Impact Factor ISSN: 1449-2288
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(4):505-517. doi:10.7150/ijbs.18834 This issue Cite
Research Paper
Targeting HSP90-HDAC6 Regulating Network Implicates Precision Treatment of Breast Cancer
Shiyi Yu1, 2*, Xiuxiu Cai1, 2*, Chenxi Wu1, 2*, Yan Liu1, 2*, Jun Zhang1, 2*, Xue Gong1, 2, Xin Wang1, 2, Xiaoli Wu3, Tao Zhu3, Lin Mo4, Jun Gu4, Zhenghong Yu5 ![]() , Jinfei Chen6, 7
, Jinfei Chen6, 7 ![]() , Jean Paul Thiery8, 9, 10
, Jean Paul Thiery8, 9, 10 ![]() , Renjie Chai2,11,12
, Renjie Chai2,11,12 ![]() , Liming Chen1, 2
, Liming Chen1, 2 ![]()
1. Jiangsu Key Laboratory for Molecular and Medical Biotechnology, College of Life Science, Nanjing Normal University, Nanjing 210046, P.R. China;
2. The Key Laboratory of Developmental Genes and Human Disease, Ministry of Education, Institute of Life Science, Southeast University, Nanjing 210096, P.R. China;
3. Institute of Immunology and CAS Key Laboratory of Innate Immunity and Chronic Disease, Innovation Center for Cell Biology, School of Life Sciences and Medical Center, University of Science and Technology of China, Hefei 230027, P. R. China;
4. Department of General Surgery, Jinling Hospital, Medical School of Nanjing University, Nanjing 210002, P. R. China;
5. Department of Medical Oncology, Jinling Hospital, Medical School of Nanjing University, Nanjing 210002, P. R. China;
6. Department of Oncology, Nanjing First Hospital, Nanjing Medical University, 210006, P. R. China;
7. Collaborative Innovation Center for Cancer Personalized Medicine, Nanjing Medical University, 210006, P. R. China;
8. Cancer Science Institute, National University of Singapore, 14 Medical Drive, 117599, Singapore;
9. Institute of Molecular and Cell Biology, A*STAR, 61 Biopolis Drive, 138673, Singapore;
10. Department of Biochemistry, Yong Loo Lin School of Medicine, National University of Singapore, 8 Medical Drive, 117596, Singapore;
11. Co-Innovation Center of Neuroregeneration, Nantong University, Nantong 226001, China;
12. Jiangsu Provincial Clinical Key Discipline and Laboratory of Otology, Nanjing 210008, China.
* These authors contribute equally to this work.
Received 2016-12-20; Accepted 2017-2-7; Published 2017-4-8
Abstract

Breast cancer is the leading cause of women death. Heat shock protein 90 (HSP90) and Histone deacetylase 6 (HDAC6) are promising anti-cancer drug targets. However, it's still unclear the applicability of anti-HSP90 and anti-HDAC6 strategies in precision treatment of breast cancer. In current study, we found that triple negative breast cancer (TNBC) cells, compared to T47D, an ERα+ breast cancer cell line, exhibited 7~40 times lower IC50 values, stronger cell cycle perturbation, increased cell apoptosis and stronger inhibition of cell migration upon 17-DMAG treatment, while T47D, compared to TNBC cells, expressed higher HDAC6 and showed stronger anti-cancer response upon treatment of Tubacin. Mechanically, 17-DMAG treatment inhibited a complex network consists at least ERK, AKT, and Hippo pathway in TNBC cells, and higher expression of HDAC6 inhibited HSP90 activity via deacetylating HSP90. Furthermore, we found higher HDAC6 expression level in tamoxifen-resistance T47D than that in T47D, and Tubacin treatment suppressed the growth of tamoxifen-resistant cells in vivo. Our data suggested that anti-HSP90 and anti-HDAC6 are promising strategies to treat TNBC and ERα+ breast cancers respectively, and anti-HDAC6 can be considered during treatment of tamoxifen-resistance breast cancers.
Keywords: Breast cancer, TNBC, HSP90, HDAC6, tamoxifen resistance.
Introduction
Breast cancer is the leading cause of women death with greater than 1,300,000 new cases and 450,000 deaths each year worldwide [1]. Breast cancer can be classified as ERα+, HER2+ and triple negative breast cancer (TNBC) based on the expression of ERα, progesterone receptor (PR) and human epidermal growth factor receptor-2 (HER2) [2]. TNBC lacks expression of ERα, PR and HER2. Since there is no targeted therapy available for TNBC, TNBC patients show poorer prognostic outcomes compared with ERα+ and HER2+ breast cancer patients [3]. Although clinical outcomes of ERα+ and HER2+ breast cancer patients have been greatly improved with anti-ERα and anti-HER2 therapies respectively, the cure rate of ERα+ patients is limited by drug resistance, poor clinical responses to current targeted drugs and tumor recurrence during treatment [4, 5]. Take tamoxifen (a widely used anti-ERα drug) as an example, almost 50% of ERα+ breast cancer patients do not respond to tamoxifen, even patients with initial response eventually develop tamoxifen resistance, leading to tumor progression and patient death [6]. Thus, discovering effective therapies on TNBC and target-drug-resistant ERα+ breast cancer is a major challenge during treatment of breast cancer.
Heat shock protein 90 (HSP90) and Histone deacetylase 6 (HDAC6) are considered to be promising targets for cancer therapy [7]. Under physiological condition, HSP90 serves as a molecular chaperone that is crucial for the stability and function of numerous proteins to maintain cellular protein homeostasis and cell survival [8]. In tumor cells, HSP90 exhibits much higher ATPase activity compared to their counterparts in normal cells [9], and is important for the stabilities and functions of many oncogenic proteins [10]. Inhibition of HSP90 is likely to suppress multiple oncogenic signaling pathways simultaneously, and is regarded as a promising anti-cancer strategy [11]. HDAC6, an unique deacetylase in HDAC family, exhibits cytoplasmic localization and ability to de-acetylate HSP90 [12]. Inactivation or knockdown of HDAC6 led to HSP90 hyper-acetylation and loss of HSP90 chaperone activity [13]. However, it's still unclear whether and how anti-HSP90 and anti-HDAC6 strategy can be applied in treatment of breast cancer.
In current study, we found that although HSP90 inhibitor 17-DMAG reduced the viabilities of all investigated breast cancer cells, TNBC cells were much more sensitive to 17-DMAG than T47D, an ERα+ breast cancer cell line. Elevated HDAC6 expression level was observed in T47D in comparison with TNBC cells. As expected, T47D was more sensitive to HDAC6 inhibitor Tubacin treatment than TNBC cells did, and treatment of 17-DMAG in combination with Tubacin showed synergetic effects on T47D. Interestingly, tamoxifen-resistant T47D (T47D-TAR) cells exhibited elevated HDAC6 compared to its parental T47D cells. Tubacin shows effective anti-cancer effect on investigated tamoxifen-resistant breast cancer cells in vivo. Taken together, our data provided important clues for precision treatment of breast cancer using anti-HSP90 and anti-HDAC6 strategies.
Material and methods
Cell culture and reagent
BT549 and Hs578T cell lines were obtained from American Type Cell Collection (ATCC) in 2012, MDA-MB-231 was bought from ATCC in 2014. MCF7 and T47D were kind gifts from Dr. Tao Zhu. All were authenticated via the short tandem repeat (STR) typing in 2015, and used within 6 months of receipt or after cell authentication for current study. BT549, Hs578T cell lines were cultured in Dulbecco's modified essential medium (DMEM) (Life Technologies, Carlsbad, CA) , MCF7 and T47D cells were grown in RMPI 1640 medium in 37℃ incubator supplemented with 5% CO2. The Tam-resistant cell line T47D-TAR cell line was generated by exposing T47D to tamoxifen (1μM) for > 12 months. ERα was significantly decreased in T47D-TAR cell line compared with its parental cells, indicating the loss of ERα function in T47D-TAR [14]. T47D-TAR was then maintained in RMPI 1640 supplemented with 1μM tamoxifen. MDA-MB-231 cells were grown in Leibovitz's L15 mediumin 37℃ with no CO2. All cell lines were supplemented with 10% fetal bovine serum (HyClone, NY, USA) and 1% penicillin-streptomycin solution (Life Technologies). 17-DMAG, Tubacin, fulvestrant were purchased from Selleck Chemicals, and tamoxifen was bought from Sigma-Aldrich.
RNA interference
ERα siRNA pool or control siRNA (Santa Cruz Biotechnology, Dallas, TX) was transfected into T47D using LipofectamineRNAi MAX (Invitrogen), remained for 72 hours and then subjected to protein or RNA extraction.
For YAP silencing, all cell lines were first seeded in 96-well plate, then transfected with control siRNA or YAP siRNA1 or YAP siRNA2 (GenePharma, Shanghai, China) by LipofectamineRNAiMAX (Invitrogen), sustained for 72 hours.
Tamoxifen and fulvestrant treatment
T47D cells were seeded in 6-well plates and cultured in phenol red-free medium without serum overnight. On the next day, the medium was removed and replaced with phenol red-free medium containing 10nM E2 (Sigma-Aldrich) with or without 1μM tamoxifen and 0.1μM fulvestrant for 24 hours.
Cell viability assay
The anti-proliferative effect of YAP siRNA, 17-DMAG and Tubacin was evaluated using CCK-8 kit (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's instructions. Briefly, cells were seeded in 96-well plate with DMSO or various concentrations of drugs for 72 hours. After that, 10ul CCK-8 solution was added into each well in 96-well plate, sustained for 2 hours, and absorbance at 450nm was measured to reflect cell viability.
Cell cycle and cell apoptosis assay
For the cell cycle assay, cells were harvested by trypsinization and fixed with 70% ethanol at 4°C overnight. Cells were then stained with propidium iodide and the cell cycle distribution was analyzed using a BD FACSCalibur flow cytometer (BD Biosciences). Cell apoptosis assay was performed using Annexin-V/Dead Cell Apoptosis Kit (Invitrogen) and analyzed on a BD FACSCalibur flow cytometer (BD Biosciences, Franklin Lakes, NJ).
Determination of synergism and IC50
The medium-effect method was applied to analyze the dose-response of single drug or drugs in combination. The synergistic effect of drugs in combination was determined according to the definition of Chouand Talalay [15]. Combination index (CI) was used to reflect the effects of two drugs at different concentrations. CI values of <1, =1 and >1 indicate synergistic, addictive and antagonistic effect respectively. Software compusyn (ComboSyn, Inc., Paramus, NJ) was used to calculate CI and IC50 (cells were inhibited to 50% compared with control group).
Western Blotting
Cells lysates were prepared using RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) with protease/phosphatase inhibitor cocktail (cell signaling technology; Beverly, MA). Antibodies for YAP, phosho-YAP (Ser127), ERK1/2, phospho-ERK1/2 (Thr202/Tyr204), AKT, phospho-AKT (Ser473), ERα, HDAC6 and HSP90 were purchased from cell signaling technology (Beverly, MA). GAPDH mouse monoclonal antibody was obtained from Kangchen (Shanghai, China). Mouse monoclonal antibodies against acetylated α-Tubulin and α-Tubulin were from Sigma-Aldrich. Anti-mouse and anti-rabbit secondary antibodies were bought from Proteintech (Chicago, IL). Briefly, protein lysates were separated by SDS-PAGE, transferred to PVDF membranes, and immunoblotted with the respective antibodies as indicated above and in the figures. Blots were developed with SuperSignal West Femto Maximum Sensitivity Substrate (Pierce/Thermo Scientific, Rockford, IL) and the images were obtained by ImageQuant LAS 4000 (GEhealthcare, Little Chalfont, England).
Immunofluorescence labeling
For immunofluorescence studies, T47D and MDA-MB-231 cells were cultured on glass slides in 24-well plates for 24 hours. Inhibitors were added into the well and cultured for another 24 hours. Prior to staining, the wells were washed with PBS, the cells were then fixed with 4% paraformaldehyde for 30 min, then permeabilized and blocked with 0.1% Triton X-100 in 1% BSA for 1h at room temperature. Mouse monoclonal anti-tubulin (Sigma-Aldrch) was used as primary antibody and Alexa Fluor 488 secondary antibody (Invitrogen) was employed to detect fluorescence. The nuclei were stained with DAPI (Vector Laboratories, Cambridgeshire, UK). Representative images were captured using the Leica DM5000 B microscope (Leica Microsystems, Buffalo Grove, IL).
Nocodazole-induced microtubule de-polymerization assay
MDA-MB-231 and T47D were cultured on glass slides in 24-well plates for 24 hours, and then treated with 0.1μM 17-DMAG or 1μM Tubacin or 0.1μM 17-DMAG and 1μM Tubacin for 24 hours. After that, cells were exposed to 3μM nocodazole for 2 hours. The cells were then fixed and processed for immunostaining.
Preprocessing of 46 breast cancer cell lines gene expression data
We retrieved the expression data of 46 breast cancer cell lines from previously published work [16]. Comparison of HDAC6 expression between ERα+ breast cancer cell lines and Basal breast cancer cell lines was made by applying Graphpad.
Wound-healing assay
Wound-healing assay was performed to evaluate cell migration activity. Cells were seeded onto the 12-well plates and grew towards confluence, wounds were made with 10ul tips. After washing with PBS for 3 times, the cells were then cultured in 1% FBS containing medium. The images were captured by a microscope (Carl Zeiss Meditec, Jena, Germany) at different time points. The wound areas were calculated employing Image Pro Plus 6.0 software (Media Cybernetics).
Immunohistochemistry
Breast tumor samples were collected from patients surgically treated for breast cancer at Nanjing General Hospital of Nanjing Military Command between 2008 and 2012. This patient series included women aged from 25 to 82. Immunohistochemistry was performed using the Max VisionTMkit (Maixin Biotech, Fuzhou, China) following manufacturer's instructions. Briefly, all species were fixed with 10% formalin, embedded, re-waxed and rehydrated according to the standard procedures. Afterwards, antigen retrieval was achieved by heating with high temperature. 3% H2O2 was added to the slides to block the activity of endogenous peroxidase. The slides were then incubated with anti-YAP antibody for 1 hour at 37°C. After incubating with primary antibody, the slides were washed with PBS and incubated with secondary antibody for 30 minutes at room temperature. The signal was visualized by incubation of slides in DAB solution. Haematoxylin was used for nuclei visualization.
In vivo mouse model
All animal experiments were conducted in accordance with the instructional standard guideline of Southeast University for animal experiments. Female athymic STOCK-Foxn1nu/Nju 4-week-old mice were obtained from Model Animal Research Center of Nanjing University. Upon tumor initiation, MDA-MB-231 (5×106) or T47D-TAR (1×107) cells were suspended in 100µl PBS and then mixed with 100µl Matrigel (BD Biosciences, Bedford, MA), after thorough mixing, cells were subcutaneously injected into the armpit of mice. For MDA-MB-231 tumor models, one week after implantation, animals were treated with either 17-DMAG (25mg/kg/2 days, i.p.) or PBS for 18 days (n=4 per group). On the other hand, T47D-TAR tumor models were administrated with Tubacin or PBS for 9 days (25mg/kg/2 days, i.p.) after 4 weeks of tumor growth (n=4 per group). The tumor size was measured using a caliper and tumor volume was calculated by the following formula:
Volume = 0.5 × Length × Width2
Statistical analysis
All data were analyzed with Prism 5.0 (GraphPad Software Inc., San Diego, CA) and expressed as means ± SD. The differences between two groups were statistically analyzed using student's t-test, the differences were considered to be significant at a p-value<0.05.
Results
TNBC cells were more sensitive to HSP90 inhibition than T47D
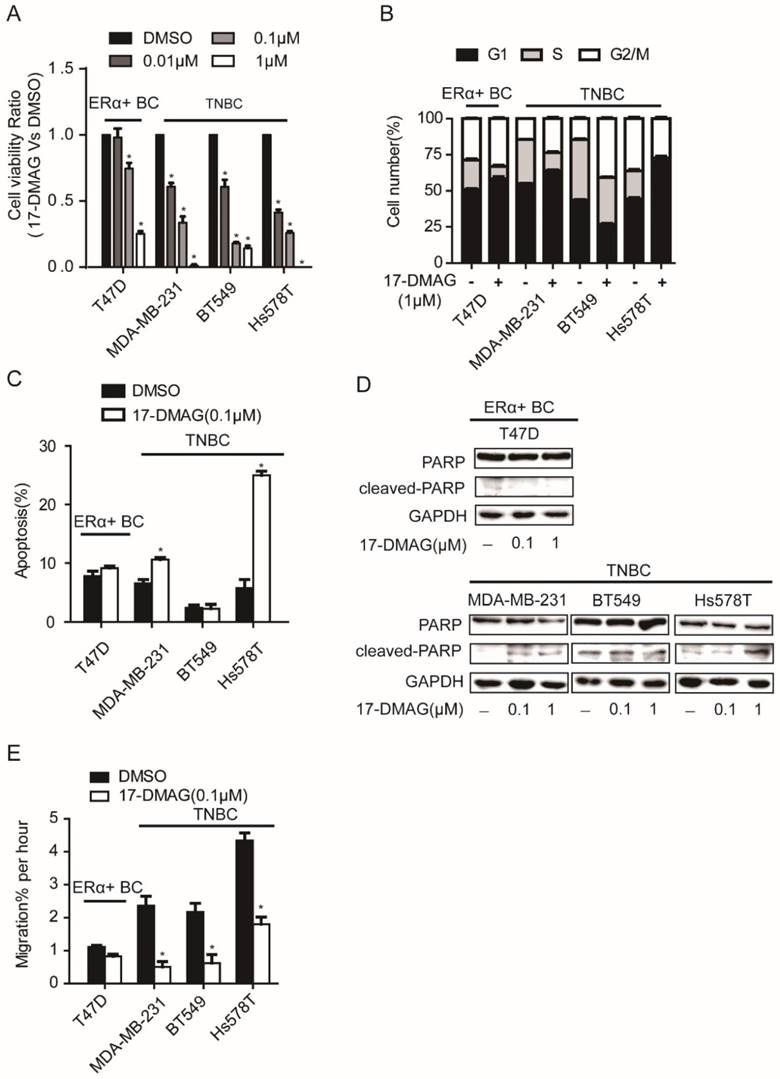
HSP90 inhibitor, 17-DMAG, inhibits the activity of HSP90 via binding with N-terminal domain of HSP90 [17]. After the determination of IC50 of 17-DMAG in a panel of breast cancer cell lines, we found that TNBC cell lines (MDA-MB-231, BT549 and Hs578T) showed lower IC50 values than ERα+ breast cancer cell lines (MCF7 and T47D) (Table 1). When TNBC cell lines (MDA-MB-231, BT549 and Hs578T) and an ERα+ breast cancer cell line (T47D) were subjected to cell viability assay upon 17-DMAG treatment, TNBC cell viability was strongly decreased, while T47D showed relatively moderate response (Figure 1A). Consistently, compared to T47D, upon 17-DMAG treatment, TNBC cells showed stronger cell cycle perturbation, increased cell apoptosis with increased PARP cleavage and stronger inhibition of cell migration (Figure 1B-E). Taken together, our results suggest that TNBC cells were more sensitive to HSP90 inhibition than ERα+ breast cancer cells.
The IC50 value for 17-DMAG in ERα+ BC and TNBC cell lines
| Cell line | IC50(nM) | |
|---|---|---|
| ERα+ BC | T47D | 327.7 |
| MCF7 | 119.8 | |
| TNBC | MDA-MB-231 | 45.7 |
| BT549 | 17.1 | |
| Hs578T | 8.4 |
Effects of HSP90 inhibitor 17-DMAG treatment on A. cell viability, B. cell cycle, C. apoptosis, D. cleavage of PARP, E. cell migration of breast cancer cells. Each group was compared with control group, *P<0.05.
T47D with elevated HDAC6 expression was sensitive to HDAC6 inhibitor Tubacin treatment
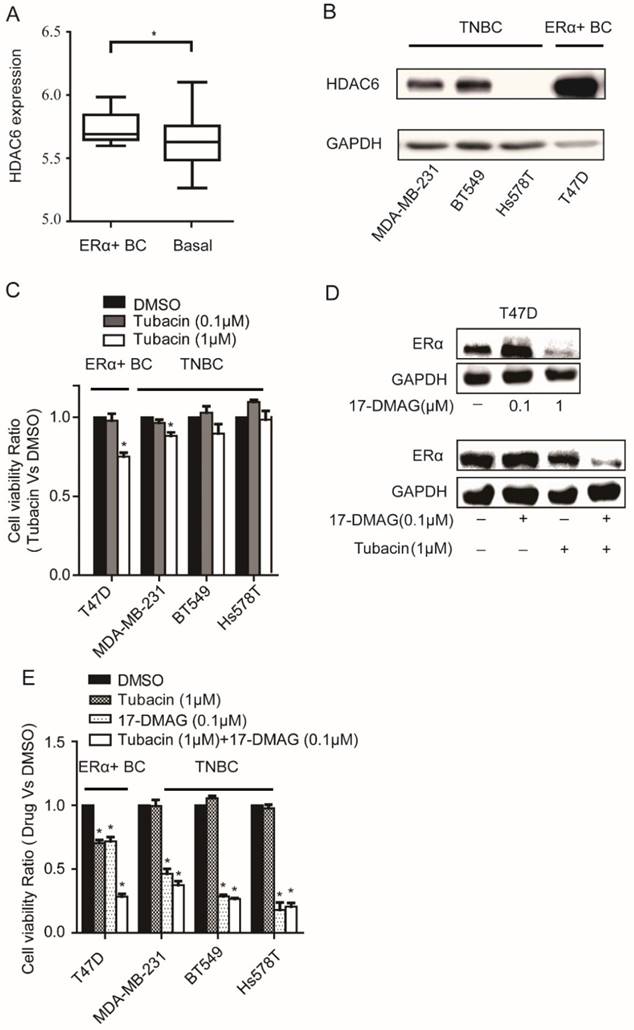
We found that HDAC6 expression was significantly elevated in ERα+ breast cancer cells compared with basal breast cancer cells by analyzing the expression data of 46 breast cancer cell lines [16] (Figure 2A). The elevated expression level of HDAC6 in T47D compared to those in TNBC cells was further confirmed at protein level (Figure 2B). As expected, compared to TNBC cells, T47D was more sensitive to HDAC6 inhibitor Tubacin treatment (Figure 2C). Inhibition of HSP90 could reduce ERα protein level in cancer cells [18, 19]. In T47D cell line, treatment of 0.1μM 17-DMAG in combination with 1μM Tubacin but not 0.1μM 17-DMAG alone was able to lead to a sharp decrease of ERα protein level (Figure 2D). These findings explained the observations that combination of HDAC6 inhibition and HSP90 inhibition treatment showed synergetic effects on T47D not TNBC cells (Figure 2E, Table 2).
Combination of HDAC6 and HSP90 inhibition showed synergistic effect on ERα+ BC T47D cells
| Cell line | 17-DMAG (µm) | Tubacin (µm) | CI | Effect |
|---|---|---|---|---|
| 0.05 | 1 | 1.06 | Addictive | |
| T47D | 0.10 | 1 | 0.62 | Synergism |
| 0.25 | 1 | 0.45 | Synergism | |
| 0.50 | 1 | 0.60 | Synergism |
Analysis of HDAC6 expression in breast cancer cells and their sensitivity to HSP90 and/or HDAC6 inhibition A. Comparison the expressions of HDAC6 mRNA in ERα+ breast cancer cells to those in basal breast cancer cells. B. Comparison the expressions of HDAC6 protein in ERα+ breast cancer cells (T47D) to those in TNBC cells (MDA-MB-231, BT549 and Hs578T). C. Upon 0.1μM and 1μM HDAC6 inhibitor Tubacin treatment for 72 hours, ERα+ breast cancer cells (T47D) showed stronger reduction of cell viability than TNBC (MDA-MB-231, BT549 and Hs578T) cells did. D. HDAC6 inhibition enhanced decrease of ERα in T47D towards HSP90 inhibition. E. Effects on cell viabilities of breast cancer cells upon inhibition of HSP90 or HDAC6 or both HSP90 and HDAC6. Each group was compared with control group, *P<0.05.
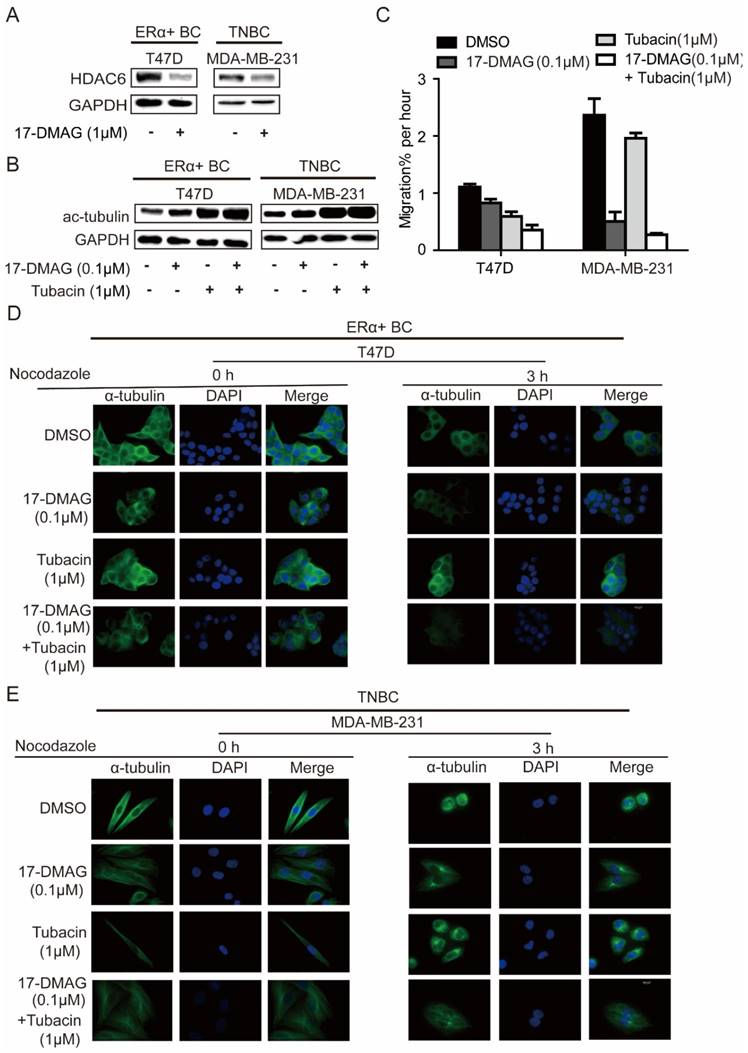
In investigated cell lines (MDA-MB-231 and T47D), HSP90 inhibition using 17-DMAG led to decrease of HDAC6 and increase of acetylated tubulin (Figure 3A, B), in comparison, HDAC6 inhibition using Tubacin increased acetylated tubulin more significantly (Figure 3B). Upon treatment of nocodazole, a known microtubule-depolymerizing agent, microtubules in both MDA-MB-231 and T47D were extensively depolymerized and accumulated at the periphery of the nucleus, while 17-DMAG and/or Tubacin antagonized nocodazole effects and stabilized microtubule network (Figure 3D-E). Migration abilities of MDA-MB-231 and T47D cells were largely repressed by 17-DMAG and Tubacin respectively (Figure 3C). Since HSP90 inhibition reduced cancer cell mobility through HDAC6 degradation and hyper-acetylation of tubulin [20], these findings indicate that inhibition of HSP90 and/or HDAC6 inhibited cell migration via inhibiting tubulin deacetylation to enhance microtubule stability. Unexpectedly, synergistic effect of 17-DMAG and Tubacin on cell migration was only observed in T47D but not MDA-MB-231 (Figure 3C). This observation might be due to the low expression level of HDAC6 in MDA-MB-231. Taken together, our data indicated that the relative insensitivity of 17-DMAG and sensitivity of Tubacin treatment in T47D might be due to the elevated expression of HDAC6 in T47D compared to TNBC cells.
Effects on microtubule cytoskeleton network stabilization of breast cancer cells upon inhibition of HSP90 or HDAC6 or both HSP90 and HDAC6. A. Treatment of 0.1μM 17-DMAG for 24 hours decreased HDAC6 protein level in both ERα+ breast cancer cells and TNBC cells. B. Inhibition of HSP90-HDAC6 with 0.1μM 17-DMAG and 1μM Tubacin increased tubulin acetylation synergistically. C. HSP90/HDAC6 inhibition with 0.1μM 17-DMAG and 1μM Tubacin decreased cell migration in both ERα+ breast cancer cells and TNBC cells. D/E. Analysis and comparison the effects on microtubule cytoskeleton stability in breast cancer cells upon inhibition of HSP90 or HDAC6 or both HSP90 and HDAC6. Each group was compared with control group, *P<0.05.
Hippo pathway contributes to the anti-cancer effects induced by both HSP90 and HDAC6 inhibition
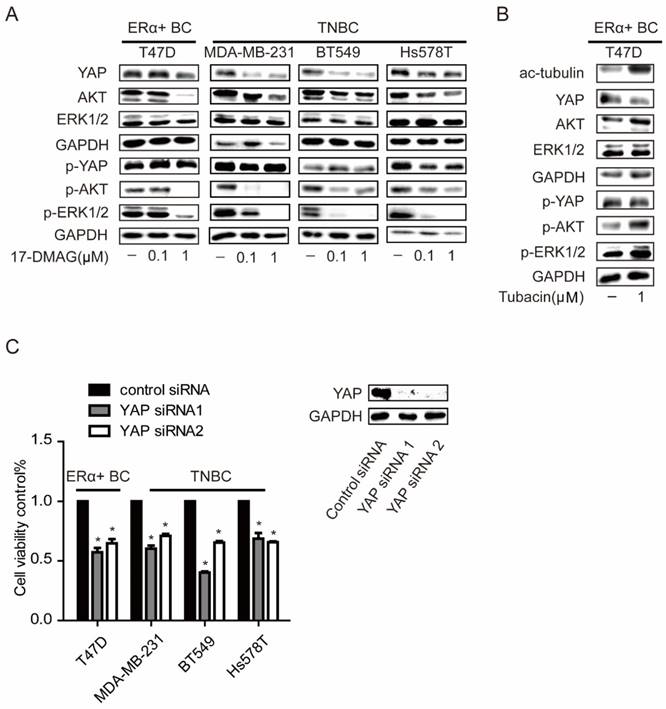
Our previous work has reported a crosstalk between ERK, AKT and Hippo pathways [21]. There was also a report which indicated that treatment of HSP90 inhibitor in TNBC cells would lead to inactivation of ERK and AKT pathways by decreasing the protein levels of several receptors [22]. In TNBC cells, p-ERK and p-AKT were strongly inhibited and YAP protein level was significantly decreased with 0.1μM 17-DMAG, while these events only can be observed in T47D, an ERα positive breast cancer cell line, when 1μM 17-DMAG was applied (Figure 2C). Compared to HSP90 inhibition, HDAC6 inhibition decreased YAP protein level with increased AKT and ERK activities (Figure 4A). YAP was overexpressed in tumors from both TNBCs and ERα+ breast cancer patients (Supplementary Figure 1A-B & Supplementary Table 1). Consistently, silencing YAP with siRNAs led to decrease of cell viabilities in both TNBC cells and T47D (Figure 4C and Supplementary Figure 2). These results together suggested that higher susceptibility of AKT, ERK and Hippo pathway to HSP90 inhibition in TNBC cells contributes to higher sensitivity of TNBC cells to HSP90 inhibition compared to T47D, while T47D sensitive to HDAC6 inhibition via modulating Hippo pathway not ERK and AKT pathway.
Effects on ERK, AKT and Hippo pathway in breast cancer cells upon inhibition of HSP90. A. Investigation of AKT (p-AKT), ERK (p-ERK), and YAP (p-YAP) levels in TNBC cells and ERα positive breast cancer cells upon 0.1μM and 1μM HSP90 inhibitor 17-DMAG treatment. B. Inhibition of HDAC6 with 1μM Tubacin led to a decrease of YAP with an increase in tubulin acetylation, AKT and ERK phosphorylation. C. Effects on cell viability of breast cancer cells upon YAP silencing using siRNAs. Each group was compared to control group, *P<0.05.
Tamoxifen-resistance T47D (T47D-TAR) sensitive to HDAC6 inhibition via modulating Hippo pathway
HDAC6 was reported to be an estrogen-regulated gene and was positively regulated by estrogen in MCF7 [23]. Surprisingly, HDAC6 expression was increased upon treatment with either tamoxifen or fulvestrant, which are ERα antagonists (Figure 5A). In addition, silencing ERα with siRNAs in T47D also led to increase of HDAC6 expression level (Figure 5B). These results raised a possibility that T47D-TAR might show reduced expression of ERα and elevated expression of HDAC6 compared to its parental T47D. This hypothesis was confirmed with the observation of ERα and HDAC6 protein level in T47D-TAR compared to T47D (Figure 5D).
Effects on HDAC6 expression in breast cancer cells upon inhibition or depletion ERα and in tamoxifen-resistant breast cancer cells A. Inhibition of ERα with 1μM tamoxifen or 0.1μM fulvestrant for 24 hours up-regulated HDAC6 at protein level in ERα positive breast cancer cells. B. Silencing ERα with siRNA elevated HDAC6 protein levelin ERα+ breast cancer cells. C. Inhibition of ERα with 1μM tamoxifen or 0.1μM fulvestrant for 24 hours elevated YAP expression at protein level. D. Higher HDAC6 and YAP expression with lower ERα and acetylated tubulin in T47D-TAR cells compared to those in their parental T47D cells.
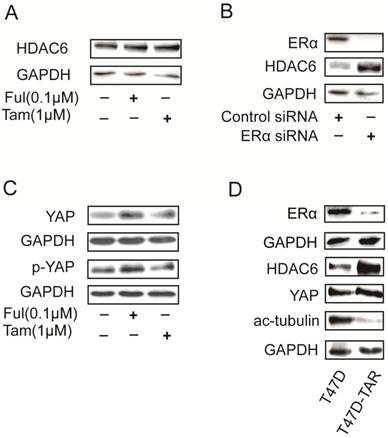
Interestingly, the elevated expression of YAP was also observed upon inhibition of ERα in T47D (Figure 5C), and in T47D-TAR compared to T47D (Figure 5D).
Taken together, our data suggested that elevated HDAC6 and YAP might contribute to the establishment of T47D-TAR cells, both HDAC6 and YAP inhibition could be regarded as promising strategy for treatment of tamoxifen-resistant breast cancer patients.
The data presented above herein supports a model whereby HSP90-HDAC6 regulating network involving Hippo pathway implicates precision treatment of breast cancer (Figure 6A-C). Xenograft mouse models further comfirmed that 17-DMAG and Tubacin effectively inhibited tumor growth of MDA-MB-231 and T47D-TAR cells respectively (Figure 6D-E).
Proposed model for the mechanism of HSP90-HDAC6 regulating network and implication in breast cancer treatment A-C. Proposed molecular mechanism driving breast cancer cells sensitive to HSP90 inhibition and HDAC6 inhibition. D. HSP90 inhibitor 17-DMAG inhibited tumor growth in TNBC cell xenograft mouse model. E. HDAC6 inhibitor Tubacin inhibited tumor growth in tamoxifen-resistant breast cancer cell xenograft mouse model. Each group was compared with control group, *P<0.05.
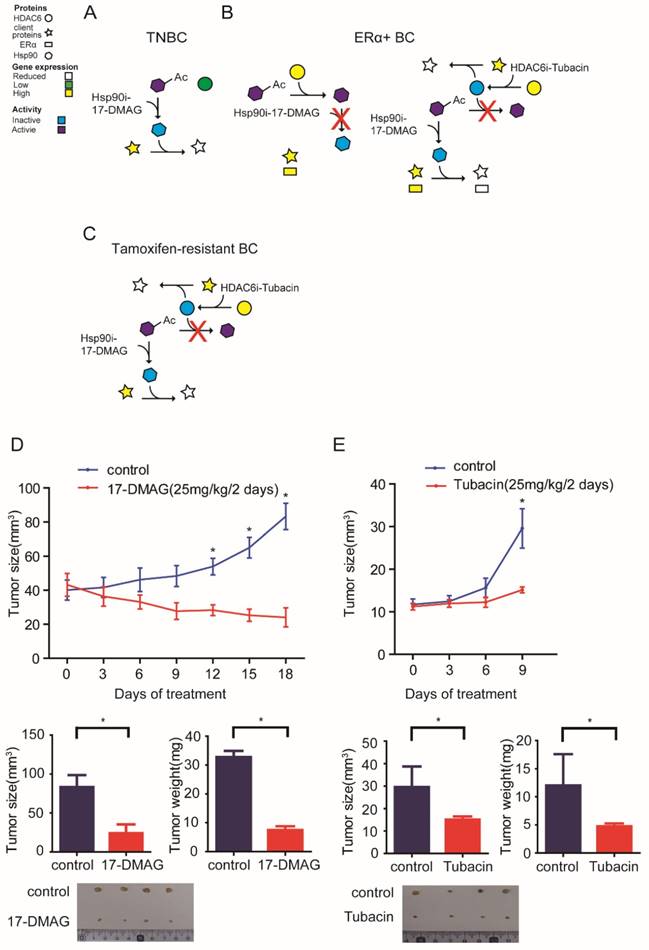
In summary, based on our findings, it is recommended to consider anti-HSP90 strategy for patients with TNBCs, anti-HDAC6 strategy for ERα+ or tamoxifen-resistant breast cancers, anti-YAP strategy for patients with either TNBCs, ERα+ or tamoxifen-resistant breast cancers.
Discussion
Targeted therapies are the foundation to achieve success in precision treatment of cancer patients [24]. Although targeted therapies have already improved clinical outcomes of breast cancer patients, TNBC patients are still lacking targeted therapies and treatments of ERα positive breast cancer patients are challenged with resistance or non-response to currently available targeted therapies [3, 25].
HSP90 and HDAC6, which play crucial roles in promoting oncogenesis via regulating a complex signaling network, were both suggested as promising targets for cancer therapies [7]. Although HSP90 inhibitors were implicated to show notable preclinical effects in the treatment of TNBCs [26, 27], and ERα+ breast cancer [28]. However, it's still unclear how to apply anti-HSP90 and anti-HDAC6 strategy in precision treatment of breast cancer. In the present study, we provided in vitro and in vivo evidences to show that anti-HSP90 strategy is more applicable for treatment of TNBC patients and anti-HDAC6 strategy might be recommended for ERα+ and tamoxifen-resistant breast cancer patients.
HDAC6 acetylated HSP90 to inhibit the chaperone function of HSP90 [29]. We found that inhibition of HSP90 led to decrease of HDAC6 protein level and both HDAC6 and HSP90 regulated microtubulin network stability via modulating the tubulin acetylation (Figure 3A-E). Our findings provided additional supporting evidences for the interplay between HSP90 and HDAC6.
Although both HSP90 and HDAC6 were believed to modulate AKT and ERK pathways, HSP90 and HDAC6 inhibition showed various even the controversial effects on AKT and ERK pathways in different malignant cell types. For HSP90, inhibition of HSP90 by 17-AGG led to inhibition of ERK and AKT pathways in colon adenocarcinoma cell lines [30], while inhibition of HSP90 by GA was reported to activate AKT in myoblasts [31] and ERK in Hela cells [32]. In our investigated breast cancer cell lines, we found that the effects of inhibition of HSP90 differed between TNBCs and T47D. Treatment of investigated breast cancer cells with 0.1μM 17-DMAG only triggered significant decrease of p-ERK and p-AKT in TNBC cells not T47D (Figure 4A). For HDAC6, silencing of HDAC6 prevents oncogenic Shp2-induced ERK hyper-activation in MDCK epithelial cells [33], HDAC6 inhibition leads to AKT de-phosphorylation and inactivation in U87MG glioblastoma and PC-3 prostate cancer cells [34], while HDAC inhibitor LBH589 treatment down-regulated HDAC6 and sustained ERK activation in LNCaP and PC-3 prostate cancer cells [35]. We discovered that inhibition of HDAC6 by Tubacin activated ERK and AKT via increasing p-ERK and p-AKT in T47D (Figure 4B).
Recent study indicated that inhibition of HSP90 inhibited Hippo pathway and activated YAP via depletion LATS1 and LATS2, two regulators of Hippo pathway [36]. We found that inhibition of HSP90 by 0.1μM 17-DMAG decreased YAP protein level in TNBCs but not T47D (Figure 4A), suggesting that compared to T47D, Hippo pathway are more sensitive to HSP90 inhibition in TNBC cells. To our knowledge, it's largely unknown whether inhibition of HDAC6 modulates Hippo pathway. We found that inhibition of HDAC6 in T47D led to decrease of YAP protein level, indicating the involvement of Hippo pathway in HDAC6 regulating network (Figure 4B). Taken together, although HSP90-HDAC6 regulating network involves ERK, AKT and Hippo pathway, involvement of these pathway in HSP90-HDAC6 regulating network differed between TNBCs and T47D. These findings might give reasonable explanations for the observation that TNBCs were more sensitive to HSP90 inhibition than T47D was. It's worth to mention, our study also indicated that YAP as an component of HSP90-HDAC6 regulating network might be a promising common target for TNBCs, ERα+ and tamoxifen-resistant breast cancers.
ERα was known to be a HSP90 client protein [19]. HDAC6 inhibition didn't affect ERα protein level in T47D. Unexpected, combination of HDAC6 and HSP90 inhibition triggered sharper decrease of ERα compared with single use of HSP90 inhibitor in T47D (Figure 2D). Since it was reported that deacetylated HSP90 is unable to bind its inhibitor [37], inhibition of HDAC6 might enhanced the effect of HSP90 inhibition on ERα via increasing the affinity of HSP90 to its inhibitor. These observations might also explain the synergetic effect of HDAC6 inhibition in combination with HSP90 inhibition in T47D (Figure 2E, Table 2).
Since tamoxifen is the most commonly drug for treatment of ERα+ breast cancer patients, and resistance-resistance is an important and common problem during clinical treatment of ERα+ breast cancer patients [25]. Besides ERα mutations [38], alteration of expression and/or activity of co-activators and co-repressors, which play pivotal roles in mediating transcriptional activation by ERα, may also contribute to tamoxifen resistance [39]. Although co-activator SRC-1 overexpression and NCoR down-regulation were suggested to contribute to tamoxifen resistance, no evidence was reported in breast tumors from tamoxifen-resistant patients [40]. ERα could not function if it was isolated from other signaling pathways, and numerous studies have validated the cross-talk between ERα and signaling pathways, including EGFR/HER2, MAPK/ERK and PI3K pathways. In current study, the expression of HDAC6 and YAP were found to be increased upon tamoxifen treatment in T47D, and T47D-TAR also exhibited elevated HDAC6 and YAP expression (Figure 5C, D). These results indicated that alteration expression of HDAC6 and YAP upon tamoxifen treatment might contribute to the induction and establishment of tamoxifen-resistance. Inhibition of HDAC6 and knockdown of YAP can be considered during treatment of tamoxifen-resistant breast cancers.
In conclusion, our current study extended the HSP90-HDAC6 regulating network and provided valuable information for precision treatment of breast cancer via targeting HSP90-HDAC6 regulating network.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was funded by the National Basic Research Program of China (973 Program, No. 2015CB965000 to L. Chen and 2013CB911300 to J. Chen), National Natural Science Foundation of China (No. 81572712 & 81272260 to L. Chen), the Fundamental Research Funds for the Central Universities (No.3231005410 to L. Chen), grants from the National Natural Science Foundation of China (No. 81572928 & 81272469 to R. Chai) and the clinical special project for Natural Science Foundation of Jiangsu Province (No. BL2012016 to J. Chen), Jiangsu Natural Science Foundation Outstanding Youth Foundation (No. SBK2016030027 to L. Chen), and the Six talent peaks project in Jiangsu Province (No. 2015-JY-002 to L. Chen).
Author's Contribution
Conception and design: L. Chen.
Development of methodology: L. Chen, S. Yu.
Acquisition of data: L. Chen, S. Yu, X. Cai, C. Wu, Y. Liu, J. Zhang, X. Gong, Z. Yu, J. Chen, R. Chai, J. Gu, L. Mo.
Analysis and interpretation of data: L. Chen, S. Yu, X. Wang, C. Wu, X. Wu.
Writing review, and /or revision of the manuscript: L. Chen, J.P. Thiery, L. Chen, S. Yu.
Administrative, technical, or material support: L. Chen, S. Yu, Z. Yu, J. Chen, R. Chai, T. Zhu, X. Wu, J. Gu.
Study supervision: L. Chen, S. Yu.
Other (project leader): L. Chen.
Ethical approval
This study was conducted in accordance with the instructional standard guideline of Southeast University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Bray F, Center MM. et al. Global Cancer Statistics. CA Cancer J Clin. 2011;61(2):69-90
2. Dent R, Trudeau M, Pritchard KI. et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429-4434
3. Yadav BS, Sharma SC, Chanana P. et al. Systemic treatment strategies for triple-negative breast cancer. World J Clin Oncol. 2014;5(2):125-133
4. Gajria D, Chandarlapaty S. HER2-amplified breast cancer: mechanisms of trastuzumab resistance and novel targeted therapies. Expert Rev Anticancer Ther. 2011;11(2):263-275
5. Garcia-Becerra R, Santos N, Diaz L. et al. Mechanisms of Resistance to Endocrine Therapy in Breast Cancer: Focus on Signaling Pathways, miRNAs and Genetically Based Resistance. Int J Mol Sci. 2013;14(1):108-145
6. Schiff R, Massarweh S, Shou J. et al. Breast cancer endocrine resistance: how growth factor signaling and estrogen receptor coregulators modulate response. Clin Cancer Res. 2003;9(1 Pt 2):447S-454S
7. Kramer OH, Mahboobi S, Sellmer A. Drugging the HDAC6-HSP90 interplay in malignant cells. Trends Pharmacol Sci. 2014;35(10):501-509
8. Wiech H, Buchner J, Zimmermann R. et al. Hsp90 Chaperones Protein Folding Invitro. Nature. 1992;358(6382):169-170
9. Kamal A, Thao L, Sensintaffar J. et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425(6956):407-410
10. Yang F, Zhang W, Shen Y. et al. Identification of dysregulated microRNAs in triple-negative breast cancer (review). Int J Oncol. 2015;46(3):927-932
11. Banerji U. Heat shock protein 90 as a drug target: some like it hot. Clin Cancer Res. 2009;15(1):9-14
12. Boyault C, Sadoul K, Pabion M. et al. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene. 2007;26(37):5468-5476
13. Aldana-Masangkay GI, Sakamoto KM. The role of HDAC6 in cancer. J Biomed Biotechnol. 2011;2011:875824
14. Li G, Wu X, Qian W. et al. CCAR1 5' UTR as a natural miRancer of miR-1254 overrides tamoxifen resistance. Cell Res. 2016;26(6):655-673
15. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27-55
16. Neve RM, Chin K, Fridlyand J. et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer cell. 2006;10(6):515-527
17. Chen X, Gardner ER, Gutierrez M. et al. Determination of 17-dimethylaminoethylamino-17-demethoxygeldanamycin in human plasma by liquid chromatography with mass-spectrometric detection. Journal of chromatography B, J Chromatogr B Analyt Technol Biomed Life Sci. 2007;858(1-2):302-306
18. Lang SA, Klein D, Moser C. et al. Inhibition of heat shock protein 90 impairs epidermal growth factor-mediated signaling in gastric cancer cells and reduces tumor growth and vascularization in vivo. Mol Cancer Ther. 2007;6(3):1123-1132
19. Bagatell R, Khan O, Paine-Murrieta G. et al. Destabilization of steroid receptors by heat shock protein 90-binding drugs: a ligand-independent approach to hormonal therapy of breast cancer. Clin Cancer Res. 2001;7(7):2076-2084
20. Chan KC, Ting CM, Chan PS. et al. A novel Hsp90 inhibitor AT13387 induces senescence in EBV-positive nasopharyngeal carcinoma cells and suppresses tumor formation. Mol Cancer. 2013:12
21. Yu S, Cai X, Wu C. et al. Adhesion glycoprotein CD44 functions as an upstream regulator of a network connecting ERK, AKT and Hippo-YAP pathways in cancer progression. Oncotarget. 2015;6(5):2951-2965
22. Caldas-Lopes E, Cerchietti L, Ahn JH. et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci U S A. 2009;106(20):8368-8373
23. Asthana J, Kapoor S, Mohan R. et al. Inhibition of HDAC6 deacetylase activity increases its binding with microtubules and suppresses microtubule dynamic instability in MCF-7 cells. J Bio Chem. 2013;288(31):22516-22526
24. Druker BJ. David A. Karnofsky Award lecture. Imatinib as a paradigm of targeted therapies. J Clin Oncol. 2003;21(23 Suppl):239s-245s
25. Garcia-Becerra R, Santos N, Diaz L. et al. Mechanisms of resistance to endocrine therapy in breast cancer: focus on signaling pathways, miRNAs and genetically based resistance. Int J Mol Sci. 2012;14(1):108-145
26. Caldas-Lopes E, Cerchietti L, Ahn JH. et al. Hsp90 inhibitor PU-H71, a multimodal inhibitor of malignancy, induces complete responses in triple-negative breast cancer models. Proc Natl Acad Sci U S A. 2009;106(20):8368-8373
27. Proia DA, Zhang CH, Sequeira M. et al. Preclinical Activity Profile and Therapeutic Efficacy of the HSP90 Inhibitor Ganetespib in Triple-Negative Breast Cancer. Clin Cancer Res. 2014;20(2):413-424
28. Wen J, Yeo S, Wang C. et al. Autophagy inhibition re-sensitizes pulse stimulation-selected paclitaxel-resistant triple negative breast cancer cells to chemotherapy-induced apoptosis. Breast Cancer Res Treat. 2015;149(3):619-629
29. Bali P, Pranpat M, Bradner J. et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: a novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem. 2005;280(29):26729-26734
30. Hostein I, Robertson D, DiStefano F. et al. Inhibition of signal transduction by the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin results in cytostasis and apoptosis. Cancer Res. 2001;61(10):4003-4009
31. Yun BG, Matts RL. Hsp90 functions to balance the phosphorylation state of Akt during C2C12 myoblast differentiation. Cell Signal. 2005;17(12):1477-1485
32. Donze O, Abbas-Terki T, Picard D. The Hsp90 chaperone complex is both a facilitator and a repressor of the dsRNA-dependent kinase PKR. EMBO J. 2001;20(14):3771-3780
33. Tien SC, Chang ZF. Oncogenic Shp2 disturbs microtubule regulation to cause HDAC6-dependent ERK hyperactivation. Oncogene. 2014;33(22):2938-2946
34. Chen CS, Weng SC, Tseng PH. et al. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. J Biol Chem. 2005;280(46):38879-38887
35. Chuang MJ, Wu ST, Tang SH. et al. The HDAC inhibitor LBH589 induces ERK-dependent prometaphase arrest in prostate cancer via HDAC6 inactivation and down-regulation. PLoS One. 2013;8(9):e73401
36. Huntoon CJ, Nye MD, Geng L. et al. Heat shock protein 90 inhibition depletes LATS1 and LATS2, two regulators of the mammalian hippo tumor suppressor pathway. Cancer Res. 2010;70(21):8642-8650
37. Barrott JJ, Haystead TA. Hsp90, an unlikely ally in the war on cancer. FEBS J. 2013;280(6):1381-1396
38. Robinson DR, Wu YM, Vats P. et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45(12):1446-1451
39. Lu R, Hu X, Zhou J. et al. COPS5 amplification and overexpression confers tamoxifen-resistance in ERalpha-positive breast cancer by degradation of NCoR. Nat Commun. 2016;7:12044
40. Ring A, Dowsett M. Mechanisms of tamoxifen resistance. Endocr Relat Cancer. 2004;11(4):643-658
Author contact
![]() Corresponding authors: Liming Chen Email:chenliming1981edu.cn TEL: +86-25-83790995 Fax:0086-25-83790972; Renjie Chai Email: rjchai821116com; Jean Paul Thiery Email: bchtjpedu.sg; Jinfei Chen Email: jinfeichencom; Zhenghong Yu Email: 13327800182cn.
Corresponding authors: Liming Chen Email:chenliming1981edu.cn TEL: +86-25-83790995 Fax:0086-25-83790972; Renjie Chai Email: rjchai821116com; Jean Paul Thiery Email: bchtjpedu.sg; Jinfei Chen Email: jinfeichencom; Zhenghong Yu Email: 13327800182cn.