2 augments intracellular iron. (A) Growth-arrested human PASMCs were treated with 10 ng/ml Lcn2 or control medium for 24 h. Intracellular iron levels were measured using the fluorescent probe PG-SK (10 μM, 10 min). For saturating the intracellular iron pool as a positive control, the cells were treated with 100 μM FeSO4 for 10 min. The cells incubated with 2'-DPD (5 mM, 20 min) as a negative control. Representative images are shown. (B) Growth-arrested human PASMCs were treated with 10 ng/ml Lcn2, 10 μM DFO, 10 μM FeSO4, or control medium for 24 h. Quantification of intracellular free iron pool were monitored using a cell-permeable calcein-AM green acetoxymethyl ester (1 mM, 30 min), whose fluorescence is stoichiometrically quenched upon binding to cellular labile iron pools. The decrease in fluorescence was used to calculate the increase in free iron pool. The fluorescence of the cells was monitored with a flow cytometry using standard set of filters for green (FL1) fluorescence. A total of 20,000 events were collected and analyzed. (n=4; *, p < 0.05).
Lcn2 augments intracellular iron levels, leading to ER stress
It has been shown that iron can mediate ER stress in a variety of cell types and Lcn2 has the capability of binding to iron. We next examined the intracellular iron levels in human PASMCs using the cell-permeable fluorescent probe PG-SK, which can be quenched by binding intracellular free iron. The cells treated with FeSO4 and 2'-DPD were used as positive and negative controls and clearly showed decreased and increased levels of fluorescence, respectively (Fig. 4A). The PG-SK fluorescence was attenuated in human PASMCs exposed to 10ng/ml Lcn2 for 24 h compared with control (Fig. 4A), which indicates that the intracellular iron levels were increased in Lcn2-treated group. We detected the fluorescence of the cells by flow cytometry using another iron sensitive probe, calcein-AM, to quantify the intracellular free iron pool. With the same results as PG-SK, Lcn2 significantly attenuated the calcein-AM fluorescence in human PASMCs compared with control (Fig. 4B). Iron chelation (10 μM deferoxamine, DFO) significantly increased the calcein-AM fluorescence in human PASMCs. DFO + Lcn2 has the same effect as DFO alone and abrogated the effect of Lcn2 on the fluorescence of calcein-AM. 10 μM FeSO4, however, more significantly decreased the calcein-AM fluorescence in human PASMCs than Lcn2 did (Fig. 4B).
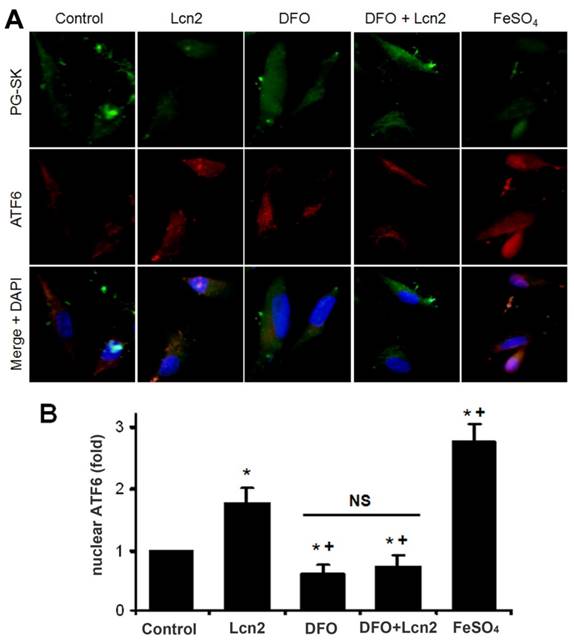
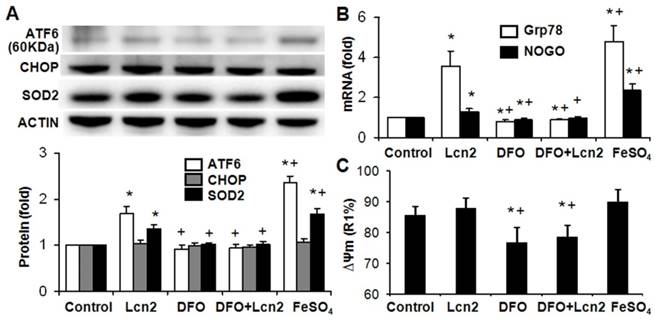
The data above indicate that Lcn2 could augment the levels of iron in human PASMCs, which motivated us to determine whether there is a direct functional significance of this change. We analyzed the effect of intracellular iron levels on the amount of nuclear ATF6 by co-staining with anti-ATF6 antibody, PG-SK and DAPI. As is shown in Fig. 5, we confirmed that individual cells treated with Lcn2 exhibiting decreased PG-SK fluorescence also showed increased nuclear ATF6 fluorescence. 10 μM DFO increased the PG-SK fluorescence and decreased the nuclear ATF6 fluorescence. DFO + Lcn2 has the same effects as DFO alone and abrogated the effects of Lcn2 on the fluorescence of PG-SK and nuclear ATF6. 10 μM FeSO4 decreased the PG-SK fluorescence and increased the nuclear ATF6 fluorescence more significantly than Lcn2 did. These data suggest that the augmented intracellular iron may promote the ATF6 nuclear localization. To confirm the mechanistic role of altered intracellular iron levels on ER stress, we next examined the other ER stress markers as well as cleaved ATF6. As is shown in Fig. 6, Lcn2 induced the significantly increased ATF6 cleavage and SOD2 expression (Fig. 6A), the significantly increased transcription of GRP78 and NOGO (Fig. 6B), and no significant changes in the CHOP expression (Fig. 6A) and the ΔΨm (Fig. 6C). 10 μM DFO has no significant effects on the ATF6 cleavage (Fig. 6A), but significantly decreased the ΔΨm (Fig. 6C) and the transcription of GRP78 and NOGO (Fig. 6B). DFO + Lcn2 has the same effects as DFO alone and abrogated the effects of Lcn2 on the aforementioned ER stress markers (Fig. 6, A, B and C). 10 μM FeSO4 produced similar but more significant changes than Lcn2 did (Fig. 6, A, B and C). There were no significant changes in the CHOP expression among these groups. These data suggest that the augmented intracellular iron is involved in the ER stress promoted by Lcn2 in human PASMCs.
Lcn2 promotes human PASMC proliferation by augmenting intracellular iron
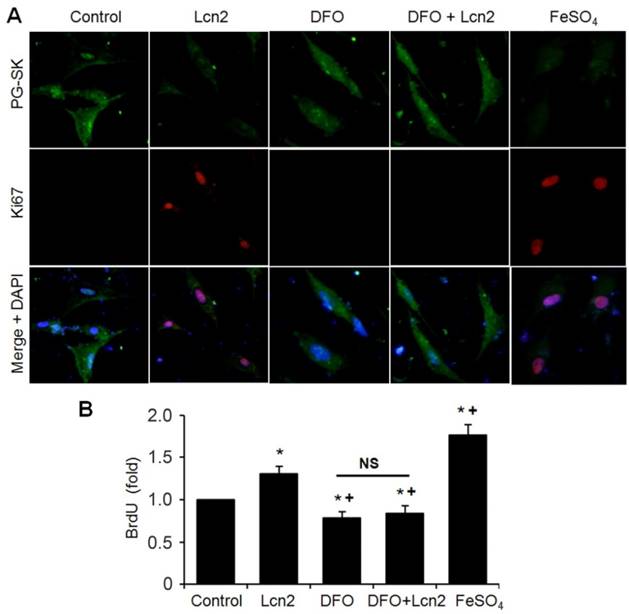
We next performed studies to examine whether Lcn2 promotes human PASMC proliferation by augmenting intracellular iron. We analyzed the relationship between the levels of intracellular iron and the human PASMC proliferation by co-staining with Ki67 and PG-SK. As is shown in Fig. 7A, there was PG-SK fluorescence and very little Ki67 staining in the control group. The Lcn2-treated group exhibited decreased PG-SK fluorescence and increased Ki67 staining. The DFO-treated group exhibited increased PG-SK fluorescence and no visible Ki67 staining. DFO + Lcn2 has the same effects as DFO alone and abrogated the effects of Lcn2 on the fluorescence of PG-SK and Ki67. 10μM FeSO4 produced less PG-SK fluorescence and more Ki67 staining than Lcn2 did. These data suggest that the augmented intracellular iron may promote the expression of Ki67, a marker of cell proliferation. We then detected the human PASMC proliferation quantitatively using the BrdU incorporation analyses. As is shown in Fig. 7B, the Lcn2-treated group exhibited significantly increased human PASMC proliferation compared with control. The DFO-treated group exhibited significantly decreased proliferation compared with the control and DFO abrogated the proliferative effect of Lcn2 on human PASMCs. 10 μM FeSO4 more significantly increased the human PASMC proliferation than Lcn2 did. These data suggest that Lcn2 promotes human PASMC proliferation (at least partly) via augmentation of the intracellular iron.
Lcn2 promotes the cleavage and nuclear localization of ATF6 by augmenting intracellular iron. Growth-arrested human PASMCs were treated with 10 ng/ml Lcn2, 10 μM DFO, 10 μM FeSO4, or control medium for 24 h. (A) Immunofluorescent staining and microscope for PG-SK (green) and ATF6 (red) were monitored, and the nuclei were stained with DAPI in blue. (B) Quantitative analysis of nuclear ATF6 localization is shown. At least 50 cells per group per experiment from five randomly selected fields in each individual experiment are analyzed. (n=4; *, p < 0.05 compared with control; +, p < 0.05 compared with Lcn2 group; NS, not significant).
Lcn2 promotes ER stress by augmenting intracellular iron. Growth-arrested human PASMCs were treated with 10 ng/ml Lcn2, 10 μM DFO, 10 μM FeSO4, or control medium for 24 h. (A) Total protein was extracted and subjected to Western blotting analyses, a representative blot and quantitative analysis are shown. (B) Total RNA was extracted and reverse transcribed to cDNA, quantitative determination of GRP78 and NOGO expression using real-time PCR are shown. (C) Quantitative analysis of ΔΨm using JC-1 staining and flow cytometry are shown in the histogram. (n=4; *, p < 0.05 compared with control; +, p < 0.05 compared with Lcn2 group).
Lcn2 promotes human PASMC proliferation by augmenting intracellular iron. (A) Growth-arrested human PASMCs were treated with 10 ng/ml Lcn2, 10 μM DFO, 10 μM FeSO4, or control medium for 24h. Representative immunofluorescent staining and microscope for PG-SK (green) and Ki67 (red) were monitored, and the nuclei were stained with DAPI in blue. (B) Growth-arrested human PASMCs in 96-well plates were treated with 10 ng/ml Lcn2, 10 μM DFO, 10 μM FeSO4, or control medium for 48 h. Cell growth was monitored using BrdU incorporation assay, data are normalized to the control group (at least 6 wells per group per experiment, n =4 experiments; *, p < 0.05 compared with control; +, p < 0.05 compared with Lcn2 group; NS, not significant).
Discussion
In this study, we showed that there were increased ATF6 cleavage and greatly increased Lcn2 expression in a standard rodent PH model, MCT-PH in rats. We showed for the first time that Lcn2, a small secreted protein, promoted human PASMC ER stress and proliferation in vitro. The effects of Lcn2 promoted ER stress include augmented proteolytic cleavage and nuclear localization of ATF6, up-regulated transcription of GRP78 and NOGO, increased expression of SOD2, and mildly increased ΔΨm. The mechanism by which Lcn2 promoted ER stress is involved in augmented intracellular iron and iron chelator (DFO) abrogated the Lcn2-promoted ER stress and proliferation.
Recent studies have shown that Lcn2 is over-expressed in cancers of diverse histological origin and it facilitates tumorigenesis by promoting survival, growth, and metastasis [22,28-30], indicating the pivotal role of this molecule in several physiological or pathological conditions. Lannetti et al showed that the ability of Lcn2 to bind and transport iron inside the cells mediate the proliferation and growth of thyroid carcinoma cell line [22]. Up-regulated expression of Lcn2 has been shown in the lungs of rats treated with MCT and in CHD-PH patients compared with the CHD-nonPH subjects [23]. In this study, we examined augmented ATF6 cleavage coexists with the up-regulated Lcn2 in the lungs of rats treated with MCT (Fig. 1). These results indicated a probability that the ability of Lcn2 controlling iron uptake play a role in PH. Using Ki67 staining, we demonstrated that recombinant Lcn2 could promote human PASMC proliferation (Fig. 2, A and B), an important component of small pulmonary arterial remodeling leading to PH. This observation was confirmed using measurement of BrdU incorporation during DNA synthesis (Fig. 2C).
Although Lcn2 can promote cell proliferation, little is known regarding its mechanisms, particularly in human PASMCs. Recent studies have found that ER stress is a common cellular response in many known PH-triggering processes, including hypoxia, viral infections, BMPRII mutation, inflammation and Notch induction [8-13]. Excessive and prolonged ER stress activates transcription factors like CHOP, usually triggering cell apoptosis [31]. However, mild to moderate ER stress could help cells adapt to a changing environment and reestablish ER function. These adaptive mechanisms involve activation of ATF6, a protein typically associated with antiapoptotic and prosurvival signaling, which induce expression of genes that enhance the folding capacity of proteins in the ER and promote ER-associated protein degradation to remove misfiled proteins [7,32]. ER stress in the pulmonary circulation results in the activation of ATF6, causing up-regulation of GRP78 and Nogo [14]. NOGO induction in PH leads to mitochondrial inhibition and a switch of cellular energy metabolism to a glycolytic phenotype that promotes proliferation and suppresses apoptosis [14,15]. Increased ER stress has been reported in several rodent PH models [6,14,15]. Indeed, attenuating ER stress in animal experiments prevents the PH progression with suppressed proliferation and induced apoptosis in PASMCs [6]. In this study, we examined greatly increased Lcn2 expression accompanied with significantly increased ATF6 cleavage in lungs of rats treated with MCT compared with control (Fig. 1). These reports and our results suggest that Lcn2 may play a role in ER stress in PH. We then examined the effects of Lcn2 on ER stress in human PASMCs in vitro. Results showed that Lcn2 promoted the cleavage and nuclear localization of ATF6, up-regulated the transcription of GRP78 and NOGO, mildly inhibited the mitochondrial function, and had no significant effect on CHOP expression (Fig. 3). These results suggest that Lcn2 could promote a proliferative ER stress response in cultured human PASMCs.
Lcn2 was initially shown to combat bacterial infection via the impedance of bacterial iron sequestration [21]. Iron is an important cofactor in the progression of chronic diseases. Iron could modulate ER stress-associated pathways and ER stress could control iron metabolism [16-19]. It has been shown that Iron chelators can inhibit the development of pulmonary vascular remodeling induced by hypoxia [20]. Our results confirmed that Lcn2 augmented the intracellular iron in human PASMCs using fluorescent staining and flow cytometry analysis (Fig. 4). We also showed that increased levels of intracellular iron promoted ER stress and proliferation and interestingly, iron chelator abrogated the roles of Lcn2 on the ER stress and proliferation in human PASMCs (Fig. 5-7). These results suggest that Lcn2, via augmentation of intracellular iron, promoted the ER stress and proliferation in cultured human PASMCs.
Lcn2 is an acute phase protein and proinflammatory marker [33,34]. It is expressed at very low concentrations in healthy children and is markedly elevated in the case of epithelial damage [35,36]. Its small size and secreted nature motivated its investigation as a diagnostic and prognostic biomarker in numerous diseases [37]. In our previous study, we revealed significantly increased Lcn2 in the plasma of CHD-PH patients and in the lung of MCT-rats compared with respective controls [23]. In pediatric cardiac surgery, plasma Lcn2 has been reported as an early biomarker for diagnosis of acute kidney injury following cardio-pulmonary bypass [38]. El Karoui et al reported that up-regulated ATF4 expression could induce over expression of Lcn2 in proteinuria-induced kidney lesions [39]. There is no report about the origins of increased Lcn2 in PH till now. The inflammation and injury of vascular endothelium in PH probably play roles in the augmentation of Lcn2 expression. Further studies on the mechanisms of Lcn2 up-regulation will help to elucidate the pathophysiology of PH.
In summary, this study provides evidence that Lcn2 plays a role in human PASMC ER stress and proliferation via augmentation of intracellular iron. The definite role and mechanism of Lcn2 in PH deserves further study. It will help to explore novel therapeutic strategies based on attenuating PASMC proliferation in pulmonary hypertension.
Acknowledgements
This work was supported by National Natural Science Foundation of China (81470424).
Conflict of interest
None.
References
1. Runo JR, Loyd JE. Primary pulmonary hypertension. Lancet. 2003;361:1533-1544
2. Burg ED, Remillard CV, Yuan JX. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: Pharmacotherapeutic implications. Br J Pharmacol. 2008;153:S99-S111
3. Perros F, Montani D, Dorfmuller P, Durand-Gasselin I, Tcherakian C, Le Pavec J. et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:81-88
4. Rajagopalan N, Simon MA, Suffoletto MS, Shah H, Edelman K, Mathier MA. et al. Noninvasive estimation of pulmonary vascular resistance in pulmonary hypertension. Echocardiography. 2009;26:489-494
5. Abe K, Toba M, Alzoubi A, Ito M, Fagan KA, Cool CD. et al. Formation of plexiform lesions in experimental severe pulmonary arterial hypertension. Circulation. 2010;121:2747-2754
6. Peter Dromparis, Roxane Paulin, Trevor H Stenson, Alois Haromy, Gopinath Sutendra, Evangelos D Michelakis. Attenuating Endoplasmic Reticulum Stress as a Novel Therapeutic Strategy in Pulmonary Hypertension. Circulation. 2013;127:115-125
7. Chunyan Xu, Beatrice Bailly-Maitre, John C. Reed. Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Invest. 2005;115:2656-2664
8. Michelakis ED, Wilkins MR, Rabinovitch M. Emerging concepts and translational priorities in pulmonary arterial hypertension. Circulation. 2008;118:1486-1495
9. Voelkel NF, Cool CD, Flores S. From viral infection to pulmonary arterial hypertension: A role for viral proteins?. AIDS. 2008;22:S49-S53
10. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900-917
11. Sehgal PB, Mukhopadhyay S, Patel K, Xu F, Almodóvar S, Tuder RM, Flores SC. Golgi dysfunction is a common feature in idiopathic human pulmonary hypertension and vascular lesions in SHIV-nef-infected macaques. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009;297:L729-L737
12. Sobolewski A, Rudarakanchana N, Upton PD, Yang J, Crilley TK, Trembath RC, Morrell NW. Failure of bone morphogenetic protein receptor trafficking in pulmonary arterial hypertension: Potential for rescue. Hum. Mol. Genet. 2008;17:3180-3190
13. Takahashi K, Adachi K, Yoshizaki K, Kunimoto S, Kalaria RN, Watanabe A. Mutations in NOTCH3 cause the formation and retention of aggregates in the endoplasmic reticulum, leading to impaired cell proliferation. Hum. Mol. Genet. 2010;19:79-89
14. Sutendra G, Dromparis P, Wright P, Bonnet S, Haromy A, Hao Z. et al. The role of Nogo and the mitochondria-endoplasmic reticulum unit in pulmonary hypertension. Sci Transl Med. 2011;3:88ra55
15. Dromparis P, Sutendra G, Michelakis ED. The role of mitochondria in pulmonary vascular remodeling. J Mol Med (Berl). 2010;88:1003-1010
16. Tan T, Crawford D, Jaskowski L, Subramaniam V, Clouston A, Crane D. et al. Excess iron modulates endoplasmic reticulum stress-associated pathways in a mouse model of alcohol and high-fat diet-induced liver injury. Lab Invest. 2013;93:1295-1312
17. Rodvold J, Mahadevan N, Zanetti M. Lipocalin 2 in cancer: when good immunity goes bad. Cancer Lett. 2012;316:132-138
18. Lu JJ, Chen SM, Zhang XW, Ding J, Meng LH. The anti-cancer activity of dihydroartemisinin is associated with induction of iron-dependent endoplasmic reticulum stress in colorectal carcinoma HCT116 cells. Invest New Drugs. 2011;29:1276-1283
19. Vecchi C, Montosi G, Zhang K, Lamberti I, Duncan SA, Kaufman RJ, Pietrangelo A. ER stress controls iron metabolism through induction of hepcidin. Science. 2009;325:877-880
20. Wong CM, Preston IR, Hill NS, Suzuki YJ. Iron chelation inhibits the development of pulmonary vascular remodeling. Free Radic Biol Med. 2012;53:1738-1747
21. DH Goetz, MA Holmes, N Borregaard, ME Bluhm, KN Raymond, RK Strong. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell. 2002;10:1033-1043
22. Lannetti A, Pacifico F, Acquaviva R, Lavorgna A, Crescenzi E, Vascotto C. et al. The neutrophil gelatinase-associated lipocalin (NGAL), a NFkB-regulated gene, is a survival factor for thyroid neoplastic cells. Proc. Natl Acad. Sci. USA. 2008;105:14058-14063
23. Wang G, Liu X, Meng L, Liu S, Wang L, Li J. et al. Up-Regulated Lipocalin-2 in Pulmonary Hypertension Involving in Pulmonary Artery SMC Resistance to Apoptosis. Int J Biol Sci. 2014;10:798-806
24. Wang G, Ma N, Meng L, Wei Y, Gui J. Activation of the Phosphatidylinositol 3-Kinase/Akt Pathway is Involved in Lipocalin-2-promoted Human Pulmonary Artery Smooth Muscle Cell Proliferation. Mol Cell Biochem. 2015;410:207-213
25. Meng L, Liu X, Zheng Z, Li J, Meng J, Wei Y, Hu S. Original rat model of high kinetic unilateral pulmonary hypertension surgically induced by combined surgery. J Thorac Cardiovasc Surg. 2013;146:1220-1226
26. Xu G, Ahn J, Chang S, Eguchi M, Ogier A, Han S. et al. Lipocalin-2 induces cardiomyocyte apoptosis by increasing intracellular iron accumulation. J Biol Chem. 2012;287:4808-4817
27. Asensio-Lopez M, Sanchez-Mas J, Pascual-Figal D, Torrec C, Valdes M, Lax A. Ferritin heavy chain as main mediator of preventive effect of metformin against mitochondrial damage induced by doxorubicin in cardiomyocytes. Free Radic Biol Med. 2014;67:19-29
28. Hsin IL, Hsiao YC, Wu MF, Jan MS, Tang SC, Lin YW. et al. Lipocalin 2, a new GADD153 target gene, as an apoptosis inducer of endoplasmic reticulum stress in lung cancer cells. Toxicol Appl Pharmacol. 2012;263:330-337
29. J. Yang, D.R. Bielenberg, S.J. Rodig, R. Doiron, M.C. Clifton, A.L. Kung, et al. Lipocalin 2 promotes breast cancer progression. Proc. Natl Acad. Sci. USA. 2009;106:3913-3918
30. T. Berger, C.C. Cheung, A.J. Elia, T.W. Mak. Disruption of the Lcn2 gene in mice suppresses primary mammary tumor formation but does not decrease lung metastasis. Proc. Natl Acad. Sci. USA. 2010;107:2995-3000
31. Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880-885
32. Schewe DM, Aguirre-Ghiso JA. ATF6alpha-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc Natl Acad Sci USA. 2008;105:10519-10524
33. Nilsen-Hamilton M, Liu Q, Ryon J, Bendickson L, Lepont P, Chang Q. Tissue involution and the acute phase response. Ann NY Acad Sci. 2003;995:94-108
34. Wang Y, Lam KS, Kraegen EW, Sweeney G, Zhang J, Tso AW. et al. Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin Chem. 2007;53:34-41
35. Schmidt-Ott KM, Mori K, Kalandadze A, Li JY, Paragas N, Nicholas T. et al. Neutrophil gelatinase-associated lipocalin-mediated iron traffic in kidney epithelia. Curr Opin Nephrol Hypertens. 2006;15:442-449
36. Zap pitelli M, Washburn KK, Arikan AA, Loftis L, Ma Q, Devarajan P. et al. Urine neutrophil gelatinase-associated lipocalin is an early marker of acute kidney injury in critically ill children: a prospective cohort study. Crit Care. 2007;11:R84
37. Chakraborty S, Kaur S, Guha S, Batra SK. The multifaceted roles of neutrophil gelatinase associated lipocalin (NGAL) in inflammation and cancer. Biochim Biophys Acta. 2012;1826:129-169
38. Fadel FI, Abdel Rahman AM, Mohamed MF, Habib SA, Ibrahim MH, Sleem ZS. et al. Plasma neutrophil gelatinase-associated lipocalin as an early biomarker for prediction of acute kidney injury after cardio-pulmonary bypass in pediatric cardiac surgery. Arch Med Sci. 2012;8:250-255
39. El Karoui K, Viau A, Dellis O, Bagattin A, Nguyen C, Baron W. et al. Endoplasmic reticulum stress drives proteinuria-induced kidney lesions via Lipocalin 2. Nat Commun. 2016;7:10330
Author contact
![]() Corresponding authors: Yingjie Wei, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100037, People's Republic of China. Tel: 86-10-60866393. E-mail: weiyingjieorg. Ning Ma, Beijing Children's Hospital, Capital Medical University, Beijing, 100045, People's Republic of China. Tel: 86-10-59616736. E-mail: echo_mncom.
Corresponding authors: Yingjie Wei, State Key Laboratory of Cardiovascular Disease, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, 100037, People's Republic of China. Tel: 86-10-60866393. E-mail: weiyingjieorg. Ning Ma, Beijing Children's Hospital, Capital Medical University, Beijing, 100045, People's Republic of China. Tel: 86-10-59616736. E-mail: echo_mncom.
Received 2016-9-29
Accepted 2016-11-8
Published 2017-1-15