Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Molecular Mechanisms Underlying...
Strategies of Overcoming PARPi...
Conclusion and Perspectives
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(2):198-208. doi:10.7150/ijbs.17240 This issue Cite
Review
Reverse the Resistance to PARP Inhibitors
Yevgeniy Kim1*, Aleksei Kim1*, Ainur Sharip1*, Aigul Sharip1*, Juhong Jiang2, Qing Yang1, Yingqiu Xie1 ![]()
1. Department of Biology, Nazarbayev University, School of Science and Technology, Astana, 010000, Republic of Kazakhstan;
2. Department of Pathology, the First Affiliated Hospital, Guangzhou Medical University, Guangzhou, China.
* Co-first authors.
Received 2016-8-17; Accepted 2016-9-9; Published 2017-2-17
Abstract

One of the DNA repair machineries is activated by Poly (ADP-ribose) Polymerase (PARP) enzyme. Particularly, this enzyme is involved in repair of damages to single-strand DNA, thus decreasing the chances of generating double-strand breaks in the genome. Therefore, the concept to block PARP enzymes by PARP inhibitor (PARPi) was appreciated in cancer treatment. PARPi has been designed and tested for many years and became a potential supplement for the conventional chemotherapy. However, increasing evidence indicates the appearance of the resistance to this treatment. Specifically, cancer cells may acquire new mutations or events that overcome the positive effect of these drugs. This paper describes several molecular mechanisms of PARPi resistance which were reported most recently, and summarizes some strategies to reverse this type of drug resistance.
Keywords: PARP, PARP inhibitor, drug resistance, DNA repair
Introduction
Poly (ADP-ribose) polymerases (PARPs) comprise an extended superfamily of seventeen proteins with different cellular functions including spindle pole formation, cell cycle regulation, cell death, inflammation, adaptive immunity, and DNA repair [1]. More importantly, PARPs are well characterized for the crucial function in DNA breaks repair. For example, PARP1 repairs single-strand breaks (SSBs) through Base Excision Repair (BER) pathway [2]. In this process, PARP1 binds to the site of SSB taking part in synthesis of Poly (ADP-ribose) polymer molecules (PARs) from NAD+ and ADP-ribose molecules [3]. Notably, these polymers contain a big negative charge which increases the electrostatic repulsion between the negative phosphate groups allowing the tight chromatin organization to get looser and recruit other BER proteins such as DNA glycosylases, DNA polymerases, and DNA ligases [4, 5].
Over the past few decades, PARP1 enzyme gained attractions as a promising target for chemotherapeutics. PARP inhibition results in a significant amount of double-strand breaks (DSBs) through the mechanism of interfering with replication fork at the site of DNA damage [2-4]. For this reason, PARP inhibitors (PARPi) can target those cells with impaired DNA DSB repair related to homologous recombination (HR) [2-4]. Researchers propose that inhibition of PARP1 could be enhanced further by applying a synthetic lethal concept [6]. This concept states that two mutations occurring within either tumor suppressors or proto-oncogenes might lead to cell death, whereas a single mutation within just one of two genes appears to be less dramatic [6]. An example of synthetic lethal concept related to PARP inhibitors is observed in cells with defected BRCA1 and BRCA2. The clinical trials of PARPi show great promising underway in ovarian cancer [7]. For instance, in Phase I and II clinical trials Pfizer and AstraZeneca pharmaceutical companies tested effects of AG014699, PF01367338, Olaparib, and AZD2281 PARP1 inhibitors in breast cancer with mutations in BRCA genes [4, 8]. Despite the promising preliminary results, a prolonged treatment of cancer with PARP inhibitors is frequently associated with resistance to this therapy. For example, it has been shown that BRCA2 gene with secondary mutations shows drug resistance to cisplatin and PARPi [9, 10]. Therefore, many ongoing studies aim to understand the fundamentals as well as details of this phenomenon in order to increase the efficiency of the PARPi therapy. This review article will discuss the updated novel mechanisms of resistance to PARP chemotherapy and different strategies to reverse the evasion of the PARPi drugs.
Molecular Mechanisms Underlying the Resistance to PARP inhibitors
Even though approaches to treat some types of cancer have been improved in recent years, majority of patients are treated with standardized therapies, which usually underestimate the unique features of relapsed tumors [11]. Alternatively, targeted therapies provide the more efficient strategy. One of the good examples of targeted therapies is inhibition of PARP enzymes in certain types of tumor characterized by deficiency in homologous recombination pathway in DNA repair process [12]. However, like many treatments, tumors frequently acquire resistance to PARP inhibitors. As drugs targeting PARP enzymes become more widespread, the problem of resistance remains more significant and alarming [2]. Consequently, to make chemotherapy more effective, careful study and analysis of details of molecular mechanisms leading to tolerance of PARP inhibitors is very important.
One of the popular PARPi drug, Olaparib, has shown its effectiveness in ovarian, breast, prostate, and pancreatic cancers [13]. Researchers designed PARP-inhibitor-resistant (PIR) cells to study PARP inhibitor resistance mechanisms in more details [14]. Multiple approaches have been used to identify the kinetic property of PAPRi such as RNA inference (RNAi) screening, genome editing, knockin mouse models to understand the resistance mechanisms [15]. Comprehensive study of these molecules would make PARPi resistance mechanism clearer and meaningful which should definitely increase the efficacy of PARPi [16]. Studies on mechanisms of resistance to PARPi derive largely from cell culture, animal, and patient models. One example comes from an in vivo model where it is observed that cancer cells treated with Olaparib drug develop resistance via partial restoration of homologous recombination molecular pathway [17]. On molecular level, resistance to PARPi is often related to the concept of synthetic lethality [18]. The principles of synthetic lethality explain well the drug resistance problem. For example, BRCA genes and PARP gene could be thought as a synthetic lethal pair [19]. PARPi resistance caused by secondary mutation in BRCA genes emerged by a synthetic lethal interaction between those molecules [20]. Followings are details of the previous and updated findings of molecular pathways contributed to the PARPi resistance.
Resistance by new BRCA1 Deletion Isoforms: Rdd-BRCA1/RING-less BRCA1) and exon11 deletion splice variant
BRCA1 is a predisposed marker for breast cancer if mutated. BRCA1 has multiple functions in cell cycle arrest, cell proliferation, HR mediated DSB repair and Ubiquitination as an RING type of E3 ligase [21]. Full length BRCA1 contains N-terminal RING domain, coiled-coil domain and C-terminal domains (BRCT) [21]. Through coil domain, BRCA1 forms a complex with BRCA2-RAD51 for DSB repair. Through BRCT domain, BRCA1 can bind phosphorylated proteins such as C-terminal 1 interaction protein 1(CtIP) [21]. BRCA1-associated RING domain 1 (BARD1) is a binding partner for stabilization of BRCA1 and the E3 ligase activity [21]. It has been found that BRCA1 truncated forms play essential roles in PARPi resistance. BRCT domain deletion mutant maintains integrity of RAD51 binding region for DNA repair as a mechanism of PARPi resistance [22]. Moreover, single amino acid mutation at RING domain C61G not only disrupts the BRCA1 function as a tumor suppressor but also gains a function of promoting the PARPi resistance and decreases the sensitivity of cancer cells to DNA damage drug platinum [23]. However, another RING domain mutation of I26A does not abolish the binding ability to BARD1, tumor suppressing and E3 ligase activity [24].
Two similar new discoveries just published while we were revising this review manuscript. One study showed that novel RBCA1 deletion forms can mediate PARPi resistance including RING-domain-deficient BRCA1 (Rdd-BRCA1) and different variants [25-27]. Both of the two studies used SUM1315MO2 cell line which was developed from a skin metastasis of ductal carcinoma [25]. SUM1315MO2 cells harbor the BRCA1185delAG allele [25]. One study applied drug resistant cells generated from continuing PARPi Rucaparib/cisplatin treatment [26]. The authors found that acquired PARPi resistance was not induced by secondary lethal mutation but by N-terminal RING domain deletion of Rdd-BRCA1 [26]. Further analysis identified the translation start amino acid is Met-297 but not others due to favor of the RNA secondary structure [26]. Functionally, Rdd-BRCA1, independent on BARD1, induces PARPi resistance both in cell lines and xenograft model. Rdd-BRCA1 also expressed in ovarian carcinomas patient samples [26]. Another similar study also used engineering mice with conditional knockout of wild type of BRCA1 but harbor Brca1185delAG alleles in epithelium with the secondary p53 knockout cancer model [27]. BRCA1185delAG mutation generates only 11 amino acid early stop of translation of BRCA1 protein and results in loss of RING domain [25]. It has been shown in this study that a new gene named RING-less Brca1 highly expresses in knockout Brca1185delAG allele. A man-made early stop mutation of Brca1185stop generated in this study showed high resistance to the PARPi in mouse model [27]. Further analysis in human breast cancer cell line SUM1315MO2 identified a protein band of loss of RING domain of BRCA1 [27]. Brca1 formed foci with Rad51 upon γ irradiation in cells generated from Brca1185stop mouse mammary tumors but not in wild type BRCA1 controls [27]. Thus clinical relevant mutation of BRCA1185delAG mediates drug resistance to PARPi [27]. Therefore, RING domain plays essential roles in prevention of drug resistance. However, how RING domain can exert the drug sensitization and through which signaling pathways are largely unknown. As RING domain plays essential roles in E3 ligase activity, ubiquitin system might fine tune the drug sensitivity through inhibition of HR mediated DSB repair. In addition, BRCA1-Δ11q is a deletion of almost whole exon 11 as a splice variant. Using CRISPR/Cas9 genome editing mediated downregulation studies the authors found that BRCA1-Δ11q protein can mediate PARPi resistance with DNA damage drug cisplatin in a mouse model [28]. However, the detailed mechanisms are unclear.
Resistance by Epigenetic Re-expression of BRCA1
Given that BRCA1 is essential to PARPi sensitivity; epigenetic re-expression of BRCA1 may confer the drug resistance to PARPi. The most recent report published just while we were writing this paper showed the epigenetic resistance of PARPi through BRCA1 using a precision oncology approach of patient-derived xenograft (PDX) models [29]. These mice developed PARPi Olaparib drug resistance in 83% of 18 mice treated with Olaparib [29]. Further studies showed that 73% of drug resistant PDX tumors were BRCA1 positive due to re-expression of BRCA1 by loss of methylation in BRCA1 promoter detected in 70% of 23 resistant tumors [29]. Detected RAD51 foci, BRCA1 gene fusions and target locus amplification in those drug resistant tumors suggest that the HR mediated genome arrangement may re-activate BRCA1 transcription through epigenetic regulation for PARPi drug resistance [29].
Replication Fork Stabilization
Another novel mechanisms reported just while we were writing the manuscript is that replication machinery stabilization contributes to drug resistance. In details, a DNA damage response protein, Pax2 transactivation domain-interacting protein (PTIP) which can form nuclear foci for DSB of DNA, can destabilize MRE11 nuclease in stalled replication forks [30]. Thus PTIP defective can stabilize nascent DNA strands by prohibiting degradation. This mechanism is found in BRCA1/2 deficient cells and contributes to PARPi and cisplatin resistance [30]. Recently, another report consistently found the same mechanism of PARPi resistance that similar to PTIP, PARP one allele loss can prohibit recruiting Mre11 nuclease to the stalled replication forks in Brca2 null mice [31]. Thus PARPi Olaparib can overcome drug resistance in BRCA2-deficient cells [31]. However, loss of PTIP or PARP does not restore HR pathways [30, 31].
Intragenic Deletion and Genetic Reversion of BRCA1/2
Some PARP-resistant cell lines often possess a deletion within the BRCA2 gene [14]. A second mutation might restore an open reading frame (ORF) of the mutant BRCA2 [14, 32]. One study showed that intragenic deletion of the c.6174delT caused restoration of the ORF that led to the synthesis of new isoforms of BRCA2 which most likely contributed to the restoration of DNA repair through HR [14]. Thus the reverse mutations in defective BRCA1 and BRCA2 genes contribute to resistance for HR-deficiency therapy. Some secondary mutations could correct the original violation and by that means make tumors resistant to treatments and increased the competence to HR to some extent [33]. For example, Sakai and coworkers observed secondary mutations within the BRCA2 gene that restored ORF allowing the HCC1428, BRCA2-defective cells to be resistant to PARPi drugs [33].
Regulation by microRNA
MicroRNA (miRNA) is a type of non-coding RNA which inhibits expression of target genes contributing to activation or repression of signaling pathways in cancer cells. Recently Choi and his coworkers identified that expression of miR-622 mediated PARPi resistance through suppression of non-homologous end joining pathway (NHEJ). Based on results, overexpression of miR-622 is associated with decreased expression of genes such as 53BP1, Ku70, and Ku80 [34]. However, miR-622 induces PARPi resistance by directly targeting mRNAs of Ku70 and Ku80 but not 53BP1 in BRCA1-mutant ovarian cells [34]. The researchers revealed that this miRNA blocked the conventional NHEJ pathway that leads to activation of HR repair mechanism by accumulation of Mre11 foci at the sites of DSBs [34]. Thus, miR-622 elevation may limit NHEJ and promote the HR pathway by balancing between NHEJ and DSB repair pathways which mediates resistance to PARPi. Moreover, other studies showed that miR-96, miR-107 miR-222 suppress DNA repair protein, RAD51 mRNA thereby disrupting DSB repair mediated through HR [35]. In addition, RNAi mediated library screening with PARP inhibitors identified that miR-182 is a miRNA that directly inhibits BRCA1 transcript [36]. In a xenograft model with injection of miR-182 overexpressing breast cancer MDA-MB-231 cells, the tumor growth was decreased upon 10 days treatment with PARPi of Olaparib [36]. Researchers further showed that miR-182 deceases HR efficiency and promotes cell sensitivity to DNA damage induced by radiation or chemical compound [36].
Phosphorylation of PARP1 by c-Met
Several pre-clinical studies report that proto-oncogene c-Met contributes to PARP inhibitor resistance in triple-negative breast cancer (TNBC) cells [37]. However, c-Met is overexpressed in different cancers including but not limited to ovarian, breast, bladder, and colorectal and usually associated with poor prognosis [38]. Furthermore, cells with overexpression of c-Met show high resistance to PARP inhibitors [37]. On the contrary, cell lines with knockdown of c-Met tend to be more responsive to inhibition of PARP enzymes [37]. Specifically, Duo and his coworkers found that c-Met phosphorylates PARP1 at Tyr907 site in different TNBC cell lines including HCC1937, MDA-MB-157, and MDA-MB-453. Phosphorylated PARP at Tyr907 shows higher enzyme activity and lower binding activity to PARPi than that of non-phosphorylated PARP, which promotes tumor cells to develop PARPi resistance [37]. Overall, the researchers proposed that patients with TNBC, who was positive for pY907-PARP1 and c-Met, tend to be at greater risk for developing resistance to PARP inhibitors [37].
Overexpression of HOX Family
HOX gene family members are master regulators of organism development including embryo and axis in animals. Recently Esposito and her colleagues demonstrated that HOX proteins play a role in resistance to PARP inhibitors [39]. The researchers found that different HOXs participate in DNA repair process. For example, HOXB9 activated TGF-β signaling pathway that may recruit ATM to the sites of DSBs in cells lines from breast tumors [39]. They studied the role of HOXA9 in resistance to PARPi in PARPi-sensitive AML1-ETO and PML-RARα-transformed cell lines by overexpressing HOXA9 [39]. Specifically, AML1-ETO and PML-RARα are two transcription factors expressed in acute myeloid leukemia (AML) [39]. Importantly, these two fusion oncoproteins appear to be very sensitive for PARPi [39]. According to the results, higher levels HOXA9 lead to more RAD51 being accumulated at the foci formation during HR repair in cancer cells [39].
Overview of Mechanisms from Previous Reviews: drug transporters, mTOR, cell inflammation pathways, PARP itself and loss of 53BP1
Usually drug efflux machinery plays essential roles in directly resisting the action of drugs. ATP binding cassette (ABC) transporter family is a large family of drug efflux pump. For example, one member of ABC family, P-glycoprotein (Pgp) transports cellular molecules, nutrients, drugs, and toxins. Pgp is encoded by ABCB1 (also known as MDR1) gene, which is found to be upregulated in some cancer cells having resistance to chemotherapy [40]. The resistance to Olaparib is associated with increased expression of the ABC genes [41-45]. The resistance can be reversed by applying Pgp inhibitor Tariquidar combined with PARPi, AZD2281 [42]. Thus, non-Pgp-substrate type of PARPi is more promising in effective treatment such as Veliparib and CEO-8983 [43]. Researchers also found EMT contributes to Pgp induced PARPi resistance [45]. Furthermore, it is still under investigation whether overexpression of Pgp is the main resistance mechanism apart from BRCA1 gene restoration and loss of 53BP1 [46]. In addition, mTOR pathway contributes to resistance to PARPi. Specifically, phosphorylating S6 ribosomal protein would lead to PARPi resistance in BRCA1-deficient cancer cell lines [47]. Substitutive mutation of S6 ribosomal protein of all five phosphor-sites including S235, S236, S240, S244, and S247 could restore sensitivity of tumor cells to PARPi via inhibition of S6 phosphorylation [47]. Mechanistically, S6 phosphorylation causes PARPi resistance by activating mTOR pathway and restoring HR in BRCA-deficient cancer cells [47-48]. Changes in PARP1 expression levels in cancer cells also lead to resistance to PARPi. Elevation of PARP1 mediates resistance to PARPi in BRCA1-deficient HCC1937 cells and is related to worse outcomes in patients with breast cancer [49-52]. Cell inflammatory pathways also may contribute to drug resistance of PARPi [53]. Particularly, Knockdown or inhibition of NF-κB signaling component restored sensitivity to PARP inhibitors [53-55]. Moreover, 53BP1 loss which partially recovered HR was observed in some PARP resistant cancer cells through competitive binding to the repair machinery complex [56-58]. Based on previous reviews, molecular mechanisms associated with resistance to PARPi can be found in more literatures [59-66].
Strategies of Overcoming PARPi Drug Resistance
Mild Hyperthermia
One of the promising solutions to deal with the issue of inefficiency of long-term application of PARP inhibitors in tumor cells is hyperthermia. Hyperthermia has been widely used solely or in combination with other methods for cancer treatment [67]. Hyperthermia acts on cancer cells in several pathways. For example, elevation of body temperature causes membrane damage, protein denaturation, inhibition of cell proliferation and DNA repair [69-72]. All of these effects lead to cell death by either apoptosis or necrosis [69-72]. In addition, hyperthermia activates anti-tumor immune signaling [69]. Mild hyperthermia has been used to overcome resistance to PARP inhibition. In the experiment of the effects of hyperthermia and PARP inhibitors on mice carrying BRO-derived tumors, the elevation in temperature or treatment with PARPi, PJ-34 or Olaparib alone did not decrease tumor growth but the combination with hyperthermia significantly inhibited tumor growth [73].
Hyperthermia promotes cancer cells to be sensitive to PARP inhibitors by inducing synthetic lethality [73]. Normally, cells use HR repair pathway for DSBs. It has been shown that HR starts from formation of a protein complex at DNA repair foci; this complex is made up three interacting proteins including Mre11, Rad51, and NBS1 [74]. This complex acts as an exonuclease creating 3' overhangs at the both sites of breakage that stabilized by replication protein A (RPA) protein [74]. Then, RAD51 mediates strand invasion process by binding to the 3' overhangs displacing the RPA protein from them and finding complementarity between the invading strand and homologous chromosome [74]. However, cells, which are exposed to high temperatures, cannot rely on HR pathway [75]. Specifically, HR inhibition is mediated by several mechanisms, including inactivation of RPA, degradation of BRCA1 and BRCA2, and decrease in MRE11 gene expression [73]. More importantly, when HR is not activated DSBs get accumulated which usually can lead to apoptosis [73].
Despite its positive effects on sensitizing tumors to PARP inhibition, cells can develop thermotolerance [76]. This thermotolerance can be caused by heat-shock response [67]. The latter is mediated by heat-shock proteins (HSPs). One of the major players in this response is HSP90. Researchers try to inhibit this protein to minimize its effect on thermotolerance. Krawczyk and colleagues highlight that the combination of mild hyperthermia, Olaparib, and HSP90 inhibitor, 17-(dimethylaminoethylamino)-17-demthoxygeldanamycin (17-DMAG) led to a complete loss of tumor growth [73]. For instance, all of the mice treated with this combination survived during the course of the treatment. On the contrary, survival of mice received treatment of hyperthermia and PARP inhibitors was about 36% [73].
Histone Deacetylase Inhibitors
Another promising solution to the problem of PARP inhibitor resistance is to use histone deacetylase inhibitors. Acetylation and deacetylation of histones is an important mechanism of posttranslational regulation of gene expression [77]. Histone Acetyltransferase (HAT) enzyme catalyzes the reaction of adding the acetyl groups to lysine residues on histone complexes. On the contrary, the removal of the group is performed by another enzyme named histone deacetylase (HDAC) [78]. In addition to histones, HAT and HDAC also act on transcription factors, DNA-binding and repair proteins, molecules involved in signal transduction, and chaperons, thus regulating activities of these molecules [78]. In healthy cells, activities of HATs and HDACs are under stringent control which leads to the proper balance between acetylation and deacetylation of substrates [79]. When the balance is out of control, series of consequences can arise [79]. For example, it has been reported in several articles that improper balance between reaction of adding and removing the acetyl groups has a positive correlation with different forms of cancer such as prostate, breast, colorectal, and gastric cancers [79-82]. The main feature of irregularity of acetylation and deacetylation is the overexpression of HDACs. Treatment with HDAC inhibitors (HDI) sensitizes cancer cells to PARP inhibition. Specifically, HDI were found to enhance responsiveness of cells with triple negative breast cancers to PARP-1inhibitors [5, 83-84]. HDI-treated cells become responsive to PARP inhibition because of BRCAness effect [5]. First, HDI blocks deacetylation of the HSP90 heat shock protein which leads to the hyperacetylation and inhibition of HSP90. As a result, several client proteins of HSP90 including BRCA1, RAD52, ATR, and CHK1 cannot interact with it [5, 85]. The absence of interaction with HSP90 leads to the degradation of these proteins [5, 85]. BRCA proteins are actively used in HR pathway to join DSBs in DNA molecule [86]. As it is expected cells have to rely on some alternative pathways such as NHEJ when BRCA genes are not functional. When treated with PARP inhibitors, cells become unable to repair SSBs [86]. SSBs are then transformed into DSBs, which are normally repaired by BRCA proteins [86]. As BRCA1 is not expressed in HDI-treated cells, cells have to rely mostly on NHEJ which is much less accurate than HR and causes genome instability [86]. Eventually, due to excessive SSBs and build-up of DSBs, cells become unable to preserve the integrity of their genome and die by apoptosis [86]. One study showed that HDI treatment by vorinostat or valproic acid in DU145 and PC3 prostate cancer cells decreases DNA repair genes expression such as RAD51, CHK1, BRCA1, RAD21 through transcription factor E2F1 [87]. In triple-negative breast cancer, treatment of HDI induces BRCAness and inhibits stemness for sensitizing PARP inhibitor [83-84]. Moreover, recent studies showed that HDI suberoylanilide hydroxamic acid (Vorinostat, SAHA) and PARPi, PJ34, synergistically induce cell death in anaplastic thyroid carcinoma and leukemia cells [88-89].
Targeting Heat Shock Protein 90 and BRCA1 Stabilization
To inactivate HSP90 protein is also an exciting method to solve the issue of PARP inhibitor resistance and it has been undergoing clinical trials [22, 90]. HSP90 functions as a chaperone molecule, which assistants in folding of different proteins such as epidermal growth factor receptors (EGFR), Protein Kinase B (AKT), MET, and Raf-1 kinase for acquiring functional 3D structures [91]. Furthermore, it is observed that the HSP90 chaperone stabilizes many tumor suppressors and proto-oncogenic proteins derived from the mutated genes, thus preventing their ubiquitin-proteasome degradation [89]. Such chaperone-mediated stabilization of oncogenic proteins is generally correlated with resistance to chemotherapy treatment [7]. For example, Curtin and colleagues detected a high level of expression of a mutated BRCA1 and HSP90 as well as 53BP1 loss in a cisplatin-resistant MDA-MB-436 cancer cell line [7].
DNA repair focus is usually the first event observed during DNA repair through HR pathway [92]. One of the active participants of DNA repair via HR mechanism is BRCA1 [85]. However, the exact mechanism underlying how BRCA1 mediates HR is still under the investigation [93]. Nevertheless, researchers found that BRCA1 acts as a mediator and co-factor of this process by binding to CtIP to produce resection at 5' broken ends [93]. Clearly mutated or truncated BRCA1 cannot perform this function properly [5]. Cancer cells usually circumvent this problem by activating the HSP90 that stabilizes BRCA1 protein to form a complex with the Partner and Localizer of PALB2-BRCA2-RAD51 recombinase (PALB2-BRCA2-RAD51) at the site of DNA repair focus [5]. Consequently, CtIP has unrestricted access to DNA breaks with the subsequent process of the strand invasion and DNA repair during HR pathway [1]. The key role of the HSP90 in the prevention of ubiquitin-proteasome degradation of many oncogenes has attracted a great attention in the field of chemotherapy [92]. Currently different inhibitors of the HSP90 are undergoing preclinical studies and clinical trials such as isoxazole derivative VER-52296/NVP-AUY922 and purine derivative BIIB021 [94-96]. Recent study suggests that free XRCC1 is a substrate of HSP90 and associates with DNA polymerase β complex for stabilization to determine the DNA repair construct and BER fate [97]. Thus, scientists propose that targeting both PARP1 and HSP90 would activate NHEJ pathway that usually causes genomic instability and cell death [98]. A more recent pre-clinical study showed that combination of PARPi Olaparib and HSP90 inhibitor, AT13387 in patient derived xenograft mouse model of ovarian cancer showed synergistic efficiency in disruption of tumor growth in BRCA1-mutated/PARPi-resistant model and phase I clinical trial of drug combination has been on designing [99]. While a large number of proteins are HSP90 clients and HSP90 inhibitors may have off-target effect on crosstalk signaling pathways of client proteins, in personalized treatment, it should be cautious to use combined HSP90 inhibitors and PARPi.
Inhibition of Cyclin-Dependent Kinase 1
Cyclin-dependent kinase 1 (CDK1) (another name is CDC2) is a Ser/Thr kinase [100]. This protein is often called M-phase promoting factor (MPF) and functions in G1/S and G2/M phases of cell cycle [100]. To review, cells can progress through the phases of cell cycle due to periodic production and degradation of cyclins, which function as partners of CDK1 [100]. For example, production and maintenance of cyclin A2 is critical to prevent the re-replication of DNA in S phase [101]. One main function of CDK1 is to participate in HR repair pathway [102]. Specifically, this kinase phosphorylates BRCA1 to promote the proper formation of DNA repair foci [102]. This focus also includes a variety of proteins and regulators such as abraxas-RAP80 complex, PALB2, BRCA2, and RAD51 [103, 104]. In addition, CDK1 enhances the production of ATP in mitochondria by phosphorylating the mitochondrial complex I in the electron transport chain [105]. The high ATP production allows cells to meet the elevated energy consumption in order to repair DNA and avoid apoptosis [105].
A cell usually controls the activity of CDK1 levels by p53 or phosphorylation by CDK-activating kinase (CAK) [100,106,107]. One promising drug RO-3306 inhibits CDK1 10-fold more selectively than with CDK2 and 50-fold more selectively than with CDK4 [100, 108]. It is attractive to block CDK1 activity to enhance the efficiency of PARP inhibition therapy. One study showed that CDK1 knockdown by inducible shRNA sensitizes PARPi AG14361 inhibition by elevation of chromosome aberrations and cell death [109]. In mouse model of lung cancer induced by Kras/Trp53 mutations, combined Cdk1 and Parp inhibition decreased tumor size and increased survival [109].
Inhibition of BARD1 disrupts BRCA1
To promote export of BRCA1 to the cytoplasm can be an interesting method to overcome resistance to PARP inhibitors [110]. Many proteins, RNAs, and other regulatory molecules cross the nuclear membrane by active transport using importin or exportin receptors [111]. Nuclear export usually relies on one of the seven well studied exportin receptors, specifically Exportin 1 or Chromosome Region Maintenance 1 protein (XPO/CRM1) receptor [112]. For example, many regulatory RNA molecules and over 200 different proteins like Foxo and BRCA1 can pass through the nuclear membrane with the assistance of XPO/CRM1 exportins [112-113]. At the molecular level, Ran GTPase should recognize the specific Nuclear Export Signal Sequence (NES) on these proteins before binding to XPO/CRM1 and forming a ternary complex, which then should be exported to the cytoplasm, where Ran GTP is hydrolyzed back to Ran GDP releasing the cargo [112].
The appropriate localization of BRCA1 is critical for cellular processes because it plays a different role depending on its cellular localization [111]. In particular, BRCA1 participates in the HR process inside the nucleus, promotes microtubule nucleation, and prevents centrosome duplication by binding to the BARD1 [111]. Researchers concluded that BARD1 stabilizes and confines BRCA1 to the nucleus [114]. BARD1 could bind to the N -terminal RING domain as well as the NES of BRCA1 [115]. Interestingly, BRCA1-BARD1 interaction usually prevents the transport of BRCA1 by Ran GTPase and XPO/CRM1 pathway [113, 116]. Specifically, scientists highlight that about 75% of nuclear BRCA1 makes stable complexes with BARD1 [115]. Furthermore, researchers showed that the depletion of nuclear BARD1 increases the export of BRCA1 from the nucleus [115, 117]. Overall, it seems both attractive and promising to block BARD1 in combination with the PARP inhibition therapy. For instance, recent studies showed that in colon cancer, BARD1 splice variant, BARD1β which has deletions of RING domain, can disrupt BRCA1 E3 ligase activity, and prevent BRCA1 nuclear localization [118]. Most importantly, overexpression of BARD1β, a dominate-negative form of BARD1, abolishes HR and RAD51 foci and can sensitize colon cancer cells to PARPi [118]. Thus targeting BARD1 can be an alternative approach to interfere BRCA1 to overcome PARPi resistance.
p53 stabilizer reverses PARPi drug resistance
It has been shown that p53 is also a regulator of cytosol-nucleus translocation of BRCA1 possibly affecting BRCA1 mediated DNA repair. p53 can interact with BRCA1 to decrease nuclear BRCA1 by nuclear export [119] thereby possibly inhibiting nuclear BRCA1 mediated PARPi drug resistance. p53 mediated shuttling of BRCA1 increases cytotoxicity and sensitization to DNA damage by radiation is not through p53 mediated transcriptional activity but only dependents on their interactions [119]. It has been shown that cytosolic BRCA1 can enhance apoptosis and can cause the loss of DNA DSB repair even though the two processes are not associated [120]. p53 stabilization agent such as Nutlin-3, which inhibits MDM2 E3 ligase of ubiquitination and degradation of p53, has been widely used for enhancing p53 signaling [121]. Thus p53 stabilizer can be applied for overcoming PARPi resistance. Indeed it has been shown that Nutlin-3 increased breast cancer cells to PARP inhibitor IQD with a decrease of DSB HR [121]. Moreover, another study in wild type of p53 expressing cells of multiple myeloma showed that p53 stabilizer Nutlin-3a together with PARPi Olaparib enhanced apoptosis and PARPi sensitivity but not in p53 mutant cells [122]. Thus combinational treatment of p53 stabilizer and PARPi can be a novel avenue to enhance chemotherapy by reverse PARPi resistance [122]. While p53 regulators are abundant, targeting BRCA1 through p53 pathway regulators might generate many novel avenues to inhibition of PARPi drug resistance. Although there might be crosstalk signaling which can interfere the p53/BRCA1 feedback, stabilizing p53 to target DSB repair by BRCA1 is still promising in cancer treatment especially in precision therapy.
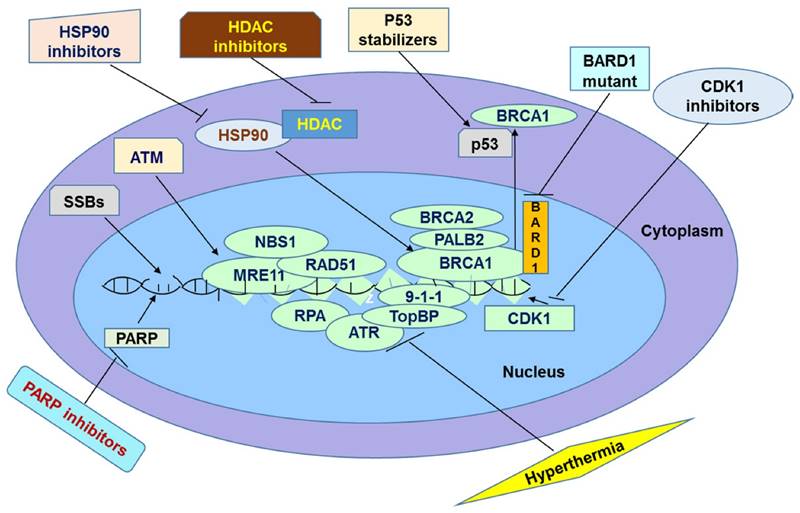
Conclusion and Perspectives
It has been widely believed that PARP inhibitors are promising agents for cancer treatment. They act by inhibiting SSBs repair pathway, which is normally initiated and mediated by PARP enzymes. Many clinical trials show that PARP inhibitors are promising in treatment of cancers. However, in some instances patients do not respond effectively to this therapy probably because of tolerance of PARP inhibitors. Particularly, tumor cells could show tolerance to this therapy by increasing efflux of the drugs through Pgp, overexpressing PARP1, reducing inhibitors' affinity for PARP1, restoring HR repair pathway, and suppressing NHEJ. In this review, we discussed several mechanisms underlying resistance as well as some promising solutions to overcome it. The methods of overcoming the resistance are based on the direct or indirect inactivation of BRCA and other HR proteins. This inactivation should end up in accumulation of DSBs leading to apoptosis. It has been speculated by several researchers that these methods should sensitize resistant cancer cells to PARP inhibition (Figure 1). When these methods are applied in combination with PARP inhibitors, their action can be synergized leading to a greater efficacy than either treatment. For example, one recent study compared the differential combinational effect to overcome PARPi resistance. The authors found that two drugs which knowingly inhibit PARPi, combined with HDAC inhibitor can efficiently inhibit drug resistance of PARPi [123]. First the authors found that HDAC inhibitor Vorinostat increases the sensitivity of cancer cells to PARPi ABT-888 in both cell lines and xenograft model by elevation of eIF2α phosphorylation but not expression levels of BRCA1, RAD51 or 53BP1[123]. Then the authors found the anti-metabolite drug 6-thioguanine (6-TG) which can inhibit proliferation in BRCA deficient cells [124] through sensitizing PARPi, can further increase phosphorylated eIF2α to induce cell death [123]. In summary, next generation PARPi of non-substrate of ABC drug transporters based cocktail therapy may provide a promising treatment avenue in clinics. However, drug-drug interactions must be investigated when using combinational therapy. The next stage of the research in this field is to examine the combined efficiency of these methods in overcoming PARP inhibitor resistance in clinical trials.
Summary of Potential Avenues to Overcome Resistance to PARPi in Chemotherapy
Acknowledgements
This work was supported by China-Kazakhstan collaboration grant (No. CK-07-09) to Yingqiu Xie and the National Natural Science Foundation of China (No. 81272901) to Juhong Jiang. The authors would like to thank the Department of Biology, SST, and Nazarbayev University for the support of acquired learning and research integrated teaching innovation of Biochemistry I course for undergraduate students. We would like to thank all the Biochemistry I course students who supported the assignment of the Biochemistry I course and the publication of their assignments.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sonnenblick A, de Azambuja E, Azim H, Piccart M. An update on PARP inhibitors - moving to the adjuvant setting. Nature Reviews Clinical Oncology. 2014;12:27-41
2. Davar D, Beumer JH, Hamieh L, Tawbi H. Role of PARP Inhibitors in Cancer Biology and Therapy. Current Medicinal Chemistry. 2012;19:3907-3921
3. Anwar M, Aslam HM, Anwar S. PARP inhibitors. Hereditary Cancer in Clinical Practice. 2015;13:4
4. Telli ML, Ford JM. PARP inhibitors in breast cancer. Clinical Advances in Hematology & Oncology. 2010;8:629-635
5. Brown JS, O'Carrigan B, Jackson SP, Yap TA. Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discovery. 2017;7:20-37
6. Yap TA, Sandhu SK, Carden CP, de Bono JS. Poly (ADP-Ribose) polymerase (PARP) inhibitors: Exploiting a synthetic lethal strategy in the clinic. CA: A Cancer Journal for Clinicians. 2011;61:31-49
7. Liu JF, Matulonis UA. What Is the Place of PARP Inhibitors in Ovarian Cancer Treatment? Current Oncology Reports. 2016;18:1-9
8. Curtin NJ, Szabo C. Therapeutic applications of PARP inhibitors: anticancer therapy and beyond. Molecular Aspects of Medicine. 2013;34:1217-1256
9. Barber LJ, Sandhu S, Chen L, Campbell J, Kozarewa I, Fenwick K. et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. The Journal of Pathology. 2013;229:422-429
10. Dhillon KK, Taniguchi T. Resistance to PARP Inhibitors Mediated by Secondary BRCA1/2 Mutations. PARP Inhibitors for Cancer Therapy 2015 (pp. 431-452) Springer International Publishing
11. Bouwman P, Jonkers J. Molecular Pathways: How Can BRCA-Mutated Tumors Become Resistant to PARP Inhibitors. Clinical Cancer Research. 2013;20:540-547
12. Underhill C, Toulmonde M, Bonnefoi H. A review of PARP inhibitors: from bench to bedside. Annals of Oncology. 2010;22:268-279
13. Lee J, Ledermann J, Kohn E. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Annals of Oncology. 2013;25:32-40
14. Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA. et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111-5
15. Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R. et al. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27:1368-1377
16. Lord C, Ashworth A. Targeted therapy for cancer using PARP inhibitors. Current Opinion in Pharmacology. 2008;8:363-369
17. Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA. et al. Loss of 53BP1 Causes PARP Inhibitor Resistance in Brca1-Mutated Mouse Mammary Tumors. Cancer Discovery. 2012;3:68-81
18. Kamal A, Shaik T, Malik M. Embracing synthetic lethality of novel anticancer therapies. Expert Opinion on Drug Discovery. 2015;10:1119-1132
19. Helleday T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Molecular Oncology. 2011;5:387-393
20. Ashworth A, Lord C, Reis-Filho J. Genetic Interactions in Cancer Progression and Treatment. Cell. 2011;145:30-38
21. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nature Reviews Cancer. 2012;12(1):68-78
22. Johnson N, Johnson SF, Yao W, Choi YE, Bernhardy AJ, Wang Y. et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proceedings of National Academy of Science USA. 2013;110(42):17041-17046
23. Drost R, Bouwman P, Rottenberg S, Boon U, Schut E, Klarenbeek S. et al. BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer Cell. 2011;20(6):797-809
24. Shakya R, Reid LJ, Reczek CR, Cole F, Egli D, Lin CS. et al. BRCA1 tumor suppression depends on BRCT phosphoprotein binding, but not its E3 ligase activity. Science. 2011;334(6055):525-528
25. Elstrodt F, Hollestelle A, Nagel JH, Gorin M, Wasielewski M, van der Ouweland. et al. BRCA1 mutation analysis of 41 human breast cancer cell lines reveals three new deleterious mutants. Cancer Research. 2006;66(1):41-45
26. Drost R, Dhillon KK, van der Gulden H, van der Heijden I, Brandsma I, Cruz C. et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. Journal of Clinical Investigation. 2016;126(8):2903-18
27. Wang Y, Krais JJ, Bernhardy AJ, Nicolas E, Cai KQ, Harrell MI. et al. RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. Journal of Clinical Investigation. 2016Aug1;126(8):3145-57
28. Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E. et al. The BRCA1-Δ11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Research. 2016;76(9):2778-90
29. Ter Brugge P, Kristel P, van der Burg E, Boon U, de Maaker M, Lips E. et al. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. Journal of National Cancer Institute. 2016:108 (11)
30. Chaudhuri AR, Callen E, Ding X, Gogola E, Duarte AA, Lee JE. et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535(7612):382-7
31. Ding X, Chaudhuri AR, Callen E, Pang Y, Biswas K, Klarmann KD. et al. Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nature Communications. 2016;7:12425
32. Ashworth A. Drug Resistance Caused by Reversion Mutation. Cancer Research. 2008;68:10021-10023
33. Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C. et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116-1120
34. Choi YE, Meghani K, Brault ME, Leclerc L, He YJ, Day TA. et al. Platinum and PARP Inhibitor Resistance Due to Overexpression of MicroRNA-622 in BRCA1-Mutant Ovarian Cancer. Cell Reports. 2016;50:429-439
35. Neijenhuis S, Bajrami I, Miller R, Lord CJ, Ashworth A. Identification of miRNA modulators to PARP inhibitor response. DNA Repair (Amst). 2013;12:394-402
36. Moskwa P, Buffa FM, Pan Y, Panchakshari R, Gottipati P, Muschel RJ. et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Molecular Cell. 2011;41(2):210-20
37. Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH. et al. Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nature Medicine. 2016;22:194-201
38. Xu C, Plattel W, van der Berg A, Rüther N, Huang X, Wang M. et al. Expression of the c-Met oncogene by tumor cells predict favorable outcome in classical Hodgskin's lymphoma. Haematologica. 2012;97:572-578
39. Esposito MT, Zhao L, Fung TK, Rane JK, Wilson A, Martin N. et al. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nature Medicine. 2015;21:1481-1490
40. Sedukhina AS, Sundaramoorthy E, Hara M, Kumai T, Sato K. Beyond resistance to PARP inhibition: Mechanisms and effective treatment options. Cancer Cell & Microenvironment. 2015;2:e821
41. Thurber GM, Yang KS, Reiner T, Kohler RH, Sorger P, Mitchison T. et al. Single-cell and subcellular pharmacokinetic imaging allows insight into drug action in vivo. Nature Communications. 2013;4:1504
42. Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA. et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proceedings of the National Academy of Sciences. 2008;105:17079-17084
43. Lawlor D, Martin P, Busschots S, Thery J, O'Leary JJ, Hennessy BT. et al. PARP Inhibitors as P-glyoprotein Substrates. Journal of Pharmaceutical Sciences. 2014;103:1913-1920
44. Durmus S, Sparidans R, van Esch A, Wagenaar E, Beijnen J, Schinkel A. Breast Cancer Resistance Protein (BCRP/ABCG2) and P-glycoprotein (P-GP/ABCB1) Restrict Oral Availability and Brain Accumulation of the PARP Inhibitor Rucaparib (AG-014699). Pharmaceutical Research. 2014;32:37-46
45. Borst P. Cancer drug pan-resistance: pumps, cancer stem cells, quiescence, epithelial to mesenchymal transition, blocked cell death pathways, persisters or what? Open Biology. 2012;2:120066
46. Lord C, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nature Medicine. 2013;19:1381-1388
47. Sun CK, Zhang F, Xiang T, Chen Q, Pandita TK, Huang Y. et al. Phosphorylation of ribosomal protein S6 confers PARP inhibitor resistance in BRCA1-deficient cancers. Oncotarget. 2014;5:3375-3385
48. Xiang T, Jia Y, Sherris D, Li S, Wang H, Lu D. et al. Targeting the Akt/mTOR pathway in Brca1-deficient cancers. Oncogene. 2011;30:2443-2450
49. Wang YQ, Wang PY, Wang YT, Yang GF, Zhang A, Miao ZH. An Update on Poly(ADP-ribose)polymerase-1 (PARP-1) Inhibitors: Opportunities and Challenges in Cancer Therapy. Journal of Medicinal Chemistry. 2016 Jul 27
50. Gilabert M, Launay S, Ginestier C, Bertucci F, Audebert S, Pophillat M. et al. Poly(ADP-Ribose) Polymerase 1 (PARP1) Overexpression in Human Breast Cancer Stem Cells and Resistance to Olaparib. PLoS ONE. 2014;9:e104302
51. Gonçalves A, Finetti P, Sabatier R, Gilabert M, Adelaide J, Borg JP. et al. Poly(ADP-ribose) polymerase-1 mRNA expression in human breast cancer: a meta-analysis. Breast Cancer Research and Treatment. 2010;127:273-281
52. Von Minckwitz G, Müller BM, Loibl S, Budczies J, Hanusch C, Darb-Esfahani S. et al. Cytoplasmic Poly(Adenosine Diphosphate-Ribose) Polymerase Expression Is Predictive and Prognostic in Patients With Breast Cancer Treated With Neoadjuvant Chemotherapy. Journal of Clinical Oncology. 2011;29:2150-2157
53. Lupo B, Trusolino L. Inhibition of poly(ADP-ribosyl)ation in cancer: Old and new paradigms revisited. Biochimica et Biophysica Acta. 2014;1846:201-215
54. Nakagawa Y, Sedukhina AS, Okamoto N, Nagasawa S, Suzuki N, Ohta T. et al. NF-kB signaling mediates acquired resistance after PARP inhibition. Oncotarget. 2015;6:3825-3839
55. Li C, Chen S, Yue P, Deng X, Lonial S, Khuri FR. et al. Proteasome Inhibitor PS-341 (Bortezomib) Induces Calpain-dependent I B Degradation. Journal of Biological Chemistry. 2010;285:16096-16104
56. Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H. et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nature Structural & Molecular Biology. 2010;17:688-695
57. Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A. et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell. 2010;141:243-254
58. Feng L, Fong KW, Wang J, Wang W, Chen J. RIF1 Counteracts BRCA1-mediated End Resection during DNA Repair. Journal of Biological Chemistry. 2013;288:11135-11143
59. Tangutoori S, Baldwin P, Sridhar S. PARP inhibitors: A new era of targeted therapy. Maturitas. 2015;81:5-9
60. Fojo T, Bates S. Mechanisms of Resistance to PARP Inhibitors-Three and Counting. Cancer Discovery. 2013;3:20-23
61. Morales J, Li L, Fattah FJ, Dong Y, Bey EA, Patel M, Gao J. et al. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit Rev Eukaryot Gene Expr. 2014;24:15-28
62. Mercier-Vogel L, Bodmer A, Castiglione M. PARP inhibitors: new therapeutic agents in breast and ovarian cancer. Revue Medicale Suisse. 2011;7:1137-40
63. Tinker AV, Gelmon K. The role of PARP inhibitors in the treatment of ovarian carcinomas. Current Pharmaceutical Design. 2012;18:3770-4
64. Wang W, Figg WD. Secondary BRCA1 and BRCA2 alterations and acquired chemoresistance. Cancer Biology & Therapy. 2008;7:1004-5
65. De Lorenzo SB, Patel AG, Hurley RM, Kaufmann SH. The Elephant and the Blind Men: Making Sense of PARP Inhibitors in Homologous Recombination Deficient Tumor Cells. Frontiers in Oncology. 2013;3:228
66. Deshmukh D, Qiu Y. Role of PARP-1 in prostate cancer. American Journal of Clinical and Experimental Urology. 2015;3:1-12
67. van der Zee J. Heating the patient: a promising approach? Annals of Oncology. 2002;13:1173-84
68. Toraya-Brown S, Fiering S. Local tumour hyperthermia as immunotherapy for metastatic cancer. International Journal of Hyperthermia. 2014;30:531-539
69. Lepock J. How do cells respond to their thermal environment? International Journal of Hyperthermia. 2005;21:681-687
70. Man J, Shoemake JD, Ma T, Rizzo AE, Godley AR, Wu Q. et al. Hyperthermia Sensitizes Glioma Stem-like Cells to Radiation by Inhibiting AKT Signaling. Cancer Research. 2015;75:1760-1769
71. Zhao P, Jiang H, Su D, Feng J, Ma S, Zhu X. Inhibition of cell proliferation by mild hyperthermia at 43˚ C with Paris Saponin I in the lung adenocarcinoma cell line PC-9. Molecular Medicine Reports. 2015;11:327-332
72. Eppink B, Krawczyk PM, Stap J, Kanaar R. Hyperthermia-induced DNA repair deficiency suggests novel therapeutic anti-cancer strategies. International Journal of Hyperthermia. 2012;28:509-517
73. Krawczyk PM, Eppink B, Essers J, Stap J, Rodermond H, Odijk H. et al. Mild hyperthermia inhibits homologous recombination, induces BRCA2 degradation, and sensitizes cancer cells to poly (ADP-ribose) polymerase-1 inhibition. Proceedings of the National Academy of Sciences. 2011;108:9851-9856
74. Thurn KT, Thomas S, Raha P, Qureshi I, Munster PN. Histone Deacetylase Regulation of ATM-mediated DNA Damage Signaling. Molecular Cancer Therapeutics. 2013;12:2017-2087
75. Bergs JW, Krawczyk PM, Borovski T, ten Cate R, Rodermond HM, Stap J. et al. Inhibition of homologous recombination by hyperthermia shunts early double strand break repair to non-homologous end-joining. DNA Repair. 2013;12:38-45
76. Oei AL, Vriend LE, Crezee J, Franken NA, Krawczyk PM. Effects of hyperthermia on DNA repair pathways: one treatment to inhibit them all. Radiation Oncology. 2015;10:1-13
77. Ververis K, Hiong A, Karagiannis TC, Licciardi PV. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Journal of Biologics: Targets and Therapy. 2013;7:47-60
78. Gräff J, Tsai L. Histone acetylation: molecular mnemonics on the chromatin. Nature Reviews Neuroscience. 2013;14:97-111
79. Halkidou K, Gaughan L, Cook S, Leung H, Neal D, Robson C. Upregulation and Nuclear Recruitment of HDAC1 in Hormone Refractory Prostate Cancer. The Prostate. 2004;59:177-189
80. Zhang Y, Carr T, Dimtchev A, Zaer N, Dritschilo A, Jung M. Attenuated DNA Damage Repair by Trichostatin A through BRCA1 Suppression. Radiation Research. 2007;168:115-124
81. Zhu P, Martin E, Mengwasser J, Schlag P, Janssen KP, Göttlicher M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell. 2004;5:455-463
82. Choi JH, Kwon HJ, Yoon BI, Kim JH, Han SU, Joo HJ. et al. Expression Profile of Histone Deacetylase 1 in Gastric Cancer Tissues. Japanese Journal of Cancer Research. 2001;92:1300-1304
83. Ha K, Fiskus W, Choi DS, Bhaskara S, Cerchietti L, Devaraj SG, Shah B, Sharma S, Chang JC, Melnick AM, Hiebert S, Bhalla KN. Histone deacetylase inhibitor treatment induces 'BRCAness' and synergistic lethality with PARP inhibitor and cisplatin against human triple negative breast cancer cells. Oncotarget. 2014;5:5637-50
84. Liu Y, Martin-Trevino R, Shang L, Davis A, Wicha M, Liu S, Burness M. Histone deacetylase inhibitors sensitize cancer stem cells to PARP inhibitors in triple-negative breast cancer. Cancer Research. 2015;75:4226
85. Stecklein SR, Kumaraswamy E, Behbod F, Wang W, Chaguturu V, Harlan-Williams LM. et al. BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation. Proceedings of the National Academy of Sciences. 2012;109:13650-13655
86. Underhill C, Toulmonde M, Bonnefoi H. A review of PARP inhibitors: from bench to bedside. Annals of Oncology. 2010;22:268-279
87. Kachhap SK, Rosmus N, Collis SJ, Kortenhorst MS, Wissing MD, Hedayati M. et al. Downregulation of Homologous Recombination DNA Repair Genes by HDAC Inhibition in Prostate Cancer Is Mediated through the E2F1 Transcription Factor. PLoS ONE. 2010;5:11208-11220
88. Jasek E, Gajda M, Lis GJ, Jasińska M, Litwin JA. Combinatorial effects of PARP inhibitor PJ34 and histone deacetylase inhibitor vorinostat on leukemia cell lines. Anticancer Research. 2014;34:1849-56
89. Baldan F, Mio C, Allegri L, Puppin C, Russo D, Filetti S, Damante G. Synergy between HDAC and PARP Inhibitors on Proliferation of a Human Anaplastic Thyroid Cancer-Derived Cell Line. Int J Endocrinol. 2015;2015:978371
90. Olaparib and Hsp90 Inhibitor AT13387 in Treating Patients with Solid Tumors That Are Metastatic or Cannot Be Removed by Surgery or Recurrent Ovarian, Fallopian Tube, Primary Peritoneal, or Triple-Negative Breast Cancer. https://clinicaltrials.gov/ct2/show/NCT02898207
91. Bae J, Munshi A, Li C, Samur M, Prabhala R, Mitsiades C. et al. Heat Shock Protein 90 Is Critical for Regulation of Phenotype and Functional Activity of Human T Lymphocytes and NK Cells. The Journal of Immunology. 2013;190:1360-1371
92. Quanz M, Herbette A, Sayarath M, de Koning L, Dubois T, Sun JS. et al. Heat Shock Protein 90В (Hsp90В ) Is Phosphorylated in Response to DNA Damage and Accumulates in Repair Foci. Journal of Biological Chemistry. 2012;287:8803-8815
93. Zhang J. The role of BRCA1 in homologous recombination repair in response to replication stress: significance in tumorigenesis and cancer therapy. Cell & Bioscience. 2013;3:1-14
94. Jia J, Xu X, Liu F, Guo X, Zhang M, Lu M. et al. Identification, Design and Bio-Evaluation of Novel Hsp90 Inhibitors by Ligand-Based Virtual Screening. PLoS ONE. 2013;8:1-15
95. He H, Zatorska D, Kim J, Aguirre J, Llauger L, She Y. et al. Identification of potent water soluble purine-scaffold inhibitors of the heat shock protein 90. Journal of Medicinal Chemistry. 2006;49:381-390
96. Huang KH, Veal JM, Fadden RP, Rice JW, Eaves J, Strachan JP. et al. Discovery of novel 2-aminobenzamide inhibitors of heat shock protein 90 as potent, selective and orally active antitumor agents. Journal of Medicinal Chemistry. 2009;52:4288-4305
97. Fang Q, Inanc B, Schamus S, Wang XH, Wei L, Brown AR, Svilar D, Sugrue KF, Goellner EM, Zeng X, Yates NA, Lan L, Vens C, Sobol RW. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase β. Nature Communications. 2014;5:5513
98. Taldone T, Zatorska D, Patel PD, Zong H, Rodina A, Ahn JH. et al. Design, synthesis, and evaluation of small molecule Hsp90 probes. Bioorganic & Medicinal Chemistry. 2011;19:2603-2614
99. Panagiotis Konstantinopoulos, Sangeetha Palakurthi, Qing Zeng, Shan Zhou, Joyce F. Liu, Elena Ivanova, Cloud Paweletz, Naveen Kommajosyula, Alan D. D'Andrea, Geoffrey Shapiro, Ursula A. Matulonis. In vivo synergism between PARP-inhibitor olaparib and HSP90-inhibitor AT13387 in high grade serous ovarian cancer patient derived xenografts. Journal of Clinical Oncology. 2016;34(suppl):abstre17045
100. Wang Q, Su L, Liu N, Zhang L, Xu W, Fang H. Cyclin Dependent Kinase 1 Inhibitors: A Review of Recent Progress. Computer, Materials & Continua Journal. 2011;18:2025-2043
101. Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V. et al. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proceedings of the National Academy of Sciences. 2012;109:3826-3831
102. Kobayashi H. Hereditary breast and ovarian cancer susceptibility genes (Review). Oncology Reports. 2013;30:1019-1029
103. Wiedemeyer W, Beach J, Karlan B. Reversing Platinum Resistance in High-Grade Serous Ovarian Carcinoma: Targeting BRCA and the Homologous Recombination System. Frontiers in Oncology. 2014;4:1-11
104. Krejci L, Altmannova V, Spirek M, Zhao X. Homologous recombination and its regulation. Nucleic Acids Research. 2012;40:5795-5818
105. Qin L, Fan M, Candas D, Jiang G, Papadopoulos S, Tian L. et al. CDK1 Enhances Mitochondrial Bioenergetics for Radiation-Induced DNA Repair. Cell Reports. 2015;13:2056-2063
106. Krajewska M, Heijink AM, Bisselink YJ, Seinstra RI, Silljé HH, de Vries EG. et al. Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene. 2012;32:3001-3008
107. Yuan J, Eckerdt F, Bereiter-Hahn J, Kurunci-Csacsko E, Kaufmann M, Strebhardt K. Cooperative phosphorylation including the activity of polo-like kinase 1 regulates the subcellular localization of cyclin B1. Oncogene. 2002;21:8282-8292
108. Kojima K, Shimanuki M, Shikami M, Andreeff M, Nakakuma H. Cyclin-dependent kinase 1 inhibitor RO-3306 enhances p53-mediated Bax activation and mitochondrial apoptosis in AML. Cancer Science. 2009;100:1128-36
109. Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ, Moreau LA, Unitt C, Bronson RT, Thomas HD, Newell DR, D'Andrea AD, Curtin NJ, Wong KK, Shapiro GI. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nature Medicine. 2011;17:875-82
110. Hill R, Cautain B, de Pedro N, Link W. Targeting nucleocytoplasmic transport in cancer therapy. Oncotarget. 2014;5:11-28
111. Henderson B. The BRCA1 Breast Cancer Suppressor: Regulation of Transport, Dynamics, and Function at Multiple Subcellular Locations. Scientifica. 2012;2012:1-15
112. Mendonca J, Sharma A, Kim HS, Hammers H, Meeker A, De Marzo A. et al. Selective inhibitors of nuclear export (SINE) as novel therapeutics for prostate cancer. Oncotarget. 2014;5:6102-6112
113. Tan D, Bedard P, Kuruvilla J, Siu L, Razak A. Promising SINEs for Embargoing Nuclear-Cytoplasmic Export as an Anticancer Strategy. Cancer Discovery. 2014;4:527-537
114. Brodie K, Mok M, Henderson B. Characterization of BARD1 targeting and dynamics at the centrosome: The role of CRM1, BRCA1 and the Q564H mutation. Cellular Signalling. 2012;24:451-459
115. Fabbro M. BARD1 Induces BRCA1 Intranuclear Foci Formation by Increasing RING-dependent BRCA1 Nuclear Import and Inhibiting BRCA1 Nuclear Export. Journal of Biological Chemistry. 2002;277:21315-21324
116. Sato K, Hayami R, Wu W, Nishikawa T, Nishikawa H, Okuda Y. et al. Nucleophosmin/B23 is a candidate substrate for the BRCA1-BARD1 ubiquitin ligase. Journal of Biological Chemistry. 2004;279:30919-30922
117. Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nature Structural & Molecular Biology. 2001;8:833-837
118. Ozden O, Bishehsari F, Bauer J, Park SH, Jana A, Baik SH, Sporn JC, Staudacher JJ, Yazici C, Krett N, Jung B. Expression of an Oncogenic BARD1 Splice Variant Impairs Homologous Recombination and Predicts Response to PARP-1 Inhibitor Therapy in Colon Cancer. Scientific Reports. 2016;6:26273
119. Jiang J, Yang ES, Jiang G, Nowsheen S, Wang H, Wang T. et al. p53-dependent BRCA1 nuclear export controls cellular susceptibility to DNA damage. Cancer Research. 2011;71:5546-57
120. Wang H, Yang ES, Jiang J, Nowsheen S, Xia F. DNA damage-induced cytotoxicity is dissociated from BRCA1's DNA repair function but is dependent on its cytosolic accumulation. Cancer Research. 2010;70:6258-67
121. Ireno IC, Wiehe RS, Stahl AI, Hampp S, Aydin S, Troester MA. et al. Modulation of the poly (ADP-ribose) polymerase inhibitor response and DNA recombination in breast cancer cells by drugs affecting endogenous wild-type p53. Carcinogenesis. 2014;35:2273-82
122. Sebastian S, Chesi M, Braggio E, Bergsagel L, Stewart K, Ahmann G. et al. PARP Inhibition (OLAPARIB) Enhance Melphalan and Nutlin-3a Sensitivity in TP53 Positive Multiple Myeloma. Blood. 2012;120:1846
123. Yalon M, Tuval-Kochen L, Castel D, Moshe I, Mazal I, Cohen O. et al. Overcoming Resistance of Cancer Cells to PARP-1 Inhibitors with Three Different Drug Combinations. PLoS One. 2016;11:e0155711
124. Issaeva N, Thomas HD, Djureinovic T, Jaspers JE, Stoimenov I, Kyle S. et al. 6-thioguanine selectively kills BRCA2-defective tumors and overcomes PARP inhibitor resistance. Cancer Research. 2010;70:6268-6276
Author contact
![]() Corresponding author: Dr. Yingqiu Xie, Department of Biology, Nazarbayev University, School of Science and Technology, 53 Kabanbay Batyr Ave., Astana, 010000, Republic of Kazakhstan. Phone: +7 7172 704686; Email: xieautumnuscom.
Corresponding author: Dr. Yingqiu Xie, Department of Biology, Nazarbayev University, School of Science and Technology, 53 Kabanbay Batyr Ave., Astana, 010000, Republic of Kazakhstan. Phone: +7 7172 704686; Email: xieautumnuscom.