Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(5):652-659. doi:10.7150/ijbs.19108 This issue Cite
Research Paper
Ezh2 Acts as a Tumor Suppressor in Kras-driven Lung Adenocarcinoma
Yanxiao Wang1, 2, Ning Hou2, Xuan Cheng2, Jishuai Zhang2, Xiaohong Tan2, Chong Zhang1, 2, Yuling Tang2, Yan Teng2 ![]() , Xiao Yang1, 2
, Xiao Yang1, 2 ![]()
1. E-institutes of Shanghai Universities, Shanghai Jiaotong University School of Medicine, Shanghai 200025, China;
2. State Key Laboratory of Proteomics, Genetic Laboratory of Development and Disease, Institute of Biotechnology, Beijing 100071, China.
Received 2017-1-8; Accepted 2017-3-17; Published 2017-5-16
Abstract

Previous studies have suggested that enhancer zeste homolog 2 (Ezh2), a histone methyltransferase subunit of polycomb repressive complex 2 (PRC2), acts as an oncogene in lung adenocarcinoma (ADC) development. However, we found that in human lung ADC samples, deletion and mutations of EZH2 were also frequently present, with 14% of patients harboring loss-of-function EZH2 alterations. To explore the effect of Ezh2 loss on lung tumor formation, lung epithelial Ezh2 gene was deleted in Kras-driven lung ADC mouse model. Unexpectedly, Ezh2 loss dramatically promoted Kras-driven ADC formation. KrasG12D/+;Ezh2fl/fl mice exhibited shorter lifespan, more tumor lesions and higher tumor burden than KrasG12D/+ mice, suggesting the tumor-suppressive role of Ezh2 in Kras-driven ADCs. Mechanistically, Ezh2 loss amplified Akt and ERK activation through de-repressing its target insulin-like growth factor 1 (Igf1). Additionally, Ezh2 loss cooperated with Kras mutation to exacerbate the inflammatory response, as shown by massive macrophage and neutrophil infiltrates, as well as a marked increase in tumor-associated cytokines such as IL-6 and TNF-α. Taken together, our findings revealed the tumor suppressive function of Ezh2 in Kras-driven ADCs, underlining the importance of revaluating the application of EZH2 inhibitors in a variety of cancers.
Keywords: Ezh2, tumor suppressor, Kras, lung adenocarcinoma.
Introduction
Lung cancer is the most frequent and deadliest cancer worldwide. Adenocarcinoma (ADC) accounts for the majority of lung cancer and is the first leading cause of death related to lung cancer [1, 2]. By using genetically modified mouse models, numerous genetic alterations such as Kras and p53 mutations have been proved to contribute to lung ADC formation and progression [3, 4]. However, the epigenetic dysregulation involved in lung tumor formation remains elusive.
EZH2 is the catalytic subunit of PRC2, which methylates Lys27 of histone H3 (H3K27), leading to transcriptional repression of the target genes [5]. An early indication of the oncogenic role for EZH2 comes from the clinical observation that EZH2 overexpression is correlated with poor prognosis in a variety of cancers including prostate cancer, bladder cancer, endometrial cancer and melanoma [6, 7]. Similar findings have been reported in non-small-cell lung cancer (NSCLC), as high level of EZH2 is associated with poor progression [8, 9]. In vitro studies have demonstrated that EZH2 promotes cancer cell proliferation in various cancer types including lung cancer [10]. Genetic engineering mouse models confirm that Ezh2 is a cancer driver, as overexpression of wild-type or activating mutant Ezh2 leads to the development of lymphoid (such as B cell lymphoma and myeloproliferative disorder) and solid (such as melanoma and lung ADC) malignancies by itself [11, 12]. Consequently, there are currently several clinical trials testing EZH2 inhibitors in different cancer types and more new EZH2 inhibitors are under development [13].
However, there is also evidence that EZH2 acts as a tumor suppressor in certain contexts. Studies from cancer genome sequencing reveal deletion and loss-of-function mutations of EZH2 gene in myelodysplastic syndromes (MDS), myeloproliferative neoplasms, glioblastoma and T-acute lymphoblastic leukemia (T-ALL) [14-16]. Furthermore, Ezh2 loss has been found to enhance the initiation of Runx1-mutant MDS, Kras-mutant pancreatic cancer and N1ICD-expressing breast cancer in mice [11, 17, 18]. In lung ADC formation, the complex roles of Ezh2 and PRC2 are emerging. Although forced overexpression of Ezh2 initiates lung ADCs, it does not enhance the development of Kras-driven ADCs [12]. Consistently, Ezh2 inhibition enhances the sensitivity of TopoII inhibitor in EGFG and BRIG mutant NSCLC mouse models, but has no effect in the NSCLC mouse model with Kras and p53 mutations [19]. On the other hand, two separate reports show that the decreased levels of H3K27me3 and EZH2 are associated with poor prognosis in human lung ADC patients [20, 21]. Furthermore, embryonic ectoderm development (Eed), another component of PRC2, exhibits the tumor-suppressive function in the formation of Kras-p53-double-mutant NSCLC in mice [22]. Thus, given that EZH2 inhibitors as a compelling strategy for anti-cancer therapy are undergoing clinical trials, it is critical to validate the definite function of EZH2 as either an oncogene or a tumor suppressor and the underlying mechanisms in certain genetic context of lung cancer.
In this study, we found that 14% of human ADC patients harbored heterozygous deletion and loss-of-function mutations of EZH2 gene in TCGA database. Next, we used conditional deletion of Ezh2 allele to investigate the consequence of Ezh2 loss in Kras-driven lung mouse model.
Materials and methods
Lung cancer mouse models
Loxp-stop-loxp-KrasG12D (KrasG12D/+) mice were kindly gifted by Dr. Kwok-Kin Wong and described previously [3]. Targeted Ezh2flox/flox (Ezh2fl/fl) embryonic stem (ES) cells (ID: EPD0052_2_A05) were from the European Conditional Mouse Mutagenesis (EOCOMM) Program. The Ezh2fl/fl mice were generated by microinjection of ES cells into C57BL/6J blastocysts. All animals were maintained under specific pathogen-free conditions, approved by the Animal Care and Use Committee, and handled in accordance with institutional guidelines for laboratory animals. To induce lung tumors, mice between 6 to 8 weeks were treated with adenoviral virus encoding Cre-recombinase (Ad-Cre) via nasal inhalation. 125 μl Opti-MEM containing 5x106 PFU Ade-Cre viruses was administered to KrasG12D/+, Ezh2fl/fl, KrasG12D/+;Ezh2fl/+ and KrasG12D/+;Ezh2fl/fl mice in two shots. 0.6 μl of 2M CaCl2 was added in the Opti-MEM culture medium to improve lung gene transfer.
Histology and immunohistochemistry assay
Five-micrometer sections of mouse lung tissues and other organs were placed on coated slides for haematoxylin-eosin (H&E) and immunohistochemistry staining. Each lung tissue section contained the maximum coronal planes of all five lung lobs in individual mice. For immunohistochemistry, after deparaffinization and rehydration, the sections were incubated with 3% H2O2 in distilled water to neutralize endogenous peroxidase. Antigen unmasking was performed using heat treatment with citrate solution. Then the sections were incubated overnight at 4 °C with the primary antibodies. The antibodies and dilutions used were: TTF1 (ab133737, abcam), H3K27me3 (ab6002, abcam), p-ERK (4376, Cell Signaling Technology), p-Akt (4060, Cell Signaling Technology), F4/80 (76437, Cell Signaling Technology), S100A9 (ab105472, abcam). The secondary antibodies were added and then incubated at room temperature for 1 hour before DAB reaction. Tumor numbers were counted on one H&E slide representing a complete cross section of the lungs per animal. Tumor burden was shown as the percentage of total tumor area over total lung area. Tumor area and total lung area quantification were performed by ImageJ measurements, and 10 randomly chosen fields from the lung lobes of each mouse were measured [23].
Western blot assay
Lung tumors were harvested from KrasG12D/+ and KrasG12D/+;Ezh2fl/fl mice for western blotting. Tumors were lysed in RIPA buffer containing phosphatase inhibitors and complete mini protease inhibitors. 20-30 μg of lysate proteins were separated by SDS-gel electrophoresis and transferred to PVDF membranes, which were then incubated with antibodies overnight. Western blots were imaged by Image Quant LAS 4000 mini (GE healthcare).
Quantitative Real-time PCR assay
Total RNAs were extracted from the dissociated tumors of KrasG12D/+ and KrasG12D/+;Ezh2fl/fl mice by Trizol reagent (Invitrogen) and phenol/chloroform methods. 2 μg total RNAs were reverse transcribed to cDNAs using Super RT cDNA synthesis kit. Quantitative real-time PCRs for specific genes were performed using the 7500 Fast Real-Time PCR System with the Real Master Mix containing SYBR Green and unique primers.
Gapdh forward CCTCGTCCCGTAGACAAAATG; reverse TCTCCACTTTGCCACTGCAA.
Cxcl1 forward GCTCCCTTGGTTCAGAAAATTG; reverse TCACCAGACAGGTGCCAT CA.
Cxcl2 forward CCTGCCAAGGGTTGACTTCA; reverse TTTTGACCGCCCTTGAGAGT.
Cxcl3 forward AGTGCCTGAACACCCTAC;
reverse AGTGGCTATGACTTCTGTCT.
Cxcl5 forward ACTCGCAGTGGAAAGAAC;
reverse GTGAGATGAGCAGGAAGC.
Cxcl7 forward GCCTGCCCACTTCATAAC;
reverse ACATTCACAAGGGAGATAG.
Ly6G-mus forward GAGGCAGTATTCCAAGGT; reverse GACTCAACAGGGAGACATT.
G-CSF-mus forward GGGAAGGAGATGGGTAAAT; reverse GGAAGGGAGACCAGAT GC.
Statistical analysis
The data of human lung cancers analyzed in this study were downloaded or derived from two previous TCGA studies. The DNA mutation and copy number data were downloaded from cBioPortal (www.cbioportal.org). “-2” in copy number indicates “deep deletion” , “-1” indicates “shallow deletion”, “2” in copy number indicates “amplify”, “1” indicates “gain”, and “0” indicates “diploid”. Kaplan-Meier analysis was used to evaluate survival time. The unpaired Student's t-test (two-tailed) was used to evaluate the gene expression, tumor number and tumor burden. Expression differences among three groups were analyzed by ANOVA. Statistical analysis was performed using GraphPad Prism software v5. Data are represented as mean ± SEM. All results are considered statistically significant at p < 0.05.
Results
Loss-of-function deletion and mutations of EZH2 gene in human lung ADCs
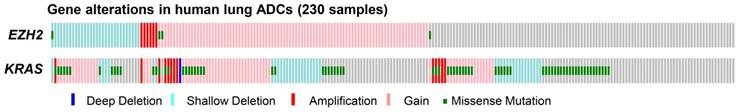
First, we analyzed the copy number variations and mutations of EZH2 in 230 human ADC samples from the previously published TCGA data [24]. We found that EZH2 gain/amplification was prevalent in human lung ADCs, as shown by EZH2 gain/amplification in 42% (97/230) of the ADC samples (Figure 1). Interestingly, heterozygous deletion and inactivating mutations of EZH2 gene were observed in 14% (33/230) of the samples (Figure 1), indicating that EZH2 loss might contribute to ADC formation and progression. Furthermore, KRAS mutations, a major oncogenic driver of ADCs, were present in 33% (75/230) of the samples (Figure 1). We also found co-occurrence of EZH2 deletion and KRAS mutations in 5% (11/230) of the samples (Figure 1), implying EZH2 loss might cooperate with KRAS mutations to contribute to ADC formation.
EZH2 alterations in human ADC samples. EZH2 alterations including gain/amplification, deletion and mutations, were found in 56% (128/230, Gain/Amplification = 97, Deletion = 30, Mutation = 4) of human lung ADC samples. KRAS alterations including gain/amplification, deletion and mutations, were found in 67% (153/230, Gain/Amplification = 77, Deletion = 40, Mutation = 75) of human lung ADC samples. The gene alterations were distinguished by colors.
Ezh2 loss promoted lung ADC development in KrasG12D/+ mice
To interrogate the role of Ezh2 loss in vivo, we generated a mouse line carrying the conditional Ezh2 allele, and the Ezh2fl/fl mice were administered with adenovirus encoding Cre recombinase (Ad-Cre) at 6-8-weeks of age. After Ad-Cre induction, Ezh2fl/fl mice did not display any pathological alterations over an average observed time of 60-weeks (n = 8), indicating that Ezh2 loss alone was insufficient to initiate pulmonary tumor.
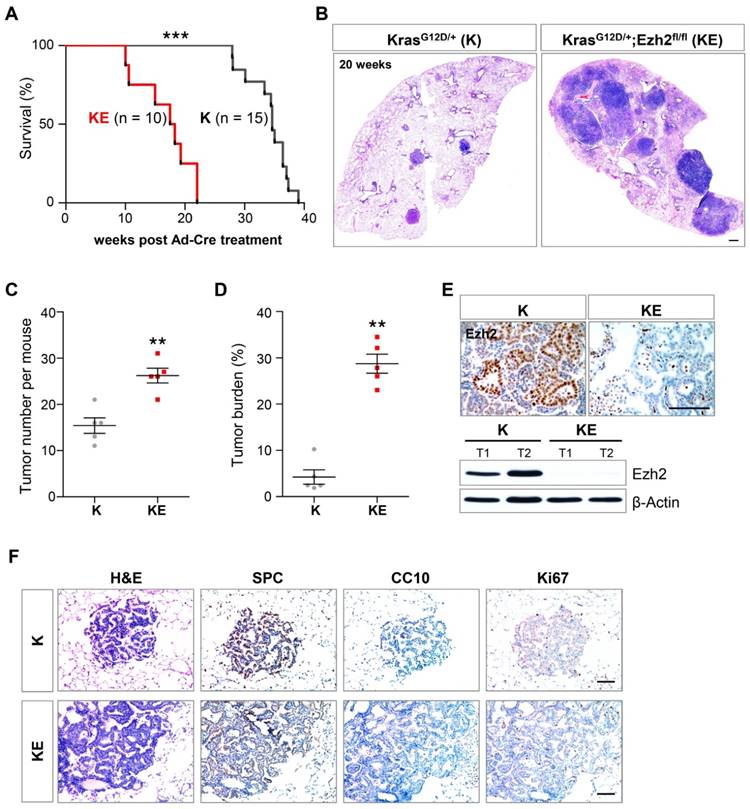
Overwhelming evidence has proved that Kras signaling is a key oncogenic driver of lung ADCs [3]. Given the KRAS gene mutations and EZH2 gene deletion were not mutually exclusive in human lung ADC samples (Figure 1), we further examined the consequence of Ezh2 loss in KrasG12D/+ (hereafter K) mice. As previously reported [3], K mice developed 10-20 lesions of adenoma/adenocarcinoma per mouse at 20 weeks post Ad-Cre treatment and died within 40 weeks (Figure 2A, 2B and 2C). In contrast, beginning at 15 weeks after recombination, KrasG12D/+;Ezh2fl/fl (hereafter KE) mice became moribund and had to be euthanized. Survival analysis revealed that KE mice showed a dramatically reduced lifespan compared with K mice (18 versus 34 weeks; p < 0.0001; Figure 2A). Histopathologic examination of the lung lobes in KE mice showed extensive tumor lesions at 20 weeks post Ad-Cre treatment (Figure 2B). Importantly, further statistical analysis revealed that KE mice presented with an approximately 2-fold increase in the total lesion number (15 versus 26; n = 5; p < 0.001; Figure 2C). Tumor burden was also dramatically increased in KE mice (4% versus 29%; n = 5; p < 0.01; Figure 2D). The Ezh2 deletion in KE tumors was confirmed by Ezh2 immunohistochemistry (IHC) and western blot (WB) analyses (Figure 2E). Moreover, KE tumors showed typical ADC features as evidenced by the expression of surfactant protein C (SPC) (Figure 2F). Immunostaining showed Ki67-positive tumor cells in Kras and Ezh2 double mutant lesions were comparable to those in Kras mutant alone lesions (Figure 2F). Furthermore, we found loss of one allele of Ezh2 enhanced the initiation of Kras-driven ADCs, as characterized by the more (20 versus 15; n = 5; p < 0.01; Figure S1A and S1B) and larger (23% versus 4%; n = 5; p < 0.01; Figure S1A and S1C) tumor lesions in KrasG12D/+;Ezh2fl/+ mice when compared with the tumors in KrasG12D/+ mice. Taken together, we demonstrated that Ezh2 loss in KrasG12D/+ mice specifically initiated lung ADCs with short latency and rapid progression, demonstrating a tumor-suppressor role of Ezh2 in lung ADC development.
Ezh2 loss accelerated the lung adenocarcinoma formation in KrasG12D/+ mice. (A) Survival curves of KrasG12D/+ (K) (n = 15) and KrasG12D/+;Ezh2fl/fl (KE) (n = 10) mice after Ad-Cre induction. The median survival period was significantly reduced from 34 weeks of K mice to 18 weeks of KE mice (p < 0.0001, Log-rank test). (B) H&E staining of the right lung lobes in K and KE mice 20 weeks post Ad-Cre inhalation. KE mice showed more tumor lesions than K mice. The scale bars represent 500 μm. (C) Quantification of the tumor lesion number per mouse in K (n = 5) and KE (n = 5) mice 20 weeks after Ad-Cre treatment (15 versus 26; p < 0.001). (D) Quantification of the tumor burden as the percentage of total tumor area over total lung area in K (n = 5) and KE (n = 5) mice 20 weeks after Ad-Cre treatment (4% versus 29%; p < 0.01). (E) IHC and WB analyses of Ezh2 expression in lung tumors of K and KE mice. The scale bars represent 100 μm. (F) H&E and IHC analyses of serial tumor sections showed that the tumor lesions of KE mice were positive for the ADC marker SPC and negative for CC10. Ki-67 staining indicated the proliferative cells in K and KE tumors. The scale bars represent 100 μm. The error bars indicate mean ± SEM and p value is calculated by unpaired Student's t test. ÕÕp < 0.01; ÕÕÕp < 0.001.
Ezh2 loss amplified Akt and ERK activation through de-repressing its target Igf1
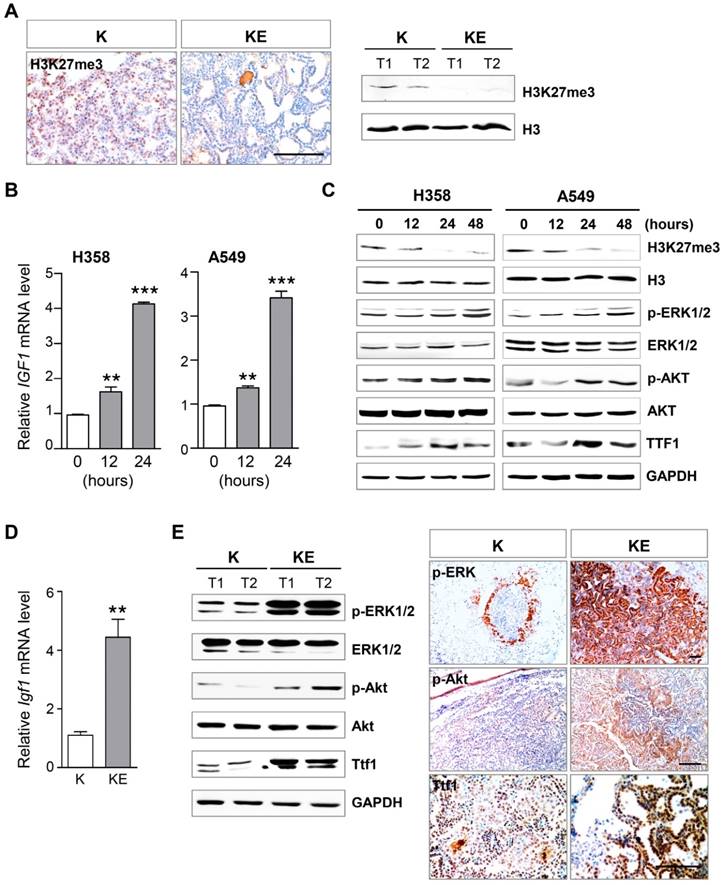
Next, we investigated the Ezh2 downstream effectors and pathways that contributed to neoplastic transformation. Because Ezh2 is crucial for the function of PRC2 complex, we first examined the H3K27me3 level in ADCs from KE mice. As expected, IHC and WB results showed that global H3K27me3 abundance was significantly reduced in KE tumors as compared to that in K tumors (Figure 3A). A previous study has demonstrated that in mouse lung development, Igf1 is an Ezh2 target that can be silenced by H3K27me3 on its promoter, and is required for inducing p63 expression, a key regulator of basal cell differentiation [25]. Given that IGF1 signaling is also involved in neoplastic transformation by its downstream pathways such as AKT and ERK, we treated two human KRAS-mutant NSCLC cell lines H358 and A549 with an EZH2-specific inhibitor GSK126 that can block the catalytic activity but does not impair PRC2 complex assembly. Notably, GSK126 treatment led to an increase in IGF1 mRNA level beginning at 12 hours in the cell lines (Figure 3B), thereby activating the downstream effectors AKT and ERK, as characterized by elevated expression of phosphorylated AKT (p-AKT) and phosphorylated ERK (p-ERK) after 24 hours of GSK126 treatment (Figure 3C). And GSK126 treatment resulted in the decreased H3K27me3 level in both lung cancer cell lines (Figure 3C). Even though IGF1 signaling was activated, GSK126 treatment did not lead to increased cancer cell proliferation, but led to ceased cell proliferation(Figure S2), implying that other EZH2 downstream effectors withstanding the effects of IGF1 signaling contributed to cell proliferation. Next, we confirmed the increased expression of Igf1 and the amplified activation of Akt and ERK in KE tumors, as compared to those in K tumors (Figure 3D and 3E). Furthermore, we found that thyroid transcription factor 1 (Ttf1), a lineage-survival oncogene in lung ADCs, was markedly upregulated in ADCs from KE mice compared with that from K mice (Figure 3E). TTF1 was also upregulated after GSK126 treatment in human lung cancer cell lines (Figure 3C). Collectively, Ezh2 loss might potentiate the effect of Kras mutation by Igf1-mediated amplifying activation of Akt and ERK, as well as inducing Ttf1 overexpression, all of which contributed to the accelerated ADC formation in KE mice.
Ezh2 loss amplified ERK and Akt activation and induced Ttf1 expression. (A) IHC and WB analyses of H3K27me3 level in KrasG12D/+ (K) and KrasG12D/+;Ezh2fl/fl (KE) ADCs. The scale bar represents 100 μm. (B) qRT-PCR analysis of IGF1 mRNA expression level after EZH2 inhibitor GSK126 treatment in H358 and A459 cell lines. (C) WB analyses of the expressions of p-ERK, p-AKT and TTF1 in H358 and A549 cell lines after GSK126 treatment. (D) qRT-PCR analysis of Igf1 mRNA expression level in K and KE ADCs. (E) IHC and WB analyses of p-ERK, p-Akt and Ttf1 levels in ADCs of K and KE mice. The scale bar represents 100 μm. The error bars indicate mean ± SEM and p values are calculated by unpaired Student's t test. ÕÕp < 0.01; ÕÕÕp < 0.001.
Ezh2 loss exacerbated inflammatory response
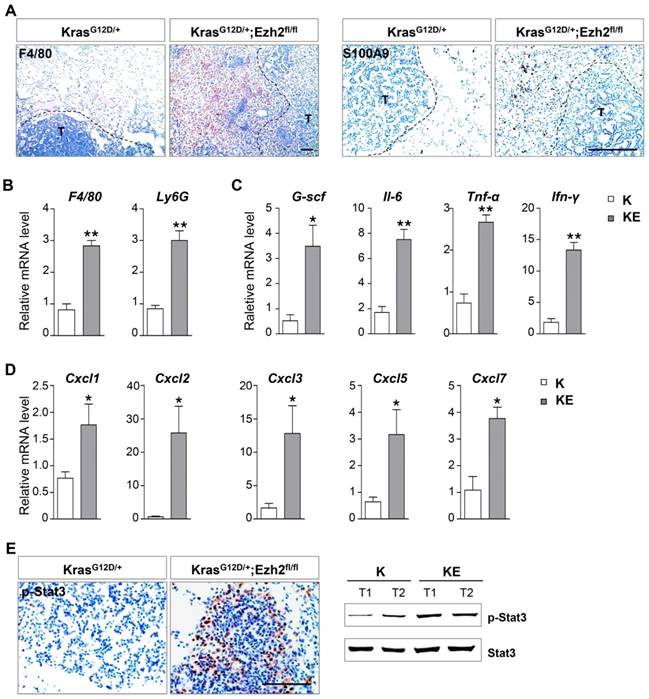
A previous study has shown that in a Kras-driven pancreatic cancer mouse model, Ezh2 loss leads to inflammatory microenvironment and thus, contributes to the acceleration of tumor progression [17]. In a lung ADC mouse model, deletion of embryonic ectoderm development (Eed) gene also cooperates with Kras and p53 mutations to induce severe inflammatory response [22]. We next examined whether Ezh2 loss led to the change of tumor stroma. In comparison with ADCs in K mice, ADC lesions in KE mice showed massive inflammatory infiltrates, which affected the alveolar airspace and effective gas exchange that might ultimately lead to respiratory failure. Immunostaining results revealed a marked increase in F4/80+ macrophages and S100A9+ neutrophils surrounding KE tumor nests (Figure 4A), which was further confirmed by quantitative real-time PCR results (Figure 4B). As expected, the mRNA levels of inflammatory-related cytokines such as Il-6, G-csf, Tnf-α and Ifn-γ, as well as the CXC-ligand family members Cxcl1, Cxcl2, Cxcl3, Cxcl5 and Cxcl7, were highly expressed in ADCs from KE mice compared to those from K mice (Figure 4C and 4D). In consistence with the upregulation of Il-6, its downstream effector Stat3 was significantly activated in the tumor cells of KE mice (Figure 4E), which thereby promoted tumor progression through survival and immune-suppression. Altogether, these data indicated that Ezh2 loss might contribute to tumor progression by inducing inflammatory response.
Ezh2 loss exacerbated inflammatory response. (A) IHC analyses of F4/80 and S100A9 of the tumor stroma in KrasG12D/+ (K) and KrasG12D/+;Ezh2fl/fl (KE) mice. The black-dotted line indicated tumor lesions (T). The scale bars represent 100 μm. (B) qRT-PCR analyses showed the increased F4/80 and Ly6G mRNA expressions in ADC lesions of K and KE mice. (C) qRT-PCR analyses of Il6, G-csf, Tnf-α and Ifn-γ in ADC lesions of K and KE mice. (D) qRT-PCR analyses of Cxcl2, Cxcl3, Cxcl5 and Cxcl7 in ADC lesions of K and KE mice. (E) IHC and WB analyses of p-Stat3 of tumors in K and KE mice. The scale bars represent 100 μm. The error bars indicate mean ± SEM and p values are calculated by unpaired Student's t test. Õp < 0.05; ÕÕp < 0.01.
Discussion
Kras mutations are the most prevalent alterations in various solid cancer types. In particular, Kras mutations are present in 33% of lung ADC patients and the most important drivers in mouse lung ADC models. Although Ezh2 transgenic mouse with overexpression of wild type Ezh2 led to lung ADCs, the oncogenic function of Ezh2 was not observed in Kras mutant background [12, 22]. They further found that Ezh2 overexpression resulted in low phosphorylation levels of Akt and ERK as compared to Kras mutant alone ADCs [12], partly explaining the inability of Ezh2 to accelerate Kras-driven ADC formation. In this study, we provided human genetic, mouse model and molecular evidence demonstrating the tumor suppressive function of Ezh2 in Kras-driven lung ADCs, pointing to the differing functions of Ezh2 in distinct genetic context of lung cancer. Implication of concurrent EZH2 deletion/mutations and KRAS mutations in human lung cancer genomic data was supported by mouse model showing the tumor-promotive effect of Ezh2 homozygous or heterozygous loss on Kras-driven ADC development. In accordance with the low levels of p-Akt and p-ERK caused by Ezh2 overexpression [12], we further found that Ezh2 loss resulted in de-repressing of Igf1, which thereby activated its downstream pathways Akt and ERK, contributing to the neoplastic transformation. Supportively, a negative correlation between low levels of Ezh2 and Akt or ERK activation is present in human lung ADC samples [12]. Notably, the similar tumor-suppressive function of Ezh2 has been shown in Kras-driven pancreatic cancer [17]. Thus, these above data showed that, in some Kras-driven cancers, at least lung ADC and pancreatic cancer, Ezh2 acted as a tumor-suppressor rather than an oncogene in tumorigenesis, supporting the notion that Ezh2 has a potent and genetic context-dependent tumor-suppressor function.
In agreement with the previous studies [10], our in vitro experiments showed that EZH2 inhibition led to reduced cell proliferation in KRAS-mutant lung NSCLC cell lines, even though EZH2 inhibition could result in pronounced activation of ERK and AKT. One possibility is that Ezh2 has dichotomous roles at different stages of lung cancer, i.e., Ezh2 could suppress the initiation of Kras-driven ADCs, but promote ADC progression. However, the emerging picture is much more complicated. A recent study has shown that although Ezh2 depletion in glioblastoma initially slows down tumor growth, the prolonged Ezh2 depletion causes a robust epigenetic switch in cell fate, resulting in altered cancer cell identity and tumor progression, arguing in favor of the tumor-suppressive function of Ezh2 as a barrier to cell identity transition during cancer progression [26]. Similar finding has emerged in the Kras-driven p53 mutant lung ADC model, co-deletion of another PRC2 component Eed leads to invasive mucinous adenomagenesis via inducing epithelial-to-mesenchymal transition program, suggesting that Eed acts as a context-dependent tumor-suppressor through regulating cell identity [22]. Therefore, it will be important to explore the in vivo definite role of Ezh2 in the progression of Kras-driven tumors.
Several small molecules inhibiting EZH2 activity are being explored in clinical trials including lung cancers [13]. However, our study showed that, instead of repressing tumors, Ezh2 inhibition could further stimulate ADC development triggered by Kras mutation, the major driver for ADCs, raising the concern of EZH2 inhibitors treatment as an anti-cancer strategy in a variety of cancers.
Supplementary Material
Supplementary figures.
Abbreviations
ADC: adenocarcinoma
EZH2: enhancer zeste homolog 2
PRC2: polycomb repressive complex 2
NSCLC: non-small-cell lung cancer
SPC: surfactant protein C
IGF1: insulin-like growth factor 1
EED: embryonic ectoderm development
TTF1: thyroid transcription factor 1
Acknowledgements
This work was supported by National Natural Science Foundation of China 81572717, 31430057, 31630093, 81272702 and 31571512, the Chinese National Key Program on Basic Research 2012CB945103 and the National Key Research and Development Program of China 2016YFC1300600.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Brambilla E, Travis WD, Colby TV. et al. The new World Health Organization classification of lung tumours. Eur Respir J. 2001;18:1059-68
2. Jemal A, Bray F, Center MM. et al. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
3. Jackson EL, Willis N, Mercer K. et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243-8
4. Winslow MM, Dayton TL, Verhaak RG. et al. Suppression of lung adenocarcinoma progression by Nkx2-1. Nature. 2011;473:101-4
5. Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343-9
6. Cerami E, Gao J, Dogrusoz U. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401-4
7. Gao J, Aksoy BA, Dogrusoz U. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
8. Behrens C, Solis LM, Lin H. et al. EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non-small cell lung carcinoma. Clin Cancer Res. 2013;19:6556-65
9. Huqun Ishikawa R, Zhang J et al. Enhancer of zeste homolog 2 is a novel prognostic biomarker in nonsmall cell lung cancer. Cancer. 2012;118:1599-606
10. Xia H, Zhang W, Li Y. et al. EZH2 silencing with RNA interference induces G2/M arrest in human lung cancer cells in vitro. Biomed Res Int. 2014;2014:348728
11. Wassef M, Rodilla V, Teissandier A. et al. Impaired PRC2 activity promotes transcriptional instability and favors breast tumorigenesis. Genes Dev. 2015;29:2547-62
12. Zhang H, Qi J, Reyes JM. et al. Oncogenic Deregulation of EZH2 as an Opportunity for Targeted Therapy in Lung Cancer. Cancer Discov. 2016;6:1006-21
13. Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128-34
14. Thomas Ernst, Andrew J Chase, Joannah Score. et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010Aug;42:722-6
15. Nikoloski G, Langemeijer SM, Kuiper RP. et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42:665-7
16. Ntziachristos P, Tsirigos A, Van Vlierberghe P. et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med. 2012;18:298-301
17. Mallen-St Clair J, Soydaner-Azeloglu R, Lee KE. et al. EZH2 couples pancreatic regeneration to neoplastic progression. Genes Dev. 2012;26:439-44
18. Sashida G, Harada H, Matsui H. et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun. 2014;5:4177
19. Fillmore CM, Xu C, Desai PT. et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature. 2015;520:239-42
20. Li Z, Xu L, Tang N. et al. The polycomb group protein EZH2 inhibits lung cancer cell growth by repressing the transcription factor Nrf2. FEBS Lett. 2014;588:3000-7
21. Chen X, Song N, Matsumoto K. et al. High expression of trimethylated histone H3 at lysine 27 predicts better prognosis in non-small cell lung cancer. Int J Oncol. 2013;43:1467-80
22. Serresi M, Gargiulo G, Proost N. et al. Polycomb repressive complex 2 is a barrier to KRAS-driven inflammation and epithelial-mesenchymal transition in non-small-cell lung cancer. Cancer Cell. 2016;29:17-31
23. William Y Kim, Samanthi Perera, Bing Zhou. et al. HIF2alpha cooperates with RAS to promote lung tumorigenesis in mice. J Clin Invest. 2009;119:2160-70
24. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543-50
25. Galvis LA, Holik AZ, Short KM. et al. Repression of Igf1 expression by Ezh2 prevents basal cell differentiation in the developing lung. Development. 2015;142:1458-69
26. de Vries NA, Hulsman D, Akhtar W. et al. Prolonged Ezh2 depletion in glioblastoma causes a robust switch in cell fate resulting in tumor progression. Cell Report. 2015 [Epub ahead of print]
Author contact
![]() Corresponding authors: tengyan0919com or yangxac.cn
Corresponding authors: tengyan0919com or yangxac.cn