Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(4):413-425. doi:10.7150/ijbs.17508 This issue Cite
Research Paper
MiR-696 Regulates C2C12 Cell Proliferation and Differentiation by Targeting CNTFRα
Han Wang1, Lei Shi1, Tingting Liang1, BinBin Wang1, WangJun Wu1, Guosheng Su2, Julong Wei1, Pinghua Li1 ![]() , Ruihua Huang1
, Ruihua Huang1 ![]()
1. Institute of Swine Science, Nanjing Agricultural University, Nanjing, 210095, China;
2. Center for Quantitative Genetics and Genomics, Department of Molecular Biology and Genetics, Aarhus University, 8830 Tjele, Denmark.
Received 2016-9-7; Accepted 2017-1-6; Published 2017-3-11
Abstract

Micro-696 (miR-696) has been previously known as an exercise related miRNA, which has a profound role in fatty acid oxidation and mitochondrial biogenesis of skeletal muscle. However, its role in skeletal myoblast proliferation and differentiation is still unclear. In this study, we found that miR-696 expressed highly in skeletal muscle and reduced during C2C12 myoblasts differentiation. MiR-696 overexpression repressed C2C12 myoblast proliferation and myofiber formation, while knockdown of endogenous miR-696 expression showed opposite results. During myogenesis, we observed an inversed expression pattern between miR-696 and CNTFRα in vitro, and demonstrated that miR-696 could specifically target CNTFRα and repress the expression of CNTFRα. Additionally, we further found that knockdown of CNTFRα suppressed the proliferation and differentiation of C2C12 cells. Taking all things together, we propose a novel insight that miR-696 down-regulates C2C12 cell myogenesis by inhibiting CNTFRα expression.
Keywords: MiR-696, CNTFRα, myoblast proliferation and differentiation.
Introduction
The development of skeletal muscle is a complicated, multistage process. First paraxial mesoderm differentiates into somites, then the latter distribute into dermomyotome. From the dermomyotome, the myogenic progenitor cells delaminate and finally activate the differentiation programme involving myoblast proliferation, differentiation and fusion [1, 2]. Endogenous genetic regulation, such as myogenic regulatory factors (MRFs) and small RNAs, exerts the most important controlling effects on myogenesis [3].
MicroRNAs (MiRNAs) are a kind of small noncoding RNAs (~22 nt in length), acting by inducing gene silencing or degradation in animals and plants [4, 5]. A series of studies have shown that miRNA is involved in various developmental and physiological processes, including cell growth and apoptosis, virus defense, hematopoiesis, organ formation, tumorigenesis, fat metabolism, and so on [6-8]. Similarly, in diverse aspects of skeletal myogenesis, extensive miRNAs have also been confirmed as important regulators [9, 10]. One class of miRNAs is muscle-specific miRNAs (myomiRs), such as miR-206, miR-1 and miR-133. Sempere et al. [11] firstly discovered high expression of miRNAs in skeletal muscle in mouse and human. Since then, lots of researchers began to study the unique function of myomiRs in regulating myoblast proliferation, differentiation and muscle fiber type transition [12-14].
Beyond those myomiRs mentioned above, many ubiquitously expressed miRNAs, such as miR-30 [15], miR-21 [16], miR-128 [17], miR-151 [18] and miR-203 [19], have been proved to have essential roles in skeletal muscle development too. Since the biological process of muscle development is complicated, which often needs multiple miRNAs to act together, it requires future studies to investigate individual roles of other vital miRNAs in skeletal myogenesis.
MiR-696 is a miRNA that is remarkable differentially expressed in gastrocnemius under exercise and immobilization intervention [20]. The function of miR-696 in skeletal muscle has been implicated in energy metabolism, mitochondrial synthesis, and in ectopic deposition of lipids which is induced by resistin [21]. On the other hand, CNTFRα is known as a CNTF-special binding component and expressed in various organs and tissues [22, 23]. The main role of CNTFRα is maintaining neuronal function [24]. Enhanced CNTFRα expression could promote the development of sensory neuron or regenerate injured spinal motor neurons [25, 26]. So far, many studies have found CNTFRα highly expressed in skeletal muscle [27, 28]. Hindlimb suspension in rats would increase the expression of CNTFRα when compared to unrestricted rats, but exogenously CNTF could prevent such changes [29, 30]. Meanwhile, it has also been found that polymorphisms in the CNTFRα gene could be associated with exercise performance in humans [31-33]. CNTF receptor signaling, which is composed of CNTFRα, LIFRβ and gp130, has been discovered to be associated with fatty-acid metabolism, muscle maintenance or regeneration and motoneuron development [34-36]. These functions are very similar to those of miR-696. In addition, exogenous CNTF has been confirmed to promote proliferation and inhibit differentiation in myoblasts [37, 38]. Through bioinformatics prediction, we hypothesized that miR-696 could inhibit myoblast proliferation and differentiation by targeting CNTFRα, thus regulating the skeletal muscle myogenesis. Therefore, we set up this experiment to verify the hypothesis. In the experiment, CNTFRα as the target of miR-696 was tested by small interfering RNA (siRNA) targeting CNTFRα in vitro. To our knowledge, our study is the first systematical investigation on the miR-696 correlation with myoblast proliferation and differentiation in myogenesis.
Materials and Methods
Animals and Cell culture
Four healthy male Balb/c mice (age, 6 weeks) were provided by Qinglongshan Laboratory Animal Company (Nanjing, China). Three samples were collected from seven tissues of each mouse. Samples were snap-frozen for real-time Q-PCR by liquid nitrogen. C2C12 cells were cultured in growth medium (DMEM; Hyclone, USA), containing 10% fetal bovine serum with 1% penicillin-streptomycin (Gibco, USA), in 5% CO2 atmosphere at 37 °C. Differentiation medium (DM), consisting of 1% penicillin-streptomycin and 2% horse serum (Gibco, USA), replaced growth medium when cell density reached 80% to 90% as described by Wei et al [39], to induce differentiation for 2, 4 and 6 days.
Plasmid construct
Fragments of the mouse CNTFRα 3'-UTR containing the miR-696 target sequence were chemically synthesized by Generay Biotech Co., Ltd., (Shanghai, China) and by using the SacI and XbaI restriction sites. And then they were cloned into the pmirGLO dual-luciferase vector (Promega). The point mutant CNTFRα plasmid was generated by replacing a 3-base substitution in the putative seed-matched sequences of CNTFRα 3'-UTR, which was also synthesized by Generay Biotech Co., Ltd., (Shanghai, China). All plasmids were verified through DNA sequencing [19].
RNA oligonucleotides and transfection
The miR-696 mimics, mimic NC duplexes, miR-696 inhibitors and inhibitor NC were synthesized by GenePharma (Shanghai, China). SiRNA against mouse CNTFRα and a NC-nonspecific duplex control were also from GenePharma (GenePharma, Suzhou, China). After the inhibitor was synthesized, the company blasted the sequence to the nucleotide database to make sure the inhibitor was non-homologous and specificity. Transfection was performed following the manufacturer's instructions of Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). All oligonucleotides are shown in Supplementary Table S1.
EdU assay
After transfection for 6 h, C2C12 myoblast cells were cultured in fresh growth medium which contained 10 mM EdU for 24 h [18]. Then, the cells were fixed, permeabilised, and stained following the manufacturer's instruction (EdU Apollo567 kit, RiboBio, Guangzhou, China). The EdU-stained cells were observed using the NIKON fluorescent microscope. Images were randomly selected from three duplicates of each treatment. Finally, both EdU-positive nuclei and total nuclei were counted.
Flow cytometry analysis of the cell cycle
C2C12 cells were cultured in GM for 24 h after transfection. Then they were harvested and washed in PBS for three times, fixed in 70% (v/v) ethanol at -20 °C overnight, and washed with PBS again. Thereafter, the cell pellet was added 50 mg/mL PI solution (Sigma Life Science, USA) and incubated for 30min at 4 °C away from light. Samples were detected using a BD flow cytometry (Becton Dickinson, USA). Cell cycle percentage was analyzed by ModFit software (Verity Software House, USA). The proliferative index was calculated as the percentage of proliferating cells from 20,000 cells [17].
Dual-luciferase reporter assay
In 12-well plates, C2C12 and HEK293T cell lines were co-transfected with 50 nM miR-696 mimics or NC with 1 μg pmirGLO-CNTFRα luciferase vector (either wild-type or mutant plasmid) following the manual of Lipofectamine 2000 reagent. Twenty-four hours after transfection, cells were harvested. Following the manufacturer's instruction (Promega), the dual-luciferase assays were performed. The relative fluorescence intensity for each sample was calculated by normalizing the signal value of renilla luciferase to firefly luciferase, and then compared with the control.
Immunofluorescence analysis
Immunofluorescence was performed after transfection and differentiation, using standard procedures as described previously [40]. Briefly, cells cultured in 12-well plates were fixed for 15 min after they were washed twice with pre cooling PBS. Thereafter the cells were permeabilized with 0.25% Triton X-100, then blocked at 4 °C overnight with fetal bovine serum (diluted in PBS). Afterwards, cells were incubated with primary antibodies (MYH, H-300, Santa Cruz Biotechnology, USA) for 1 h at room temperature, and incubated with the secondary antibody (the FITC-labeled IgG, Boster, China) at room temperature for another 1 h. The cell nuclei were stained using DAPI (Invitrogen, USA) away from light. Samples were photographed using the fluorescent microscope (Nikon, Japan). At least three replicates of each sample were calculated for fusion index by using ImageJ [19].
Quantitative real-time RT-PCR
Total RNA extraction, integrity detection, reverse transcription and real-time RT-PCR were performed as described by Shi, L et al [17]. Differential expression analysis was calculated by 2-ΔΔCt method [41]. Primer sequences were listed in Supplementary Table S2.
Western blot analysis
Proteins were generated and used in Western blot analysis as reported in previous study [42]. Cells were collected and treated by RIPA buffer with 1% PMSF (Beyotime, China) on ice for 30 min to extract total proteins. Proteins were then separated on 12% SDS-PAGE, afterwards transferred to PVDF membranes (Millipore, USA). MyHC (Santa Cruz Biotechnology, USA; 1:1000 dilution), CNTFRα, MyoG and β-actin (Abcam, USA; 1:500 dilution) were used as primary antibodies, and HRP-labeled anti-rabbit/mouse IgG (Beyotime, China; 1:5000 dilution) was used as a secondary antibody. Blots were visualized with a commercial ECL reagent (Advansta, USA), tested by a chemiluminesence detection system (Amersham, USA) and quantified by ImageJ software.
Statistical analysis
All results were showed as the mean±S.E.M. Each treatment was repeated for three times. Unpaired Student's t-test was performed to test statistical significance using SPSS 20.0 software. Two-tailed t tests were used in the analysis. Our original hypothesis was that miR-696 can affect myoblast proliferation and differentiation while our null hypothesis is that miR-696 has no effect on muscle cell proliferation or differentiation. P < 0.05 was considered as significant and P < 0.01 as highly significant.
Results
Tissue distribution of miR-696 and its expression pattern during C2C12 myoblast proliferation and differentiation
To investigate the expression profile of miR-696 in various tissues, qPCR assay was performed. Results indicated that miR-696 not only expressed the highest level in fat but also had a relatively high expression in liver, kidney and skeletal muscle (Figure 1A). Furthermore, we detected the expression of miR-696 in different types of muscles. We found that the expression level of miR-696 in soleus (SOL) muscles was lower (P<0.05) than that in extensor digitorum longus (EDL) (Figure 1B).
Tissue distribution of miR-696 and its expression pattern during C2C12 myoblast differentiation. (A) Relative expression of miR-696 in different mouse tissues. (B) Expression of miR-696 in mouse EDL and SOL muscles. (C) Morphological images of C2C12 myoblasts cultured in GM or in DM for 2, 4 and 6 days. (D) The mRNA level of MyHC and MyoG in 0, 2, 4, and 6 days of cell differentiation. The fold change was relative to day 0 of GM expression. GAPDH was used as the reference gene for Q-PCR. (E) The protein level of MyHC and MyoG in 0, 2, 4, and 6 days of cell differentiation. The fold change was relative to day 0 of GM expression.β-actin as controls for western blotting. (F) Expression of miR-696 during proliferation. The fold change was relative to 30% cell confluence. (G). Expression of miR-696 in C2C12 cells differentiated for 0, 2, 4, and 6 days. The fold change was relative to day 0 of GM expression. U6 was used as the reference gene. Results are expressed as mean ± S.E.M. (n = 3). *P<0.05; **P<0.01.
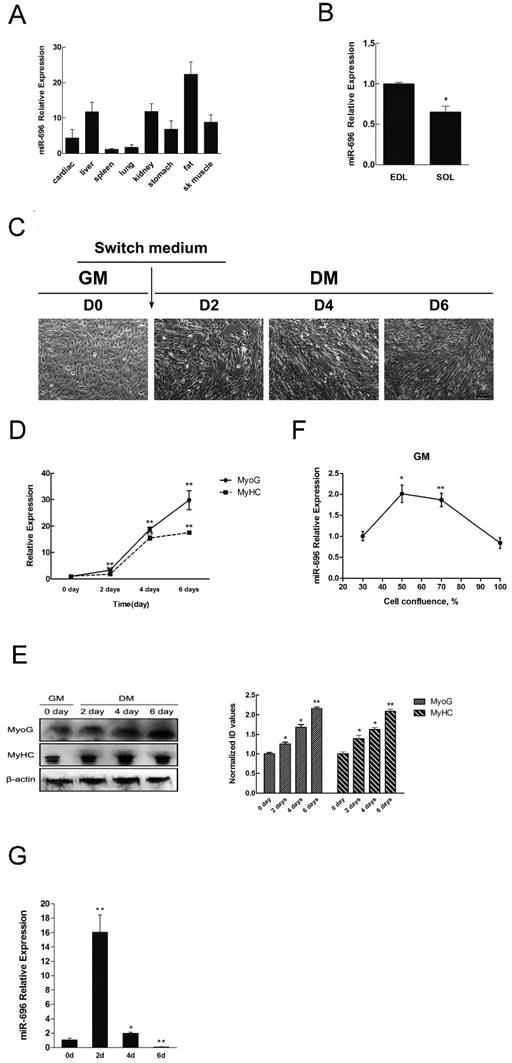
Next, we set up a C2C12 cells model to detect the expression of miR-696 during myoblast proliferation and differentiation. Typical myofibers were clearly observed after differentiation induction (Figure 1C). Through analysis of MyHC and MyoG (two markers of myogenic differentiation) in both mRNA and protein level, we further confirmed that the in vitro model of differentiation was successfully established (Figure 1D and E). As shown in Figure 1F, miR-696 levels decreased progressively during proliferation. It also declined from day 2 (D2) to day 6 (D6) during differentiation (Figure 1G). However, the expression of miR-696 on 2 day of DM was dramatically higher than 0 days of GM. Altogether, these results indicated that miR-696 might have potential roles in skeletal myogenesis.
MiR-696 inhibits C2C12 myoblast proliferation
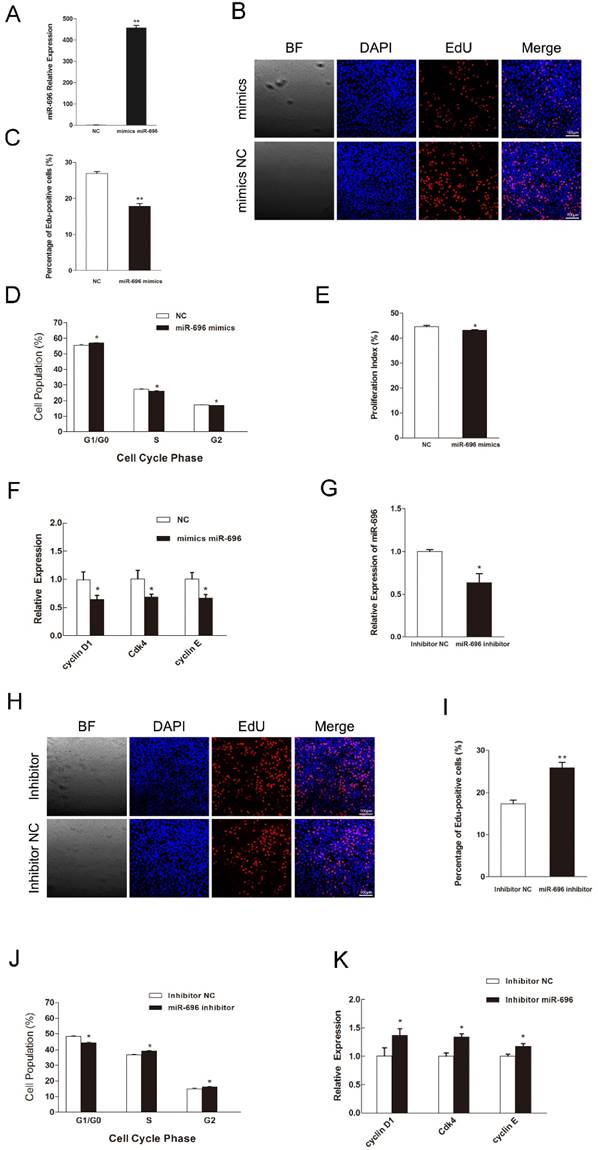
To explore the function of miR-696 overexpression in myoblast proliferation, synthetic miR-696 mimics or negative control (NC) was transfected into myoblasts cultured in GM (Figure 2A). EdU staining assay indicated that miR-696-transfected cells had less proportion of EdU-positive cells than the control cells at 24 h post-transfection (Figures 2B and C). Besides, analyzing the phase of cell cycle elucidated that miR-696 mimics transfection could significantly block C2C12 myoblasts in the G0/G1 period and have a decrease in the proliferation index, as compared to negative control (NC) (Figures 2D and E). Moreover, the expression of cell cycle activators [43], like cyclin D1, cyclin E and Cdk4, were notably lower in the miR-696 overexpression group than in the control group at 24 h after transfection (Figure 2F). We also conducted the loss-of-function study in vitro by using an inhibitor of miR-696 (Figure 2G). The result further explored the role of miR-696 in myoblast proliferation. Both the EdU-positive cells and the proportion of cells in S and G2 phase increased in miR-696 inhibitor group compared with the inhibitor NC group (Figure 2H-2J). In addition, the mRNA expression of cyclin D1, cyclin E and Cdk4 also rose obviously (Figure 2K). Collectively, these data elucidated that miR-696 could repress myoblast proliferation.
MiR-696 represses the proliferation of C2C12 cells. (A) The expression of miR-696 was detected using qPCR in myoblast transfected with miR-696 mimics or NC. (B) After transfection with miR-696 mimics or NC for 24 h, cells were fixed for EdU (red). The “large things” in Fig 2B was not a cell but some water-drops. It might because of the lid wasn't on tight caused by our carelessness. Scale bar = 100 μm. (C) The proportion of EdU-positive cells were presented. (D) C2C12 cells were collected for PI flow cytometry. (E) The proliferation index was calculated. (F) Expression of cell cycle related genes at 24 h post-transfection. (G) miR-696 expression was detected in myoblasts after transfected with miR-696 inhibitor or inhibitor NC. (H) After transfection with miR-696 inhibitor or inhibitor NC for 24 h, proliferating C2C12 cells were fixed for EdU (red). Scale bar = 100 μm. (I) The proportion of EdU-positive cells were compared between miR-696 inhibitor group and inhibitor-NC group. (J) Cell cycle distribution was detected by PI flow cytometry. (K) Expression of cell cycle related genes at 24 h post-transfection. Results are expressed as mean ± S.E.M. (n = 3). *P<0.05; **P<0.01.
MiR-696 represses the differentiation of C2C12 cells
Since myogenic differentiation is another critical biological process in skeletal myogenesis, we examined the function of miR-696 in myoblast differentiation. The expression of miR-696 was promoted by transfecting miR-696 mimics into C2C12 myoblasts. After transfection, cell differentiation was induced by substituting the growth medium with the differentiation medium. Real-time qPCR showed that the overexpression of miR-696 was significantly observed (Figure 3A). Both mRNA and protein expression of MyHC and MyoG, two major marker genes of myogenic differentiation, were dramatically lower in miR-696 mimics groups compared to the control groups at 72 h of differentiation (Figure 3B and C).
MiR-696 inhibits myogenic differentiation of C2C12 cells. (A) The expression of miR-696 were detected by qPCR in myoblast transfected with miR-696 mimics or NC duplexes at day 3 of differentiation medium (DM). (B) After transfection with miR-696 mimics or NC at GM and collected at day 3 of DM, the MyHC and MyoG mRNA expression were determined. (C) The MyHC and MyoG protein expression were determined at 3 days of DM. (D) MyHC (green) was detected with Immunofluorescence after transfection with miR-696 mimics or NC duplexes at day 3 of DM. Scale bar = 50 μm. (E) The expression of miR-696 were detected in myoblast after transfected with inhibitor miR-696 or inhibitor NC at day 3s of differentiation medium (DM). (F) After transfection with inhibitor miR-696 or inhibitor NC at GM and collected at day 3 of DM, MyHC and MyoG mRNA expression were determined. (G) The MyHC and MyoG protein expression were determined at day 3 of DM. (H) MyHC (green) was detected with Immunofluorescence after day 3 of miR-696 inhibitor or inhibitor NC transfection at DM. Scale bar = 50 μm. Results are expressed as mean ± S.E.M. (n = 3). *P<0.05; **P<0.01.
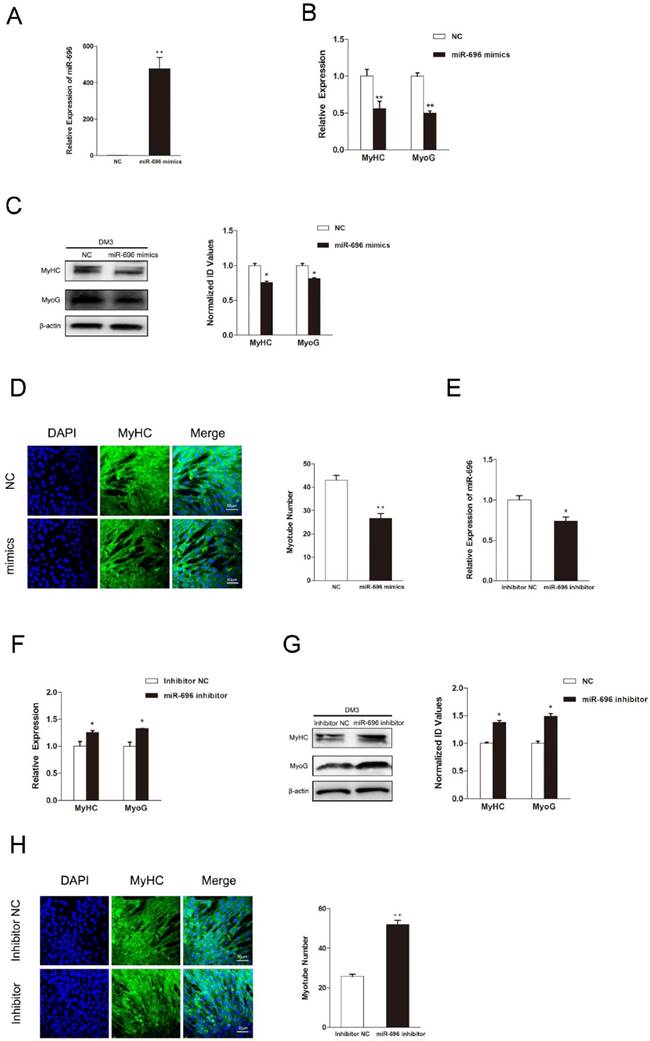
In addition, the immunofluorescence of MyHC also indicated that the miR-696-transfected myoblasts had less cells forming into myotubes than the control (Figure 3D). Furthermore, the miR-696 inhibitor was also transfected into C2C12 cells to demonstrate the loss-of-function effects of miR-696 during myoblast differentiation. As shown in Figure 3E, miR-696 expression decreased at day 3 of DM. Compared with the inhibitor NC group, a significant increase was observed in the expression of MyHC and MyoG in the miR-696 inhibitor group (Figure 3F and G). Meanwhile, the myotube number in cells transfected with miR-696 inhibitor was greater than that of control (Figure 3H). In summary, these results illustrated that miR-696 could repress myoblast differentiation.
CNTFRα is a direct target of miR-696
Despite the important role of miR-696 in skeletal myogenesis, the underlying regulatory mechanism is still unknown. In order to clarify the mechanism, we first predicted the regulatory target genes of miR-696 by several online prediction programs: TargetScan, PicTar and miRanda. Combining these procedures together yielded hundreds of putative targets for miR-696. Among them, CNTFRα was identified as a prime target because its 3'UTR had a target site for miR-696, which was a highly conserved complementarity in the seed region (Figure 4A). To verify whether CNTFRα was the direct target gene of miR-696, dual luciferase reporter assays were performed. We used the pmirGLO vector to generate a plasmid by inserting a part of the CNTFRα 3'UTRs which contained the putative miR-696 target site into the 3'UTR of the firefly luciferase gene (luc2) (Figure 4B). The firefly luciferase gene was used as a reporter gene. Less expression of firefly luciferase meant that the exogenous miRNA could combine with the cloned target sequence. Moreover, renilla luciferase gene (hRluc-neo) was treated as the control gene for normalization. Likewise, a dual luciferase with 3-base substitutions in the binding site was cloned (Figure 4B). When co-transfected miR-696 mimics or NC with the plasmid pCNTFRα into C2C12 myoblasts and HEK293T cells, we found miR-696 overexpression efficiently repressed the luciferase activity. However, compared with the mutant and control groups, no assessable inhibitory effect could be observed on the luciferase activity (Figure 4C). These results suggested that miR-696 could specifically target the expression of CNTFRα.
CNTFRα is a direct target of miR-696. (A) Sequence of miR-696 and its predicted binding region in CNTFRα 3'UTR (red). (B) The sketch map of the dual-luciferase reporter vector pmirGLO. The putative miR-696 target site of the CNTFRα 3'UTRs and mutation target site were inserted into the 3' end of the firefly luciferase gene (luc2). The expression of the Renilla luciferase (hRluc-neo) was treated as an internal normalization control. (C) C2C12 and HEK293T cells transfected with miR-696 mimics or NC were co-transfected with the pCNTFRα vector or mutant dual-luciferase vector. The relative luciferase activity was assayed 24 h later. (D) The mRNA expression of CNTFRα in C2C12 cells during proliferation. (E) The protein expression of CNTFRα in C2C12 cells during proliferation. (F) The mRNA expression of CNTFRα in 0, 2, 4, and 6 days of cell differentiation. The fold change was relative to day 0 of GM expression. GAPDH was used as the reference gene in qPCR. (G) The protein expression of CNTFRα in in 0, 2, 4, and 6 days of cell differentiation. β-actin was treated as control protein in western blotting assay. (H) After transfection for 24 h with miR-696 mimics, inhibitor miR-696, NC, or inhibitor NC for 24h, the mRNA expression of CNTFRα was detected by qPCR in proliferating C2C12 cells. (I) The protein expression of CNTFRα after transfection for 24 h with miR-696 mimics, inhibitor miR-696, NC, or inhibitor NC for 24h was detected by western blot in proliferating C2C12 cells. (J) The mRNA level of CNTFRα was determined by qPCR in myoblast transfected with miR-696 mimics, inhibitor miR-696, NC, or inhibitor NC at day 3 of differentiation medium (DM). (K) The protein level of CNTFRα in myoblast transfected with miR-696 mimics, inhibitor miR-696, NC, or inhibitor NC at day 3 of differentiation medium (DM). Results are expressed as mean ± S.E.M. (n = 3). *P<0.05; **P<0.01.
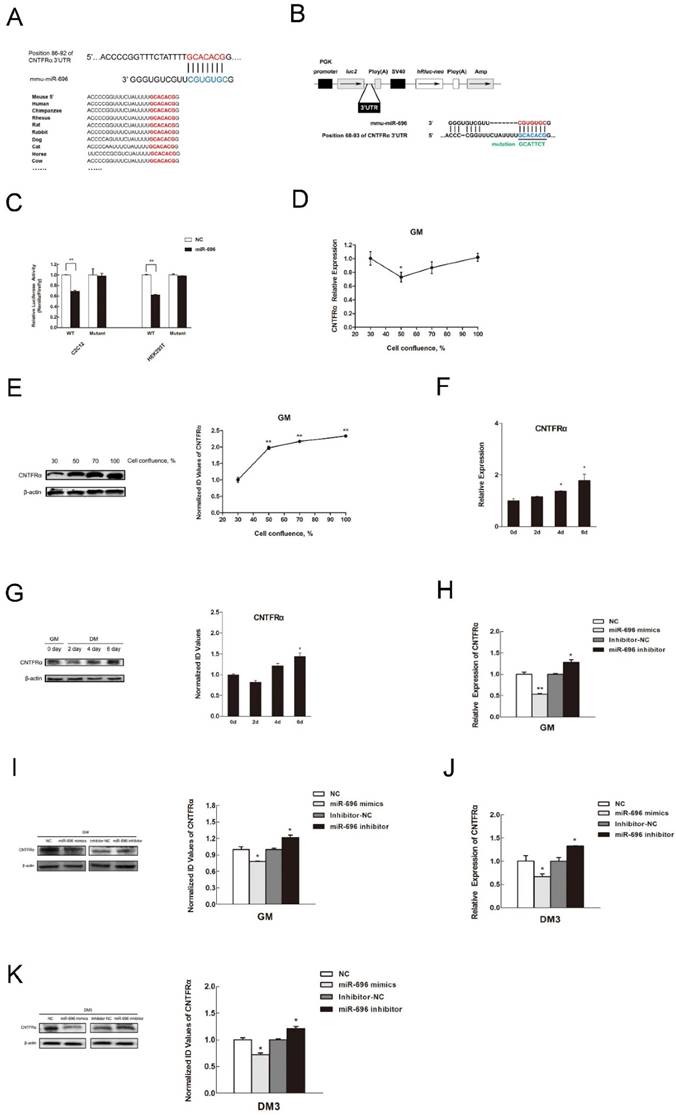
Next, we considered the role of CNTFRα in the regulation of miR-696 during myoblast proliferation and differentiation. During C2C12 cell proliferation, CNTFRα mRNA and protein level rose during cell confluence from 50% to 100%. However, from 30% to 50% cell confluence, the protein expression decreased (Figure 4D and E). Moreover, CNTFRα expression also ascended during cell differentiation from day 2 to day 6 (Figure 4F and G). To an extent, such patterns of expression were contrary to those of miR-696 in vitro. Over-expression of miR-696 in GM leads to noteworthy reduction of CNTFRα mRNA and protein expression, while the mRNA and protein level of CNTFRα obviously rose when endogenous miR-696 was suppressed (Figure 4H and I). Afterwards, we transfected miR-696 mimics into myoblasts, then induced for myogenic differentiation for 3 days. As shown in Figure 4J and K, both the mRNA and protein levels of CNTFRα were significantly repressed. Nevertheless, the inhibitor of miR-696 in DM up-regulated CNTFRα expression. Therefore, we argued that miR-696 had a critical role in suppressing CNTFRα expression during C2C12 myoblast proliferation and differentiation.
CNTFRα has a positive effect on myogenic proliferation and differentiation
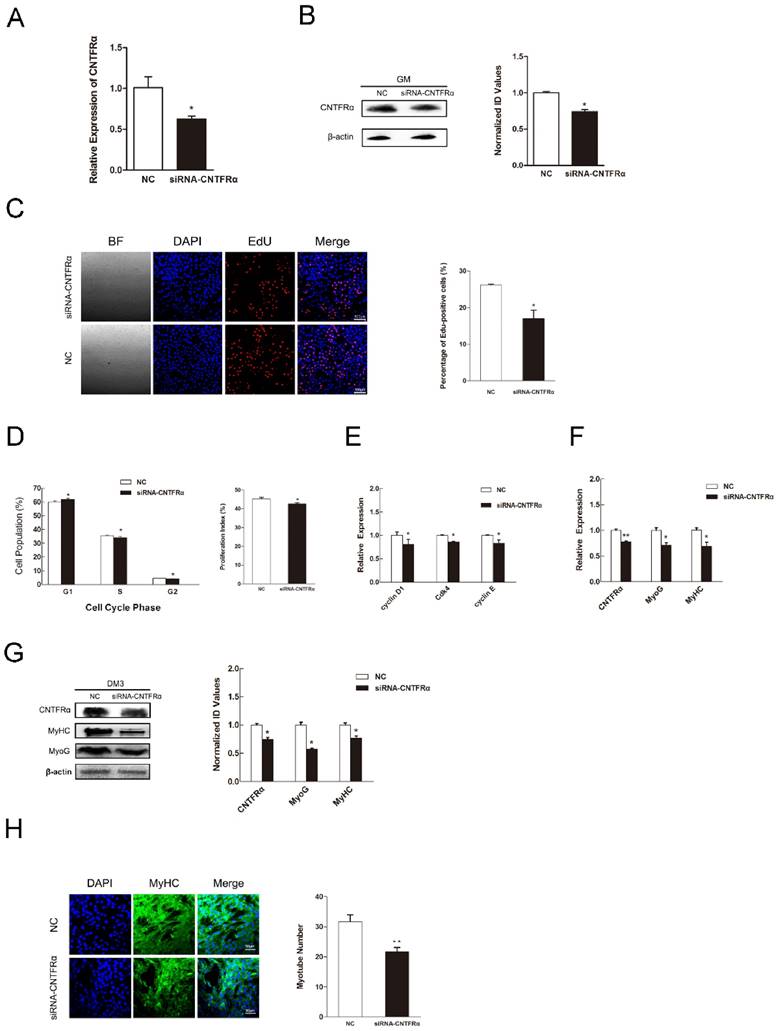
We used an anti-CNTFRα siRNA to knockdown CNTFRα expression to address its function in myogenic proliferation and differentiation. According to the designs of previous studies, we only took 24 h post-transfection as the time point of our research [44-47]. As a result, in C2C12 myoblasts, the anti- CNTFRα siRNA well suppressed the expression of CNTFRα mRNA and protein at 24 h post-transfection (Figure 5A and 5B). The siRNA group had lower proportion of proliferation cells than the NC group according to the results of EdU assay (Figure 5C). Down-regulated CNTFRα expression arrested cells in the G0/G1 period and decreased proliferation index (Figure 5D). Meanwhile, the mRNA expression of cell cycle related genes was also declined (Figure 5E). In addition, transfection of siCNTFRα during myoblast differentiation also substantially descended the expression of CNTFRα (Figure 5F and G). Two myogenic differentiation marker genes, MyHC and MyoG mRNA and protein level, were both significantly down-regulated at 3 day of DM (Figure 5F and G). And, apparent reduction was observed in the number of myotubes when CNTFRα was inhibited (Figure 5H). These outcomes were similar to miR-696 overexpression in C2C12 muscle cells. Thus, these results indicated that CNTFRα could promote myoblast proliferation and differentiation.
Down-regulation of CNTFRα inhibited myoblast proliferation and differentiation. (A) The expression of CNTFRα mRNA was detected at 24 h post-transfection in GM. (B) The expression of CNTFRα protein was detected at 24 h post-transfection in GM. (C) After transfection with siCNTFRα or NC for 24h, cells were fixed for EdU (red). Scale bar = 100 μm. The proportion of EdU-positive cells were presented. (D) The cell cycle distribution was detected by PI flow cytometry. The proliferation index was calculated. (E) Expression of cell cycle related genes at 24 h post-transfection. (F) The mRNA level of CNTFRα, MyHC and MyoG were detected by qPCR in myoblast transfected with siCNTFRα or NC at day 3 of differentiation medium (DM). (G) The protein level of CNTFRα, MyHC and MyoG were detected by western blotting in myoblast transfected with siCNTFRα or NC at day 3 of differentiation medium (DM). (H) MyHC (green) was detected with Immunofluorescence after 3 days of siCNTFRα or NC transfection at DM. Scale bar = 50 μm. Results are expressed as mean ± S.E.M. (n = 3). *P<0.05; **P<0.01.
Discussion
MiR-696 was previously known as an exercise related miRNA, which could have a function of mitochondrial biogenesis and fatty acid oxidation in skeletal muscle [20]. It has been reported that miR-696 could increase the TG content and diminish the mitochondrial content under resistin in C2C12 myoblast [21]. In present study, we first found that miR-696 was widely expressed in several tissues including mouse skeletal muscles (Figure 1A). High expression of miR-696 in skeletal muscle suggests that it may play a role in skeletal muscle myogenesis. Previous research has shown that anti-miR-696 expression could lead to a modest increase in slow MyHC [48]. However, no studies have so far been performed to determine the function of miR-696 in skeletal myoblast proliferation and its mechanism in regulating muscle cell differentiation.
In present study, we discovered the expression of miR-696 decreased during C2C12 cell proliferation and differentiation (Figure 1F and G), which indicated that miR-696 might have a negative function in skeletal muscle myogenesis. Through Edu and cell cycle test, we testified that miR-696 could repress the proliferation of C2C12 cells (Figure 2B-2E and Figure 2H-2J). According to the flow cytometry results, despite the tiny difference between miR-696 mimics group and NC group, it could be of biological significance. The similar results to our study were also reported in several previous studies [17, 49]. Besides, the genes involved in cell cycle regulation, such as cyclin D1, cyclin E and Cdk4, were down-regulated or up-regulated when miR-696 was over-expressed or inhibited (Figure 2F and K). Moreover, some studies showed that a decreased expression of those cell proliferation related genes induced cell proliferation repression by arresting muscle cells in the G0/G1 stage [43, 50]. Therefore, it can be concluded that miR-696 could play a negative role in muscle cell proliferation.
Previous researches indicated that many miRNAs could influence both the proliferation and differentiation process of skeletal muscle. Many studies also reported that some miRNAs had opposite roles in the process of proliferation and differentiation, such as miR-133 and miR-29, which repressed myoblast proliferation and promoted cell differentiation [39, 51]. On the other hand, some miRNAs were confirmed to have the same effect on the muscle cell proliferation and differentiation, such as miR-214 and miR-203 [19, 52].
Moreover, previous studies reported that skeletal muscle differentiation was usually along with the expression of myogenic marker genes, such as MyoG and MyHC [53]. In the current research, expression of these genes declined after miR-696 overexpression and rose after miR-696 inhibition respectively, which showed that miRNA-696 could also inhibit the differentiation of C2C12 cells. However, the underlying mechanisms of how miR-696 functions in skeletal myogenesis remains unclear.
Our study showed that the mRNA expression of CNTFRα was opposite to miR-696 expression during myoblast proliferation and differentiation, and the CNTFRα mRNA and protein levels were different during early period of cell proliferation and differentiation (Figure 4D-4G). These results indicated that there might exist the factors that could regulate CNTFRα expression at transcription level during these periods. Through bioinformatics analysis and dual-luciferase assay, we further certified that CNTFRα could be a direct target of miR-696. Additionally, overexpression or inhibition of miR-696 showed that miR-696 significantly inhibited the expression of CNTFRα in both myoblast proliferation and differentiation. Furthermore mimicking the overexpression of miR-696 by knockdown of CNTFRα expression inhibited the proliferation and differentiation of myoblasts, which supported our hypothesis.
It has been well known that CNTF is abundantly expressed in the cytoplasm of Schwann cell but seldom expressed in skeletal muscle or embryonic cells [54, 55]. The mature protein of CNTF lacks a signal sequence, thus it can't stimulate classical CNTF signals through autocrine modes in skeletal muscle cells in vitro [56-58]. Therefore, we propose that miR-696 targeting CNTFRα for regulating C2C12 myogenesis must be coordinated by other signaling pathways. An earlier study reported that CNTFRα knockout mice died perinatal with reduced motor neurons populations, while mice lacking CNTF showed poor muscle strength [59]. These indicate that there exists a second, developmentally important, CNTF-like ligand.
Previous works have shown that CLC, a new member of the CNTF/LIF family, could be induced by CNTFR, and activate gp130, LIFR and STAT3 signaling components on the cell surface [60]. Unlike CNTF, CLC has a leader sequence, suggesting it can be secreted via the classical secretory pathway within the cell [61]. A previous study reported that when CLC co-expressed with the soluble cytokine receptor CLF, the CLC/CLF could activate the tripartite CNTF receptor complex, but only acted on cells expressing functional CNTF receptors [62]. It was also observed that in muscle cells, CLC was co-secreted either with sCNTFR or CLF [63]. Hence, we speculate that CLC may be the possible ligand replaces CNTF to combine with CNTFRα on regulating myogenesis in C2C12 vitro model.
In our study, myoblast proliferation was inhibited when CNTFRα was interfered. In addition, the expressions of several cell proliferation related genes (cyclin D1, cyclin E and Cdk4) were also decreased, arresting C2C12 cells in the G0/G1 stage. It has been reported that the CLC/CLF composite cytokine combined with the membrane form of CNTFR could cause an obviously increase in the phosphorylation of PI3-kinase/AKT and ERK1/2 MAPK pathway in several human cell lines [62, 64]. In skeletal muscle cells, these two pathways are considered as the major signaling pathway regulating the cell proliferation [65, 66]. Either inhibition of the PI3K/Akt or ERK1/2 MAPK pathway significantly down-regulated the activity of cyclin D1, cyclin E and Cdk4, causing cell cycle retention in G1/S phase [67-71]. Taken together, we suppose miR-696 may suppress the proliferation of C2C12 myoblasts by blocking the CNTFRα receptor complex mediated PI3-kinase/AKT and ERK1/2 MAPK pathway activation.
Previous work from many laboratories indicates that activation of ERK1/2 regulates myogenic differentiation negatively and induces the repression of MyoG and MyHC expression level [72-74]. Contrarily, blockade of the PI3K/Akt pathway prevented myogenic differentiation by down-regulating the expression of myogenin and MyHC [75-77]. However, activation of the PI3K-Akt pathway could inhibit the ERK1/2 MAPK pathway, and this cross-regulation only happened on the differentiation state of the muscle cells [78]. In current research, the expression of MyoG and MyHC decreased when the expression of CNTFRα was interfered. Therefore, we assume that miR-696 may inhibit the expression of CNTFRα, thus attenuate the differentiation of C2C12 myoblasts, which may be through reducing the PI3-kinase/AKT signaling and stimulating the ERK1/2 MAPK pathway.
Additionally, in the current research, we discovered that the expression of miR-696 was much higher in extensor digitorum longus than in soleus muscle (Figure 1B), which had higher proportion of slow muscle fiber and mitochondria [79]. A previous study validated that PGC-1alpha was a target of miR-696 in regulating slow muscle fiber formation [80]. Thus we suggest that miR-696 may have an important function in muscle fiber type transitions. Meanwhile, previous study showed that CNTFRα modulated multiple downstream signaling pathways in many kinds of cells [81]. For example, in adipocytes, CNTF receptor signaling was found to activate p38 MAPK to regulate the expression of PGC-1alpha and induce mitochondrial biogenesis [82]. However, the relationship between CNTFRα and PGC-1α in muscle cells is still not clear. CNTFRα might also play an important role in muscle fiber type transformation. Therefore, our further studies will be needed to verify the mechanism of CLC/CLF composite cytokine in muscle development and discover the interaction between CNTFRα and PGC-1α under the regulation of miR-696 in detail.
In summary, our study verifies that the role of miR-696 in C2C12 cells is to inhibit proliferation and differentiation. And, CNTFRα is a direct target of miR-696. The effects of miR-696 on myogenesis are partially by inhibiting CNTFRα expression.
Supplementary Material
Supplementary tables.
Abbreviations
MyHC: myosin heavy chain; CNTFRα: ciliary neurotrophic factor receptor; MyoG: myogenin; GM: growth medium; DM: differentiation medium; EdU: 5-ethynyl-2'-deoxyuridine; DAPI: 4', 6-diamidino-2-phenylindole; PBS: Phosphate Buffered Saline; PI: Propidium Iodide; RIPA: Radio-Immunoprecipitation Assay; PVDF: polyvinylidene fluoride; PMSF: Phenylmethanesulfonyl fluoride.
Acknowledgements
This work was supported by the Natural Science Foundation of Jiangsu Province (BK20140688), Agricultural Science and Technology independent Innovation Foundation of Jiangsu Province (CX (15) 1006), Youth Science and Technology Innovation Fund of Nanjing Agricultural College in China (KYCYL201502-3) and the foundations from science and technology support program of Jiangsu Province (BE2014396).
Ethic statement
All the experiments were approved by the Animal Care and Use Committee of Nanjing Agricultural University, Nanjing, China. Each procedure in this study was conducted following the Guide for the Care and Use of Laboratory.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S. et al. The formation of skeletal muscle: from somite to limb. Journal of anatomy. 2003;202:59-68
2. Perry RL, Rudnick MA. Molecular mechanisms regulating myogenic determination and differentiation. Frontiers in bioscience: a journal and virtual library. 2000;5:D750-67
3. Buckingham M, Rigby PW. Gene regulatory networks and transcriptional mechanisms that control myogenesis. Developmental cell. 2014;28:225-38
4. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215-33
5. Hutvagner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science. 2002;297:2056-60
6. Sun W, Julie Li YS, Huang HD, Shyy JY, Chien S. microRNA: a master regulator of cellular processes for bioengineering systems. Annual review of biomedical engineering. 2010;12:1-27
7. Bhaskaran M, Mohan M. MicroRNAs: history, biogenesis, and their evolving role in animal development and disease. Veterinary pathology. 2014;51:759-74
8. Wienholds E, Plasterk RH. MicroRNA function in animal development. FEBS letters. 2005;579:5911-22
9. Guller I, Russell AP. MicroRNAs in skeletal muscle: their role and regulation in development, disease and function. The Journal of physiology. 2010;588:4075-87
10. Nguyen HT, Frasch M. MicroRNAs in muscle differentiation: lessons from Drosophila and beyond. Current opinion in genetics & development. 2006;16:533-9
11. Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V. Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 2004;5:R13
12. Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM. et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nature genetics. 2006;38:228-33
13. Chen JF, Tao Y, Li J, Deng Z, Yan Z, Xiao X. et al. microRNA-1 and microRNA-206 regulate skeletal muscle satellite cell proliferation and differentiation by repressing Pax7. The Journal of cell biology. 2010;190:867-79
14. Zhang D, Wang X, Li Y, Zhao L, Lu M, Yao X. et al. Thyroid hormone regulates muscle fiber type conversion via miR-133a1. The Journal of cell biology. 2014;207:753-66
15. Guess MG, Barthel KK, Harrison BC, Leinwand LA. miR-30 family microRNAs regulate myogenic differentiation and provide negative feedback on the microRNA pathway. PloS one. 2015;10:e0118229
16. Bai L, Liang R, Yang Y, Hou X, Wang Z, Zhu S. et al. MicroRNA-21 Regulates PI3K/Akt/mTOR Signaling by Targeting TGFbetaI during Skeletal Muscle Development in Pigs. PloS one. 2015;10:e0119396
17. Shi L, Zhou B, Li P, Schinckel AP, Liang T, Wang H. et al. MicroRNA-128 targets myostatin at coding domain sequence to regulate myoblasts in skeletal muscle development. Cellular signalling. 2015;27:1895-904
18. Wei H, Li Z, Wang X, Wang J, Pang W, Yang G. et al. microRNA-151-3p regulates slow muscle gene expression by targeting ATP2a2 in skeletal muscle cells. Journal of cellular physiology. 2015;230:1003-12
19. Luo W, Wu H, Ye Y, Li Z, Hao S, Kong L. et al. The transient expression of miR-203 and its inhibiting effects on skeletal muscle cell proliferation and differentiation. Cell Death Dis. 2014;5:e1347
20. Aoi W, Naito Y, Mizushima K, Takanami Y, Kawai Y, Ichikawa H. et al. The microRNA miR-696 regulates PGC-1{alpha} in mouse skeletal muscle in response to physical activity. American journal of physiology Endocrinology and metabolism. 2010;298:E799-806
21. Wen F, Zhang H, Bao C, Yang M, Wang N, Zhang J. et al. Resistin Increases Ectopic Deposition of Lipids Through miR-696 in C2C12 Cells. Biochemical genetics. 2015;53:63-71
22. Davis S, Aldrich TH, Valenzuela DM, Wong V, Furth ME, Squinto SP. et al. The Receptor for Ciliary Neurotrophic Factor. Science. 1991;253:59-63
23. Ip NY, Mcclain J, Barrezueta NX, Aldrich TH, Pan L, Li YP. et al. The Alpha Component of the Cntf Receptor Is Required for Signaling and Defines Potential Cntf Targets in the Adult and during Development. Neuron. 1993;10:89-102
24. Stefater MA, MacLennan AJ, Lee N, Patterson CM, Haller A, Sorrell J. et al. The anorectic effect of CNTF does not require action in leptin-responsive neurons. Endocrinology. 2012;153:2647-54
25. MacLennan AJ, Devlin BK, Neitzel KL, McLaurin DL, Anderson KJ, Lee N. Regulation of ciliary neurotrophic factor receptor alpha in sciatic motor neurons following axotomy. Neuroscience. 1999;91:1401-13
26. Rhee KD, Yang XJ. Expression of cytokine signal transduction components in the postnatal mouse retina. Molecular vision. 2003;9:715-22
27. Davis S, Aldrich TH, Valenzuela DM, Wong VV, Furth ME, Squinto SP. et al. The receptor for ciliary neurotrophic factor. Science. 1991;253:59-63
28. Steinberg GR, Watt MJ, Ernst M, Birnbaum MJ, Kemp BE, Jorgensen SB. Ciliary neurotrophic factor stimulates muscle glucose uptake by a PI3-kinase-dependent pathway that is impaired with obesity. Diabetes. 2009;58:829-39
29. Guillet C, Huchet-Cadiou C, Gascan H, Leoty C. Changes in CNTF receptor alpha expression in rat skeletal muscle during the recovery period after hindlimb suspension. Acta physiologica Scandinavica. 1998;163:273-8
30. Fraysse B, Guillet C, Huchet-Cadiou C, Camerino DC, Gascan H, Leoty C. Ciliary neurotrophic factor prevents unweighting-induced functional changes in rat soleus muscle. J Appl Physiol (1985). 2000;88:1623-30
31. Miyamoto-Mikami E, Murakami H, Tsuchie H, Takahashi H, Ohiwa N, Miyachi M. et al. Lack of association between genotype score and sprint/power performance in the Japanese population. Journal of Science and Medicine in Sport. 2017;20(1):98-103
32. Hong AR, Hong SM, Shin YA. Effects of resistance training on muscle strength, endurance, and motor unit according to ciliary neurotrophic factor polymorphism in male college students. Journal of sports science & medicine. 2014;13:680-8
33. Miyamoto-Mikami E, Fujita Y, Murakami H, Ito M, Miyachi M, Kawahara T. et al. CNTFR Genotype and Sprint/power Performance: Case-control Association and Functional Studies. International journal of sports medicine. 2016;37:411-7
34. Watt MJ, Dzamko N, Thomas WG, Rose-John S, Ernst M, Carling D. et al. CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nature medicine. 2006;12:541-8
35. Lee N, Spearry RP, Leahy KM, Robitz R, Trinh DS, Mason CO. et al. Muscle ciliary neurotrophic factor receptor alpha promotes axonal regeneration and functional recovery following peripheral nerve lesion. The Journal of comparative neurology. 2013;521:2947-65
36. Derouet D, Rousseau F, Alfonsi F, Froger J, Hermann J, Barbier F. et al. Neuropoietin, a new IL-6-related cytokine signaling through the ciliary neurotrophic factor receptor. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4827-32
37. Wang X, Wu HT, Zhang ZX, Liu SH, Yang J, Chen XP. et al. Effects of interleukin-6, leukemia inhibitory factor, and ciliary neurotrophic factor on the proliferation and differentiation of adult human myoblasts. Cellular and molecular neurobiology. 2008;28:113-24
38. Chen XP, Mao ZB, Liu SH, Liu H, Wang X, Wu HT. et al. Dedifferentiation of adult human myoblasts induced by ciliary neurotrophic factor in vitro. Molecular biology of the cell. 2005;16:3140-51
39. Wei W, He HB, Zhang WY, Zhang HX, Bai JB, Liu HZ. et al. miR-29 targets Akt3 to reduce proliferation and facilitate differentiation of myoblasts in skeletal muscle development. Cell Death Dis. 2013;4:e668
40. Jia L, Li YF, Wu GF, Song ZY, Lu HZ, Song CC. et al. MiRNA-199a-3p regulates C2C12 myoblast differentiation through IGF-1/AKT/mTOR signal pathway. International journal of molecular sciences. 2014;15:296-308
41. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods. 2001;25:402-8
42. Chen X, Huang Z, Chen D, Yang T, Liu G. MicroRNA-27a is induced by leucine and contributes to leucine-induced proliferation promotion in C2C12 cells. International journal of molecular sciences. 2013;14:14076-84
43. De Falco M, De Luca A. Involvement of cdks and cyclins in muscle differentiation. European journal of histochemistry: EJH. 2006;50:19-23
44. Wang F, Wang H, Wu H, Qiu H, Zeng C, Sun L. et al. TEAD1 controls C2C12 cell proliferation and differentiation and regulates three novel target genes. Cellular signalling. 2013;25:674-81
45. Dey BK, Pfeifer K, Dutta A. The H19 long noncoding RNA gives rise to microRNAs miR-675-3p and miR-675-5p to promote skeletal muscle differentiation and regeneration. Genes & development. 2014;28:491-501
46. Wang GQ, Wang Y, Xiong Y, Chen XC, Ma ML, Cai R. et al. Sirt1 AS lncRNA interacts with its mRNA to inhibit muscle formation by attenuating function of miR-34a. Scientific reports. 2016:6
47. Wei X, Li H, Zhang B, Li C, Dong D, Lan X. et al. miR-378a-3p promotes differentiation and inhibits proliferation of myoblasts by targeting HDAC4 in skeletal muscle development. RNA biology. 2016:1-10
48. Cheng CS, Ran L, Bursac N, Kraus WE, Truskey GA. Cell Density and Joint microRNA-133a and microRNA-696 Inhibition Enhance Differentiation and Contractile Function of Engineered Human Skeletal Muscle Tissues. Tissue engineering Part A. 2016;22:573-83
49. Wei W, Zhang WY, Bai JB, Zhang HX, Zhao YY, Li XY. et al. The NF-kappaB-modulated microRNAs miR-195 and miR-497 inhibit myoblast proliferation by targeting Igf1r, Insr and cyclin genes. Journal of cell science. 2016;129:39-50
50. Wu M, Yang G, Chen Y, Zhou X, Chen H, Li M. et al. CEP2 attenuates myoblast differentiation but does not affect proliferation. International journal of biological sciences. 2015;11:99-108
51. Feng Y, Niu LL, Wei W, Zhang WY, Li XY, Cao JH. et al. A feedback circuit between miR-133 and the ERK1/2 pathway involving an exquisite mechanism for regulating myoblast proliferation and differentiation. Cell Death Dis. 2013;4:e934
52. Feng Y, Cao JH, Li XY, Zhao SH. Inhibition of miR-214 expression represses proliferation and differentiation of C2C12 myoblasts. Cell biochemistry and function. 2011;29:378-83
53. Sabourin LA, Rudnicki MA. The molecular regulation of myogenesis. Clin Genet. 2000;57:16-25
54. Rende M, Muir D, Ruoslahti E, Hagg T, Varon S, Manthorpe M. Immunolocalization of ciliary neuronotrophic factor in adult rat sciatic nerve. Glia. 1992;5:25-32
55. Stockli KA, Lillien LE, Naher-Noe M, Breitfeld G, Hughes RA, Raff MC. et al. Regional distribution, developmental changes, and cellular localization of CNTF-mRNA and protein in the rat brain. The Journal of cell biology. 1991;115:447-59
56. Lin LF, Mismer D, Lile JD, Armes LG, Butler ET 3rd, Vannice JL. et al. Purification, cloning, and expression of ciliary neurotrophic factor (CNTF). Science. 1989;246:1023-5
57. Stockli KA, Lottspeich F, Sendtner M, Masiakowski P, Carroll P, Gotz R. et al. Molecular cloning, expression and regional distribution of rat ciliary neurotrophic factor. Nature. 1989;342:920-3
58. Davis S, Yancopoulos GD. The molecular biology of the CNTF receptor. Current opinion in cell biology. 1993;5:281-5
59. DeChiara TM, Vejsada R, Poueymirou WT, Acheson A, Suri C, Conover JC. et al. Mice lacking the CNTF receptor, unlike mice lacking CNTF, exhibit profound motor neuron deficits at birth. Cell. 1995;83:313-22
60. Plun-Favreau H, Elson G, Chabbert M, Froger J, deLapeyriere O, Lelievre E. et al. The ciliary neurotrophic factor receptor alpha component induces the secretion of and is required for functional responses to cardiotrophin-like cytokine. Embo Journal. 2001;20:1692-703
61. Shi Y, Wang W, Yourey PA, Gohari S, Zukauskas D, Zhang J. et al. Computational EST database analysis identifies a novel member of the neuropoietic cytokine family. Biochemical and biophysical research communications. 1999;262:132-8
62. Lelievre E, Plun-Favreau H, Chevalier S, Froger J, Guillet C, Elson GCA. et al. Signaling pathways recruited by the cardiotrophin-like cytokine/cytokine-like factor-1 composite cytokine - Specific requirement of the membrane-bound form of ciliary neurotrophic factor receptor alpha component. Journal of Biological Chemistry. 2001;276:22476-84
63. de Bovis B, Derouet D, Gauchat JF, Elson G, Gascan H, Delapeyriere O. clc is co-expressed with clf or cntfr in developing mouse muscles. Cell communication and signaling: CCS. 2005;3:1
64. Vlotides G, Zitzmann K, Stalla GK, Auernhammer CJ. Novel neurotrophin-1/B cell-stimulating factor-3 (NNT-1/BSF-3)/cardiotrophin-like cytokine (CLC)-a novel gp130 cytokine with pleiotropic functions. Cytokine & growth factor reviews. 2004;15:325-36
65. Coolican SA, Samuel DS, Ewton DZ, McWade FJ, Florini JR. The mitogenic and myogenic actions of insulin-like growth factors utilize distinct signaling pathways. The Journal of biological chemistry. 1997;272:6653-62
66. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261-74
67. Yang W, Zhang Y, Li Y, Wu Z, Zhu D. Myostatin induces cyclin D1 degradation to cause cell cycle arrest through a phosphatidylinositol 3-kinase/AKT/GSK-3 beta pathway and is antagonized by insulin-like growth factor 1. The Journal of biological chemistry. 2007;282:3799-808
68. Fujio Y, Guo K, Mano T, Mitsuuchi Y, Testa JR, Walsh K. Cell cycle withdrawal promotes myogenic induction of Akt, a positive modulator of myocyte survival. Molecular and cellular biology. 1999;19:5073-82
69. Lavoie JN, LAllemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44(MAPK) and negatively by the p38/HOG(MAPK) pathway. Journal of Biological Chemistry. 1996;271:20608-16
70. Weber JD, Raben DM, Phillips PJ, Baldassare JJ. Sustained activation of extracellular-signal-regulated kinase 1 (ERK1) is required for the continued expression of cyclin D1 in G(1) phase. Biochemical Journal. 1997;326:61-8
71. Schramek H. MAP kinases: From intracellular signals to physiology and disease. News Physiol Sci. 2002;17:62-7
72. Yang W, Chen Y, Zhang Y, Wang X, Yang N, Zhu D. Extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase pathway is involved in myostatin-regulated differentiation repression. Cancer research. 2006;66:1320-6
73. Lu H, Shi X, Wu G, Zhu J, Song C, Zhang Q. et al. FGF13 regulates proliferation and differentiation of skeletal muscle by down-regulating Spry1. Cell proliferation. 2015;48:550-60
74. Yokoyama T, Takano K, Yoshida A, Katada F, Sun P, Takenawa T. et al. DA-Raf1, a competent intrinsic dominant-negative antagonist of the Ras-ERK pathway, is required for myogenic differentiation. The Journal of cell biology. 2007;177:781-93
75. Xu Q, Wu Z. The insulin-like growth factor-phosphatidylinositol 3-kinase-Akt signaling pathway regulates myogenin expression in normal myogenic cells but not in rhabdomyosarcoma-derived RD cells. The Journal of biological chemistry. 2000;275:36750-7
76. Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. American journal of physiology Cell physiology. 2009;296:C1258-70
77. Sun L, Liu L, Yang XJ, Wu Z. Akt binds prohibitin 2 and relieves its repression of MyoD and muscle differentiation. Journal of cell science. 2004;117:3021-9
78. Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K. et al. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286:1738-41
79. Schiaffino S, Reggiani C. Fiber types in mammalian skeletal muscles. Physiological reviews. 2011;91:1447-531
80. Lin J, Wu H, Tarr PT, Zhang CY, Wu ZD, Boss O. et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797-801
81. Hu X, Zhao Y, He X, Li J, Wang T, Zhou W. et al. Ciliary neurotrophic factor receptor alpha subunit-modulated multiple downstream signaling pathways in hepatic cancer cell lines and their biological implications. Hepatology. 2008;47:1298-308
82. Crowe S, Turpin SM, Ke F, Kemp BE, Watt MJ. Metabolic remodeling in adipocytes promotes ciliary neurotrophic factor-mediated fat loss in obesity. Endocrinology. 2008;149:2546-56
Author contact
![]() Corresponding authors: Pinghua Li, PhD, Institute of Swine Science, Nanjing Agricultural University, Nanjing, Jiangsu 210095, China, Tel: +86-25-84395362, Fax: +86-25- 84395314, E-mail: lipinghua718edu.cn; Ruihua Huang, Institute of Swine Science, Nanjing Agricultural University, Nanjing, Jiangsu 210095, China, Tel: +86-25-84395362, Fax: +86-25- 84395314, E-mail: rhhuangedu.cn
Corresponding authors: Pinghua Li, PhD, Institute of Swine Science, Nanjing Agricultural University, Nanjing, Jiangsu 210095, China, Tel: +86-25-84395362, Fax: +86-25- 84395314, E-mail: lipinghua718edu.cn; Ruihua Huang, Institute of Swine Science, Nanjing Agricultural University, Nanjing, Jiangsu 210095, China, Tel: +86-25-84395362, Fax: +86-25- 84395314, E-mail: rhhuangedu.cn