Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(5):669-681. doi:10.7150/ijbs.19714 This issue Cite
Research Paper
PPARD is an Inhibitor of Cartilage Growth in External Ears
Zhen Zhang*, Yanyu Duan*, Zhongping Wu, Hui Zhang, Jun Ren ![]() , Lusheng Huang
, Lusheng Huang
State Key Laboratory of Pig Genetic Improvement and Production Technology, Jiangxi Agricultural University, Nanchang, 330045, China.
* These authors contributed equally to this work.
Received 2017-2-18; Accepted 2017-3-29; Published 2017-5-16
Abstract

Peroxisome proliferator-activated receptor beta/delta (PPARD) is an important determinant of multiple biological processes. Our previous studies identified a missense mutation in the PPARD gene that significantly reduces its transcription activity, and consequently causes enlarged external ears in pigs. However, the mechanisms underlying the causality has remained largely unknown. Here, we show that PPARD retards the development of auricular cartilage by accelerating the apoptosis of cartilage stem/progenitor cells (CSPCs), the terminal differentiation of cartilage cells and the degradation of cartilage extracellular matrix in the auricle. At the transcription level, PPARD upregulates a set of genes that are associated with CSPCs apoptosis and chondrogenic differentiation, chondroblast differentiation and extracellular matrix degradation. ChIP-seq identified direct target genes of PPARD, including a well-documented gene for cartilage development: PPARG. We further show that compared to wild-type PPARD, the G32E mutant up-regulates the expression of PPARG and subsequently leads to the downregulation of critical genes that inhibit cartilage growth. These findings allow us to conclude that PPARD is an inhibitor of auricular cartilage growth in pigs. The causative mutation (G32E) in the PPARD gene attenuates the PPARD-mediated retardation of cartilage growth in the auricle, contributing to enlarged ears in pigs. The findings advance our understanding of the mechanisms underlying auricular development in mammals, and shed insight into the studies of innate pinna disorders and cartilage regeneration medicine in humans.
Keywords: PPARD, ear size, cartilage growth, chondrogenesis, matrix degeneration.
Introduction
Peroxisome proliferator-activated receptor beta/delta (PPARD) is a ligand-modulated transcription factor that belongs to the nuclear receptor superfamily [1]. PPARD serves as a key and multifaceted determinant of diverse biological functions, such as lipid metabolism, embryonic development, inflammatory response, would healing, etc. [2-7]. Our previous study established a novel role of PPARD in external ear development [8]. We identified a protein-altering mutation (G32E) in an evolutionary conservation domain of PPARD that causes enlarged ears in pigs [8]. The G32E causative mutation stimulates nuclear export of PPARD, decreases ubiquitination level in the A/B domain of PPARD that is required for efficient ligand-induced transactivation and consequently reduces activity of PPARD [9]. We further show that PPARD G32E decreases the expression of β-catenin and its target genes, presumably leading to lipid production and storage in external ears and contributing to enlarged ear size in pigs [8].
The external ear is mainly composed of skin, cartilage and fat. So far, the role of PPARD in skin development and fat metabolism in the auricle has been firmly established [8-11]. However, the effect of PPARD in auricular cartilage development and the molecular mechanisms underlying the causality of PPARD G32E has remained largely unknown. To this end, we herein conducted a series of assays to show that PPARD is an inhibitor of cartilage development in the external ear. The G32E mutation attenuates the inhibition effect of PPARD, stimulates the cartilage and skin development and fat deposition in the auricle, which collectively causes enlarged ear sizes in pigs. The finding improves our understanding of mechanisms underlying external ear development in mammals, and provides insight into studies of congenital pinna defects and cartilage regeneration medicine in humans.
Materials and Methods
Measurement of ear size
The ear sizes of Erhualian and Duroc pigs were measured as previously described [8]. Briefly, photographs were taken for one ear of each animal after ear was fixed and covered with a ruler as an internal reference of the size. Ear sizes were calculated using the Qwin software (Leica). Significance was evaluated using a Student's t test.
Histochemical examination
Auricular cartilage was dissected from the root of the external ear of five Erhualian and five Duroc pigs and was then fixed in 4% paraformaldehyde. The paraffin-embedded cartilage samples were cut into 7-µm sections and placed on glass slides. Three sections for each sample were used for H&E staining and Masson-Goldner trichrome staining as previously described [12].
Cell culture and reagent
Auricular-derived primary cartilage cells were isolated from external ears of a 4-month-old Duroc × (Landrace × Large White) hybrid (hereafter DLL) pig homozygous for the wild-type allele at PPARD G32E using enzymatic digestion as previously described [9]. Cells were cultured at 37 °C in a humid atmosphere of 5% CO2 in DMEM-F12 (Gibco) medium freshly supplemented with 20% fetal bovine serum (Gibco) and antibiotics (Gibco). When the confluence reached 70%, the third-generation primary cells were incubated with 0.1 μM GW0742 while mediums were replaced with DMEM-F12 plus 10% charcoal-stripped serum (Hyclone). After incubated for 24 h, cells were collected for subsequent flow cytometry, RNA sequencing and chromatin immunoprecipitation (ChIP) sequencing analyses. The quantity and quality of isolated RNA and ChIPed DNA samples were analyzed using a Qubit Fluorometer (Thermo Fisher) and a 2100 Bioanalyzer (Agilent). Pig PK-15 cells were cultured in DMEM (high glucose, Gibco) medium supplemented with 10% fetal bovine serum (Gibco) and antibiotic solution. When cells were incubated with the PPARD agonist GW0742 (Tocris), culture mediums were replaced with DMEM plus 5% charcoal-stripped serum (Hyclone).
Flow cytometry analysis
Flow cytometry was conducted to identify cartilage stem/progenitor cells (CSPCs) using two stem cell markers CD44 and CD90. When the confluence of primary cells reached 80%, these cells were digested using EDTA-free trypsin (Gibco). After washed two times by PBS (Gibco), one million cells were resuspended in 100 μL PBS. Four experimental sets were simultaneously prepared as follows: double negative samples and single positive samples for CD44, single positive samples for CD90 and double positive samples for CD44 and CD90. Cells were incubated with 20 μL CD44 (BD pharmingen) and/or 5 μL CD90 (BD pharmingen) for 35 min on ice in darkroom. Then cells were analyzed by an Accuri C6 flow cytometer (BD pharmingen).
The apoptosis of CSPCs and chondrocytes were measured by the Annexin V/ PI apoptosis detection kit II (BD pharmingen) according to the manufacture's instruction. Briefly, one million cells were resuspended in 100 μL binding buffer after washed two times by PBS. Four experimental sets, namely double negative, single positive for annexin V and single positive for PI and double positive for annexin V and PI, were prepared by adding 5 μL PI and/or 5 μL Annexin V into binding buffer, respectively. After incubation for 30 min on ice in darkroom, the data were obtained by the Accuri C6 flow cytometer (BD pharmingen).
RNA sequencing (RNA-seq)
Total RNA was isolated from three untreated (DMSO) and three GW0742-treated primary cartilage cells using Trizol reagent (Invitrogen), and was then used to construct cDNA libraries using the TruSeq RNA Sample Preparation kit (Illumina). Single-end reads (125SE) were then generated on an Ion Proton sequencer (Life Technologies) at a depth of approximately 20 million reads. Next, trimmed clean reads were mapped to the pig reference genome (Sscrofa 10.2, ftp://ftp.ensembl.org/pub/release-78/fasta/sus_scrofa/dna/) allowing five mismatches using the SOAP2 software [13]. Gene expression levels were further calculated using the RPKM (reads per kilobase of transcript per million mapped reads) method to identify differentially expressed genes (DEGs) between treated and untreated samples as previously described [14]. A FDR of no more than 0.001 (FDR ≤ 0.001) and a difference ratio of PRKM no less than 2 (|log2Ratio|≥1) between the untreated and treated samples were adopted as the significant threshold to identify DEGs. The common DEGs among three comparison groups were further filtered by fold-change ≥ 2 and diverge probability ≥ 0.8 [15]. GO and KEGG pathway enrichment analyses of DEGs were performed using Blast2GO [16] and Cytoscape [17], respectively. The functional interactions among DEGs were retrieved from the STRING database that integrates experiments, text mining, databases and co-expression to link candidate genes into a related network [18].
ChIP sequencing (ChIP-seq)
Chromatin immunoprecipitation samples were prepared from auricle-derived primary cartilage cells using the SimpleChIP Enzymatic Chromatin IP kit (CST) according to manufacturer's instruction. Briefly, cells were harvested and chemically cross-linked with 1% formaldehyde for 10 min at 15 °C. The cell nucleus was released by sonication using an Ultrasonic Processor (SONXI). The collected nuclei were digested with Micrococcal nuclease (CST) for 20 min at 37 °C. Sheared chromatin was incubated with Anti-PPARD (Santa Cruz), Anti-Histone H3 (positive control, CST) and Anti-Rabbit IgG (negative control, CST) overnight. After enriched by ChIP-Grade Protein G magnetic beads for 2 h, immunoprecipitated DNA was treated with Proteinase K (CST) and purified with the SimpleChIP kit (CST). ChIP sequencing was performed at Beijing Institute BGI (Wuhan, China). Briefly, sequencing library was constructed from enriched DNA using the TruSeq ChIP Library Prep kit (Illumina). Final DNA libraries were validated and sequenced at 50 bp per sequence read using a HiSeq 4000 sequencer (Illumina) at a depth of approximately 20 million sequences. ChIP-seq reads were mapped to the pig reference genome (Sscrofa 10.2) by BEDtools [19]. Only the alignments with less than 2 mismatches were considered for enriched-region (peak) identification (peak calling) using the Model-based Analysis of ChIP-seq (MACS) algorithm (--bwidth=150, --gsize= 2.50e+09 --mfold=8) [20]. Only peaks with a MACS-assigned FDR of less than 10% and fewer than 100 tags in the IgG track were selected. The PeakAnalyzer software was used for peak annotation and the nearest target gene analyses [21].
Real-time quantitative PCR
Total RNA was reverse transcribed with random hexamers (Takara). Real-time quantitative-PCR was conducted on a 7900HT Real-Time PCR Detection system (ABI) using the Bio SYBR Green Master Mix (Takara) or Taqman Master Mix (ABI). Amplification efficiencies of each gene-specific primer pair was determined using a five-fold serial dilution series. Primers with an efficiency of 1.8 to 2.15 were used for further analyses (Table S8). The housekeeping gene BACT was used to normalize relative fold changes between PPARD agonists (GW0742)-treated samples and untreated (DMSO) samples. Each qPCR analysis was repeated for at least three independent biological replicates, and significance was analyzed using a Student's t test.
Dual luciferase reporter assay
The PPARD binding region in the PPARG promoter spanning Chromosome 13: 75588490 to 75588637 bp was cloned and ligated upstream of tk-pGL4.20 to generate PPARG-tk-Luc plasmids. The PPARG-tk-Luc and pRL-tk constructs were co-transfected into PK-15 cells (1×105 cells/well) together with empty, PPARD G32E mutant- or wild-type PPARD-overexpression vectors using Lipofectamine 2000 (Invitrogen) in 24-wells plates [9]. Twenty-four hours after transfection, the activity of firefly luciferase was measured using the Dual-Luciferase Reporter Assay System (Promega) on an Infinite 200 PRO multimode reader (Tecan). Relative luciferase activity was determined based on the firefly/renilla luciferase activity ratio. Each reporter plasmid was transfected in triplicate, and three independent reporter assays were performed. Significance was analyzed using a Student's t test.
Results
Phenotypic characterization of external ears in Erhualian and Duroc pigs
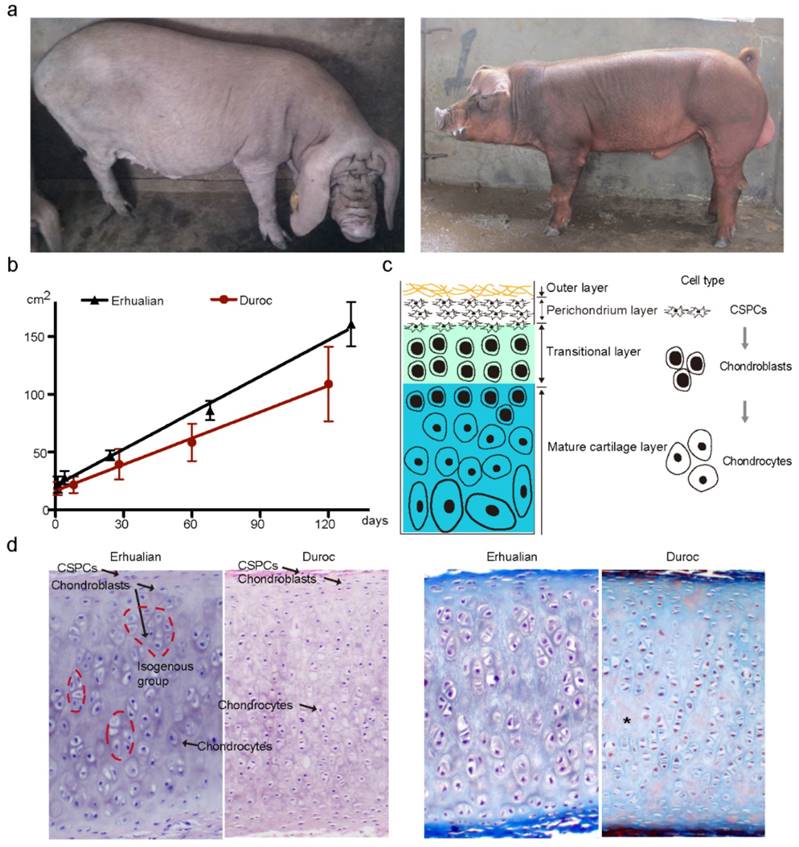
We first measured the size of external ears in one Chinese large-eared breed (Erhualian) and one European small-eared breed (Duroc) (Fig. 1a) at five time points during the period of 0 to 4 months after birth. Erhualian had significantly (P < 0.0001) larger ears than Duroc across these five time points (Fig. 1b). Indeed, the ear size exhibited a growth slope of 6.29 (i.e. 6.29 cm2 per day) in Erhualian and of 4.50 in Duroc (Fig. 1b). Then, we examined the histological properties of auricular cartilage at 120 ± 3 days in the two breeds. Like in mice and humans [22, 23], auricular cartilage in pigs is also composed of fibers and vessels in the outer layer, CSPCs in the perichondrium layer, chondroblasts in the transitional layer and chondrocytes in the mature cartilage layer (Fig. 1c). Chondroblasts express high levels of aggrecan and type II collagen, the two main components of the cartilage extracellular matrix, while mature chondrocytes express type X collagen and catabolic factors [24-26]. Compared with Duroc, Erhualian showed obvious difference in the proportion of cell types in the auricular cartilage. Erhualian had much more isogenous groups that were formed after chondroblasts divided continuously (Fig. 1d), representing an interstitial growth that occurs mainly in immature cartilage. In contrast, mature chondrocytes accounted for a majority of cartilage cells and matrix showed a certain level of degradation in Duroc (Fig. 1d), indicating that chondroblasts tend to differentiate into chondrocytes rather than to proliferate in external ears of Duroc pigs. This explains that Erhualian pigs have a thicker and denser immature cartilage layer in outer ears than Duroc.
External ear characteristics of Erhualian and White Duroc pigs. (a) Erhualian pigs (left panel) have large and fully floppy ears while Duroc pigs (right panel) have small and partly pricked ears. (b) Ear size (y axis) of Erhualian (n = 48) and Duroc pigs (n = 60) measured at five time points (x axis). Growth curves of the two breeds were compared using linear regression analysis (P < 0.0001). (c) Schematic diagram of auricular cartilage. (d) Representative sections of auricular cartilage stained with hematoxylin and eosin (left panel) and Masson-Goldner trichrome (right panel) from Erhualian and Duroc pigs at 120 ± 3 days of age. Arrows and asterisks indicate cell types and degenerating matrix, respectively.
PPARD accelerates the apoptosis and chondrogenic differentiation of CSPCs in the external ear
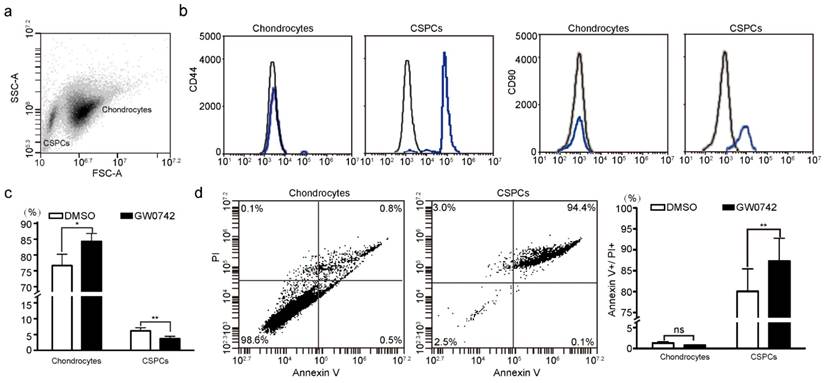
To identify the role of PPARD in chondrogenesis, we first tested the percentage of CSPCs in auricular cartilage cells in DLL pigs using flow cytometry analysis. We found that 2%-6% of cartilage-derived cells were CSPCs (Fig. 2a), which expressed mesenchymal stem cell markers CD44 and CD90 (Fig. 2b). Then, we investigated the effect of PPARD on auricular cartilage cells by evaluating its agonist (GW0742)-induced cell growth. After the treatment with GW0742 for 24 h, a larger (P < 0.05) proportion of chondrocytes and a smaller (P < 0.01) proportion of CSPCs were identified in treated cells than untreated samples (Fig. 2c), indicating that PPARD is a positive regulator of CSPCs differentiation. Further, we evaluated the apoptosis of auricular cartilage cells using the Annexin V-FITC and PI staining (see Materials and Methods) after GW0742 treatment for 24 h. The result demonstrates that PPARD activation specifically induces the apoptosis of CSPCs but not chondrocytes in the external ear (Fig. 2d). In summary, PPARD inhibits the proliferation of auricular cartilaginous cells by affecting two critical steps in chondrogenesis, i.e. PPARD accelerates the apoptosis of CSPCs and promotes the chondrogenic differentiation of CSPCs. This indicates that large-eared breeds carrying the mutant allele at the PPARD G32E site (such as Erhualian) have a larger number of CSPCs and hence a greater potential of auricle development than small-eared breeds carrying the wild-type allele at PPARD G32E (such as Duroc) due to the decreased biological activity of PPARD caused by the G32E mutation [9].
Effect of PPARD on cells in pig auricular cartilage. Scatter plot for primary cells isolated from auricular cartilage in pigs. Cells were analyzed using FACS on a flow cytometer. (b) Characterization of CSPCs. CSPCs show higher expression levels of two cell-surface markers (CD44 and CD90, blue line) than control samples (black line). Isotype control antibodies were used for control samples. (c) PPARD stimulates the differentiation of CSPCs into chondrocytes. Cells were either untreated (DMSO) or treated with 0.1 µM PPARD agonist (GW0742) for 24 h and analyzed by flow cytometry. Values are expressed as mean ± S.E. of triplicate experiments. Significance was assessed by a two-paired t-test. * P < 0.05, ** P < 0.01. (d) PPARD induces the apoptosis of CSPCs but not chondrocytes. Primary cartilage cells were untreated (DMSO) or treated with 0.1 µM GW0742 for 24 h and analyzed for phosphatidylserine exposure (Annexin V binding) and plasma membrane rupture (PI uptake) using FACS. The fluorescence intensities of Annexin V-FITC and PI are presented as dot plots. Annexin V-/PI- staining represents surviving cells, Annexin V+/PI- cells indicates early apoptosis, Annexin V+/PI+ and Annexin V-/PI+ cells indicates necrotic or apoptotic cells at the middle and terminal stage, respectively. Values are expressed as mean ± S.E. of triplicate experiments. Significance was evaluated using a two-paired t-test. ** P < 0.01.
PPARD upregulates target genes involved in CSPCs apoptosis, cartilage-derived cells differentiation and matrix degradation
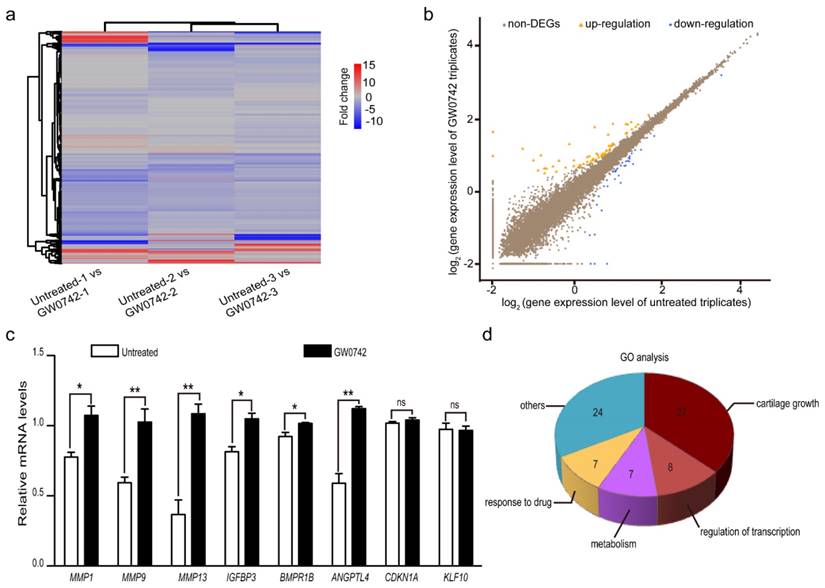
To determine the effect of PPARD on auricular cartilage growth at the transcription level, we quantified whole-genome transcriptomes of auricle-derived primary cartilage cells that were treated (n = 3) or untreated (n = 3) with GW0742 for 24 h using RNA-seq. In total, we obtained over 22 million clean reads from each library after trimming adaptor sequences, low quality reads and multiple mapped reads. The clean reads were mapped to 18,200 genes annotated in the pig reference genome (Sus Scrofa 10.2, Table S1). We then identified DEGs between the GW0742-treated and untreated groups by comparing the RNA-seq data between the two groups (Fig. 3a). A total of 98 DEGs were defined using the thresholds of false discovery rate (FDR) ≤ 0.001, difference ratio of RPKM (reads per kilobase of exon per million fragments mapped) ≥ 2 [27] and diverge probability ≥ 0.8 [15] (see Materials and Methods), including 62 up-regulated genes and 36 down-regulated genes (Fig. 3b, Table 1). To confirm the reliability of the identified DEGs, we randomly chose eight genes with RPKM values of 0 to 75 and detected their expression levels in the GW0742-treated and untreated samples using reverse-transcription quantitative PCR (RT-qPCR) (Table S2). A consistent pattern with a correlation coefficient of 0.71 was observed for these eight genes between qPCR and RNA-seq data (Figure S1). We noticed that two DEGs with a RPKM value of 0 (CDKN1A and KLF10) did not show differential expression in RT-qPCR analysis (Fig. 3c). So we excluded 14 DEGs with a RPKM value of 0 to reduce potentially false positives, and focused on the remaining 84 DEGs with a minimal PRKM value of 0.2 (Table S3) in subsequent analyses.
Differentially expressed genes (DEGs) between PPARD agonist (GW0742)-treated and untreated primary cartilage cells derived from pig external ears. (a) Heatmap shows comparison of whole-genome gene expression between the treated and untreated cells. Each column represents one pairwise comparison and each row one gene. Red color indicates upregulation and blue color indicates downregulation. (b) Scatter plots of whole-genome gene expression in the treated and untreated cells. Log2 value of each gene expression in untreated and GW0742-treated triplicates is shown in X- and Y-axis, respectively. DEGs are defined by diverged probability ≥ 0.8 and fold-change ≥ 2. Each dot represents one gene. Up-regulated genes in the treated samples are shown in orange, down-regulated genes in blue, and non-DEGs in brown. (c) Validation of RNA sequencing data by RT-qPCR. Eight DEGs were randomly selected and analyzed by RT-qPCR. Values are expressed as mean ± S.E. of triplicate experiments after normalization to BACT mRNA levels. Significance was assessed by a two-paired t-test. * P < 0.05, ** P < 0.01. (d) The enriched GO biological processes for the identified DEGs.
Differentially expressed genes identified by RNA-seq
| Comparison group 1 | Comparison group 2 | Comparison group 3 | Common DEGs | |||||
|---|---|---|---|---|---|---|---|---|
| Up | Down | Up | Down | Up | Down | Up | Down | |
| Characterized gene | 191 | 124 | 293 | 241 | 129 | 88 | 57 | 35 |
| Uncharacterized gene | 15 | 8 | 14 | 22 | 5 | 10 | 5 | 1 |
DEGs, differentially expressed genes; Up, up-regulated genes; Down, down-regulated genes.
Next, we performed the pathway enrichment analysis for these 84 DEGs. As expected, the PPAR signaling pathway was the most significantly (P = 6.00×10-06, Q = 6.96×10-04) enriched pathway (Table S4). By applying functional enrichment analysis, we noticed that 57.53% of DEGs (n = 42) were involved in cartilage growth, transcription regulation and metabolism (Fig. 3d). The top 20 DEG-enriched biological processes included positive regulation of angiogenesis (GO:0045766), positive regulation of osteoblast differentiation (GO:0045669), bone mineralization (GO:0030282), negative regulation of cells proliferation (GO:0043066) and positive regulation of apoptotic process (GO:0043065) (Table S3). These biological processes are all potentially related to the development of the auricle.
Then, we made a close examination on 15 DEGs functionally related to cartilage growth according to their GO terms and literature meaning. These 15 DEGs are involved in five processes of cartilage development (Table 2). Of them, six genes including ANGPTL4 [28], BCL6 [29], CEBPB [30-33], IGFBP3 [34], PGF [35] and RAD21 [36] could reduce the number of CSPCs in the perichondrium layer by inducing the programmed cell death of CSPCs and stimulating chondrogenic differentiation of CSPCs. IGFBP3 [34] and RAD21 [36], two well-characterized mediators for CSPCs apoptosis, were up-regulated by 2.08 and 2.76 fold in GW0742-treated cells, respectively. Another four up-regulated genes are positive regulators of chondrogenic differentiation of CSPCs, such as CEBPB [30-33] (17.32-fold) and ANGPTL4 [28] (8.92-fold). These findings were in agreement with our finding that PPARD activation led to a smaller proportion of CSPCs in auricular cartilage-derived cells after treating with GW0742 for 24 h.
Differentially expressed genes related to cartilage growth between GW0742-treated and untreated samples
| Function category | Gene symbol | Gene name | Fold change |
|---|---|---|---|
| Induce programmed cell death in CSPCs | IGFBP3 | Insulin-growth factor-binding protein 3 | 2.08 |
| RAD21 | RAD21 cohesin complex component | 2.76 | |
| Stimulate chondrogenic differentiation of CSPCs | ANGPTL4 | Angiopoietion-4 | 8.92 |
| BCL6 | B-cell lymphoma 6 protein | 4.52 | |
| CEBPB | CCAAT/Enhancer-binding protein beta | 17.32 | |
| IGFBP3 | Insulin-growth factor-binding protein 3 | 2.08 | |
| PGF | Placenta growth factor isoform 1 | 2.84 | |
| Enhance migration of CSPCs | AQP1 | Aquaporin-1 | 2.37 |
| Positive regulation of chondrocyte differentiation | BMPR1B | Bone morphogenetic protein receptor type-1B | 2.24 |
| CHI3L1 | Chitinase 3 like 1 | 2.12 | |
| CSF1 | Colony-stimulating factor 1 | 2.22 | |
| CTHRC1 | Collagen triple helix repeat-containing protein 1 | 2.14 | |
| Degrade and reduce synthesis of the cartilage extracellular matrix | ANGPTL4 | Angiopoietion-4 | 8.92 |
| BMPR1B | Bone morphogenetic protein receptor type-1B | 2.24 | |
| CSF1 | Colony-stimulating factor 1 | 2.22 | |
| CTHRC1 | Collagen triple helix repeat-containing protein 1 | 2.14 | |
| MGP | Matrix gla protein | 2.11 | |
| MMP1 | Matrix metallopeptidase 1 | 4.09 | |
| MMP9 | Matrix metallopeptidase 9 | 2.87 | |
| MMP13 | Matrix metallopeptidase 13 | 9.93 | |
| PGF | Placental growth factor | 2.84 |
All 15 genes shown in this table were up-regulated in GW0742-treated samples.
Besides the above-mentioned genes, ten DEGs are involved in cell differentiation and matrix degradation (Table 2). Of them, four genes for chondroblast differentiation were up-regulated including three positive regulators (BMPR1B [37, 38], CSF1 [39] and CTHRC1 [40]) and one secretory glycoprotein at late stage of chondrocyte maturation (CHI3L1 [41]). In cartilage plates, chondroblasts produce extracellular matrix (ECM) typically composing of type II collagen and aggrecan [42]. When ECM is degraded, the differentiation process evolves toward a terminal stage [26]. Interestingly, there were as many as nine genes (60%) in 15 DEGs that play a role in degrading ECM or reducing the synthesis of ECM (Table 2). These nine genes included three ones encoding matrix metallopeptidases (MMP1, MMP9 and MMP13) that were up-regulated in GW0742-treated cells. MMP13 [43] (9.93-fold) and MMP1 [44] (4.09-fold) preferentially digest type II collagen and aggrecan and MMP9 (2.87-fold) degrades type I collagen and aggrecan in cartilage [45]. The remaining six DEGs were also up-regulated in GW0742-treated cells and were all positive regulators of ECM degradation [37-40]. Of note, an activator for ECM remodeling, ANGPTL4, was remarkably up-regulated (8.92-fold). ANGPTL4 is responsible for inhibiting expression of type II collagen and aggrecan in cartilage and inducing the synthesis of two catabolic factors MMP1 and MMP13 [28]. In short, PPARD up-regulate a set of genes that consequently inhibit the growth of auricular cartilage by promoting the apoptosis and chondrogenic differentiate of CSPCs, diminishing the secretion of new matrix components, and irreversibly breaking down the existing matrix.
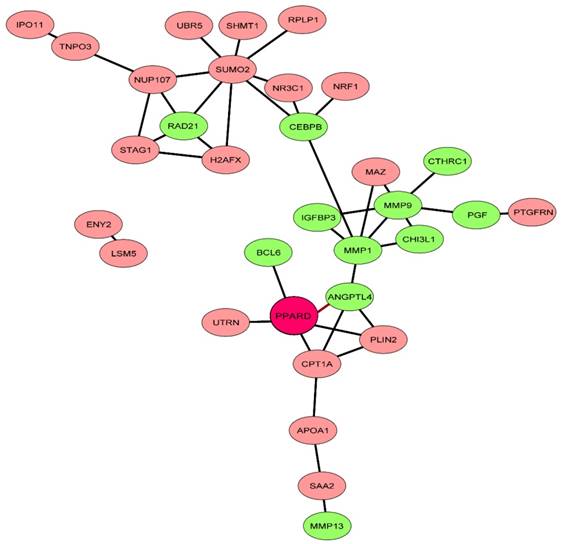
Finally, we investigated the interaction of PPARD with 71 characterized genes out of the 84 DEGs using the STRING algorithm [18]. After removing non-connected genes, 32 genes have interactions, of which 30 were associated with PPARD (Fig. 4). Interestingly, the resulting network showed a cluster of genes (n = 11) that were associated with the CSPCs apoptosis and differentiation, chondroblast differentiation and ECM degradation. This further supports that PPARD activation could slow down cartilage growth.
Network of PPARD downstream genes identified by comparative RNA-seq. The STRING database [37] was explored to uncover functional interactions (edges) among the 71 characterized DEGs (nodes) identified by RNA-seq. Eleven genes related to cartilage growth are indicated by green.
PPARD regulates chondrogenesis and matrix degeneration likely via its target gene PPARG
PPARD is a transcription factor that can directly bind to DNA and regulate the expression of adjacent genes. Here, we performed chromatin immunoprecipitation sequencing (ChIP-seq) to characterize DNA-binding sites of PPARD across the genome in auricular cartilage-derived primary cells.
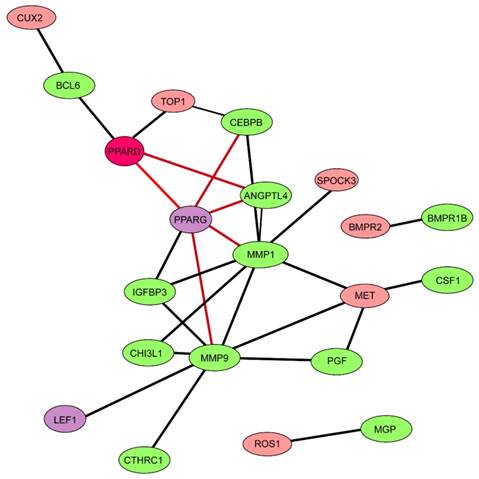
ChIP-seq yielded a total of 25,299,637 reads for PPARD-bound chromatins and 28,535,881 reads for IgG-bound chromatins (control) that were mapped to a unique location on the pig reference genome (Sscrofa 10.2) (Table S5). A total of 838 high confidence enrichment peaks (FDR < 0.1) were identified for PPARD using the MACs peak calling algorithm [20] (Table S6). By searching against the BioMart-Ensembl database [46], 87 binding regions were mapped to target genes (Table S7). Interestingly, these target genes did not overlap with any of the 84 DEGs in response to PPARD activation. It is known that cross-regulatory networks are commonly involved in biological processes, in which one transcription factor regulates the expression of other transcription factors to mediate its downstream genes [47, 48]. We speculated that these DEGs were not directly regulated by PPARD but directly modulated by one or more targets of PPARD, likely transcription factors. To test this speculation, we searched for transcription factors out of DEGs via the TRRUST database [49] and identified five transcription factors including ONECUT1, PPARG, ARNT, ETV6 and LEF1 (Table S7). One transcription factor, PPARG, has direct interactions with six DEGs related to cartilage growth as revealed by the STRING algorithm (Fig. 5). In addition, four DEGs appeared to be direct targets of PPARG in the TRRUST database, including one positive regulator of CSPCs differentiation - CEBPB [30-33], and three regulators of cartilage remodeling - ANGPTL4 [28], MMP1 [43, 44] and MMP9 [45].
Network of 15 differentially expressed genes related to cartilage growth and 87 target genes of PPARD. The network was constructed via STRING database [37]. Nodes and edges indicate genes and functional interactions among genes, respectively. Twelve genes related to cartilage growth are shown in green and two genes (PPARG and LEF1) encoding transcription factors in purple.
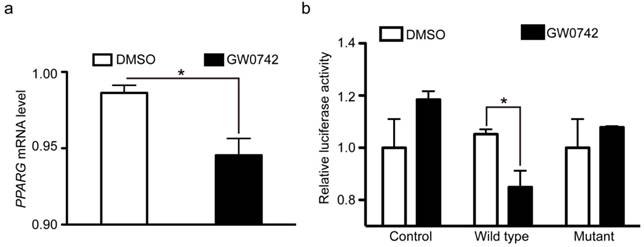
As described above, PPARD acts as a promoter of cartilage degeneration in the development of auricular cartilage. In contrast, PPARG is shown to be an inhibitor of cartilage degeneration [50, 51]. The opposite effect between PPARD and PPARG on cartilage growth indicates that they might have an intrinsic connection. Our ChIP-seq data showed that PPARD bound to the adjacent region of the PPARG gene. This led us to speculate that PPARD activation could repress PPARG expression and subsequently decreases cartilage growth. To test this hypothesis, we further examined whether PPARD represses PPARG expression at the transcription level using RT-qPCR and dual luciferase reporter assay. We first quantified the mRNA level of PPARG in pig auricular cartilage cells treated with PPARD agonist (GW0742) relative to untreated samples via RT-qPCR. A 1.2-fold (P < 0.05) decrease of PPARG mRNA was observed in the treated samples (Fig. 6a), which was consistent with our RNA-seq data (data not shown). Then, we cloned a 147 bp PPARD binding fragment downstream of the transcription start site of PPARG into the pGL4.20-tk luciferase vector. We transfected the construct and plasmids overexpressing the wild-type PPARD, the G32E mutant PPARD or empty plasmid into pig PK-15 cells and measured the luciferase reporter activity after 24 h. The wild-type PPARD but not the G32E mutant overexpression was associated with a significant decrease (1.24 fold, P < 0.05) in luciferase activity (Fig. 6b). Thus, we conclude that PPARD down-regulates the expression of PPARG by directly binding with the promoter of PPARG, while the downregulation is abolished by the G32E mutant in the PPARD gene.
PPARG mRNA expression is repressed by the wild-type PPARD but not by the G32E mutant. PPARD activation inhibits PPARG mRNA expression. Auricle-derived primary cartilage cells were untreated (DMSO) or treated with 0.1 µM PPARD agonist (GW0742) for 24 h and quantified for the mRNA level of PPARG via RT-qPCR. Values are expressed as mean ± S.E. of triplicate experiments and normalized by BACT. Significance was assessed by a two-paired t-test, * P < 0.05. (b) The PPARD G32E mutant has no effect on PPARG mRNA expression. PK-15 cells were transfected with vectors expressing pcDNA4A-His (control), wild-type PPARD (wild type), and G32E mutant (mutant) along with PPARG-tk-Luc and pRL-tk reporter vectors. Cells were incubated for 24 h with DMSO or 0.1 µM GW0742. The ratio of firefly luciferase to renilla luciferase activity was defined as relative luciferase activity. Data represent fold changes relative to empty vector-transfected and GW0742 untreated cells. * P < 0.05.
Discussion
PPARD is a nuclear hormone receptor, which has the ability to directly bind to DNA and regulate the transcription of downstream genes. PPARD has been a drug target for metabolic disorders for decades [52], and is known to play pivotal roles in cartilage development in mice and humans [53-58]. We established a novel role of PPARD in the growth of auricular cartilage in pigs in our previous studies [8, 9]. However, the underlying mechanism of this role remains largely unknown. Here we show that PPARD retards the growth of auricular cartilage by reducing the potential of chondrogenesis and degrading the existing ECM.
The ear grows rapidly from birth until puberty, but slows down after that time in humans and animals [22, 59]. The growth of auricular cartilage involves three processes: (i) new matrix is secreted by chondroblasts at cartilage surface, (ii) internal mass of cartilage increases by chondrocytes and (iii) protein polysaccharide and elastic fibers undergo degradation [23]. During these processes, the growth is mainly driven by continuously chondrogenic differentiation of CSPCs after birth. We herein performed a series of analyses to uncover the role of PPARD in the three processes. By applying flow cytometry assay, we show that PPARD accelerates the chondrogenic differentiation and apoptosis of CSPCs in auricular cartilage. This is consistent with our previous finding that the growth of auricle-derived primary cartilage cells was inhibited after 5-day treatment with PPARD agonist (GW0742) [9]. Through histochemical staining of auricular cartilage, we illustrated that large-eared Erhualian had a larger proportion of CSPCs/ chondroblasts than small-eared Duroc. Furthermore, chondroblasts, the producer of extracellular matrix, had a longer proliferative state in Erhualian in comparison with Duroc. Instead, the chondroblasts in Duroc tended to differentiate into mature chondrocytes that secreted catabolic factors to degrade the existing ECM. Taken together, PPARD has an inhibitive effect on chondrogenesis in pig external ears. We have shown that the G32E causative mutation reduces activity of PPARD [9]. Therefore, it is conceivable to observe that Erhualian carrying the mutant allele at the G32E site had a faster growth rate of auricular cartilage than Duroc carrying the wild-type allele at this site.
Next, we made an effort to uncover the molecular mechanism underlying the inhibition of the growth of auricular cartilage by PPARD. We searched for downstream genes of PPARD that are involved in ear growth by conducting comparative RNA-seq on auricle-derived primary cartilage cells treated or untreated by PPARD agonist, GW0742. As a result, we identified 84 reliable DEGs between the treated and untreated samples, including 15 well-documented genes for cartilage development. Of the 15 DEGs, two directly accelerate the apoptosis of CSPCs, five promote the chondrogenic differentiation of CSPCs, four upregulate chondroblast differentiation and nine reduce the synthesis of ECM and degrade the existing matrix. The biological functions of these DEGs are in complete agreement with our phenotypic observation that PPARD inhibits the growth of auricular cartilage [9]. To further elucidate the molecular mechanism how PPARD regulates the expression of these 15 DEGs, we performed ChIP-seq for PPARD in auricular cartilage-derived primary cells, and identified 838 high confidence enrichment peaks for PPARD, corresponding to 87 binding regions proximal to characterized genes. Unexpectedly, these target genes did not include any of the 15 DEGs for cartilage growth. Of note, five genes encode transcription factors and could be the PPARD-mediated executors that regulate the expression of the identified DEGs. We are particularly interested in one transcription factor, PPARG, as it has four target genes that are critical to cartilage growth, including CEBPB [30-33], ANGPTL4 [28], MMP1 [43, 44] and MMP9 [45]. Then, we obtained further evidence that PPARD represses the expression of PPARG and subsequently upregulates the expression of the critical genes inhibiting cartilage growth.
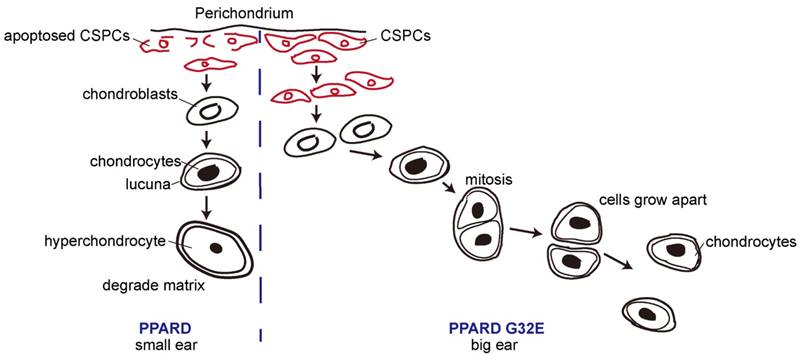
Our findings in this and previous studies [8, 9] allow us to propose a model for the conclusion that PPARD G32E causes phenotypic variation in pig external ears (Fig. 7). The G32E mutation promotes nuclear exportation and reduces ubiquitination level in the critical A/B domain of PPARD, consequently decreasing transcription activity of PPARD [9]. Individuals carrying the mutant allele (G32E), like Erhualian pigs, downregulate a set of genes that stimulate lipid production and storage in the auricle [60], and retard the apoptosis of CSPCs, terminal differentiation of chondroblasts and matrix degradation in auricular cartilage, leading to a prolonged production of proliferative chondroblasts driven by CSPC, and consequently enlarged ears. In contrast, wild-type pigs like Duroc have normal biological activity of PPARD and hence, smaller outer ears than mutant animals.
A proposed model for the conclusion that PPARD G32E stimulates cartilage development in the auricle and consequently causes enlarged external ears in pigs. The continuous chondrogenic differentiation of CSPCs in the prechondrium drives the formation of chondroblasts that secrete new matrix at cartilage surface. Chondrocytes are formed when chondroblasts embed themselves into ECM and lie in the matrix lacunae. Then chondrocytes evolve toward a terminal stage when ECM is degraded. In small-eared pigs (left panel), the wild-type PPARD accelerates the apoptosis of CSPCs and the terminal differentiation of cartilage-derived cells, causes less proliferative chondrocytes in auricular cartilage and consequently retards the growth rate of outer ears. In big-eared pigs (right panel), the G32E mutant reduces the transcription activity of PPARD [9], leading to downregulation of PPARD target genes that inhibit auricular cartilage growth. As a result, the production of proliferative chondrocytes driven by CSPCs/ chondroblasts is prolonged, and hence, the yield of ECM is increased, resulting in enlarged external ears.
In cartilage regeneration medicine, CSPCs have been shown to be a good cellular resource for elastic cartilage reconstruction [23]. For example, the grafting of perichondrocytes allow restoring reconstructed tissue with the unique elastic properties for a long term because of self-renewing CSPCs in the perichondrium layer [23, 61]. Nowadays, surgical reconstruction of craniofacial injuries or abnormalities, such as microtia, completely relies on elastic cartilage grafted from the auricle. Therefore, the development of drugs regulating the differentiation and proliferation of CSPCs could pave the way to cartilage regenerative therapies. We herein show that the activation of PPARD by treatment with the agonist (GW0742) leads to accelerated apoptosis and chondrogenic differentiation of CSPCs. Hence, PPARD provides a potential molecular target to regulate cartilage reconstruction in vitro via its agonists and antagonists [62].
Recently, the role of PPARD in cartilage disorder has attracted a lot of interests from researchers [56]. Osteoarthritis (OA) is the most common form of arthritis, characterized by altered cartilage homeostasis, accelerated chondrocyte cell death and subsequent cartilage degeneration. It has been shown that that inhibition of PPARD protects mice from OA, supporting that PPARD is a promising therapeutic target [56]. We show that PPARD activation leads to cartilage remodeling likely via downregulation of PPARG, which could result in severe OA [51]. This indicates that PPARD could also serves as a therapeutic target for OA in humans.
In summary, our findings are of scientific significance in three points. First, we establish the inhibitory role of PPARD in the development of auricular cartilage, providing further evidence that the causative mutation (G32E) in the PPARD gene contributes to large ears in pigs. This advances our understanding of the mechanisms underlying the growth of external ears in mammals, and sheds insight into studies of human ear pathologies. Second, we demonstrate that PPARD accelerates the apoptosis and chondrogenic differentiation of CSPCs, providing a potential molecular target for regulating elastic cartilage regeneration in vitro to improve long-term tissue restoration for patients with craniofacial defects. Third, we show that PPARD represses the transcription of PPARG in cartilage cells, indicating that PPARD is a potential therapeutic target for OA. Taken together, these findings help us to better understand the biology of cartilage and external ear development in mammals, and to better treat cartilage related diseases in humans.
Supplementary Material
Additional File 1Figure S1.
Tables S1-S8.
Abbreviations
ChIP-seq: chromatin immunoprecipitation sequencing; CSPCs: cartilage stem/progenitor cells; DEGs: differentially expressed genes; DLL: Duroc × (Landrace × Large White); FACS: fluorescence activated cell sorting; OA: osteoarthritis; PPARD: peroxisome proliferator-activated receptor beta/delta; RPKM: reads per kilobase of transcript per million mapped reads; RNA-seq: RNA sequencing; RT-qPCR: reverse-transcription quantitative polymerase chain reaction; STRING: Search Tool for the Retrieval of Interacting Genes/Proteins.
Acknowledgements
We are grateful to Professor Ruihua Huang and Dr. Pinghua Li at Nanjing Agricultural University and Jia Zhang at Fujian Yichun Company for their kind assistance with sample collections. This work was financially supported by the National Natural Science Foundation of China (31301950 and 31671288) and the Special Funds for Graduate Students Innovation of Jiangxi Province (YC2016-B039).
Accession Number
ChIP-seq and RNA-seq data obtained in this study are available in the NCBI GEO database (https://www.ncbi.nlm.nih.gov/geo/) under an accession number GSE92374.
Ethics Statement
This study was conducted strictly in compliance with guidelines of experimental animals established by the Ministry of Agriculture of China, and was approved by the ethics committee of Jiangxi Agricultural University (China).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Saini A, Kaushal C, Gupta P, Tyagi S, Sharma S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. 2011;2(4):236-40
2. Barish GD, Narkar VA, Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. J Clin Invest. 2006;116(3):590-7
3. Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H. et al. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell. 2003;113(2):159-70
4. Evans RM, Barish GD, Wang YX. PPARs and the complex journey to obesity. Nat Med. 2004;10(4):355-61
5. Wang YX, Zhang CL, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR. et al. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e61
6. Han C, Lim K, Xu LH, Li GY, Wu T. Regulation of Wnt/beta-catenin pathway by cPLA(2)alpha and PPAR delta. J Cell Biochem. 2008;105(2):534-45
7. Macsai CE, Foster BK, Xian CJ. Roles of Wnt signalling in bone growth, remodelling, skeletal disorders and fracture repair. J Cell Physiol. 2008;215(3):578-87
8. Ren J, Duan Y, Qiao R, Yao F, Zhang Z, Yang B. et al. A missense mutation in PPARD causes a major QTL effect on ear size in pigs. PLoS Genet. 2011;7:e1002043
9. Duan Y, Brenig B, Wu X, Ren J, Huang L. The G32E functional variant reduces activity of PPARD by nuclear export and post-translational modification in pigs. PLoS One. 2013;8:e75925
10. Sertznig P, Seifert M, Tilgen W, Reichrath J. Peroxisome proliferator-activated receptors (PPARs) and the human skin: importance of PPARs in skin physiology and dermatologic diseases. Am J Clin Dermatol. 2008;9(1):15-31
11. Feingold KR. The outer frontier: the importance of lipid metabolism in the skin. J Lipid Res. 2009;50(Suppl):S417-22
12. Rentsch C, Schneiders W, Manthey S, Rentsch B, Rammelt S. Comprehensive histological evaluation of bone implants. Biomatter. 2014:4 (1)
13. Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966-7
14. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621-8
15. Tarazona S, Garcia-Alcalde F, Dopazo J, Ferrer A, Conesa A. Differential expression in RNA-seq: a matter of depth. Genome Res. 2011;21(12):2213-23
16. Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res. 2008;36(10):3420-35
17. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M. et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008;36(Database issue):D480-4
18. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A. et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(Database issue):D808-15
19. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841-2
20. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137
21. Salmon-Divon M, Dvinge H, Tammoja K, Bertone P. PeakAnalyzer: genome-wide annotation of chromatin binding and modification loci. BMC Bioinformatics. 2010;11(1):415
22. Kobayashi S, Takebe T, Zheng YW, Mizuno M, Yabuki Y, Maegawa J. et al. Presence of cartilage stem/progenitor cells in adult mice auricular perichondrium. PloS One. 2011;6:e26393
23. Kobayashi S, Takebe T, Inui M, Iwai S, Kan H, Zheng YW. et al. Reconstruction of human elastic cartilage by a CD44+ CD90+ stem cell in the ear perichondrium. Proc Natl Acad Sci USA. 2011;108(35):14479-84
24. Steinert AF, Ghivizzani SC, Rethwilm A, Tuan RS, Evans CH, Noth U. Major biological obstacles for persistent cell-based regeneration of articular cartilage. Arthritis Res Ther. 2007;9(3):213
25. Akkiraju H, Nohe A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J Dev Biol. 2015;3(4):177-92
26. Malemud CJ. Matrix metalloproteinases: role in skeletal development and growth plate disorders. Front Biosci. 2006;11(10):1702-15
27. Salkind NJ, Rasmussen K. Encyclopedia of Measurement and Statistics. Reference Reviews. 2007 (8)
28. Mathieu M, Iampietro M, Chuchana P, Guerit D, Djouad F, Noel D. et al. Involvement of angiopoietin-like 4 in matrix remodeling during chondrogenic differentiation of mesenchymal stem cells. J Biol Chem. 2014;289(12):8402-12
29. Mrugala D, Dossat N, Ringe J, Delorme B, Coffy A, Bony C. et al. Gene expression profile of multipotent mesenchymal stromal cells: Identification of pathways common to TGFbeta3/BMP2-induced chondrogenesis. Cloning Stem Cells. 2009;11(1):61-76
30. Smink JJ, Leutz A. Instruction of mesenchymal cell fate by the transcription factor C/EBPbeta. Gene. 2012;497(1):10-7
31. Gutierrez S, Javed A, Tennant DK, van Rees M, Montecino M, Stein GS. et al. CCAAT/enhancer-binding proteins (C/EBP) beta and delta activate osteocalcin gene transcription and synergize with Runx2 at the C/EBP element to regulate bone-specific expression. J Biol Chem. 2002;277(2):1316-23
32. Smink JJ, Tunn PU, Leutz A. Rapamycin inhibits osteoclast formation in giant cell tumor of bone through the C/EBPbeta - MafB axis. J Mol Med. 2012;90(1):25-30
33. Henriquez B, Hepp M, Merino P, Sepulveda H, van Wijnen AJ, Lian JB. et al. C/EBPbeta binds the P1 promoter of the Runx2 gene and up-regulates Runx2 transcription in osteoblastic cells. J Cell Physiol. 2011;226(1):3043-52
34. Longobardi L, Torello M, Buckway C, O'Rear L, Horton WA, Hwa V. et al. A novel insulin-like growth factor (IGF)-independent role for IGF binding protein-3 in mesenchymal chondroprogenitor cell apoptosis. Endocrinology. 2003;144(5):1695-702
35. McCoy RJ, Widaa A, Watters KM, Wuerstle M, Stallings RL, Duffy GP. et al. Orchestrating osteogenic differentiation of mesenchymal stem cells-identification of placental growth factor as a mechanosensitive gene with a pro-osteogenic role. Stem Cells. 2013;31(11):2420-31
36. Chen F, Kamradt M, Mulcahy M, Byun Y, Xu H, McKay MJ. et al. Caspase proteolysis of the cohesin component RAD21 promotes apoptosis. J Biol Chem. 2002;277(19):16775-81
37. Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci USA. 2005;102(14):5062-7
38. Kobayashi T, Lyons KM, McMahon AP, Kronenberg HM. BMP signaling stimulates cellular differentiation at multiple steps during cartilage development. Proc Natl Acad Sci USA. 2005;102(50):18023-7
39. Raggatt LJ, Partridge NC. Cellular and molecular mechanisms of bone remodeling. J Biol Chem. 2010;285(33):25103-8
40. Kimura H, Kwan KM, Zhang Z, Deng JM, Darnay BG, Behringer RR. et al. Cthrc1 is a positive regulator of osteoblastic bone formation. PloS One. 2008;3:e3174
41. Rehli M, Krause SW, Andreesen R. Molecular characterization of the gene for human cartilage gp-39 (CHI3L1), a member of the chitinase protein family and marker for late stages of macrophage differentiation. Genomics. 1997;43(2):221-5
42. Kruger JP, Hondke S, Endres M, Pruss A, Siclari A, Kaps C. Human platelet-rich plasma stimulates migration and chondrogenic differentiation of human subchondral progenitor cells. J Orthop Res. 2012;30(6):845-52
43. Knauper V, Lopez-Otin C, Smith B, Knight G, Murphy G. Biochemical characterization of human collagenase-3. J Biol Chem. 1996;271(3):1544-50
44. Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci. 2006;11(1):529-43
45. Itoh T, Matsuda H, Tanioka M, Kuwabara K, Itohara S, Suzuki R. The role of matrix metalloproteinase-2 and matrix metalloproteinase-9 in antibody-induced arthritis. J Immunol. 2002;169(5):2643-7
46. Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S. et al. The Ensembl gene annotation system. Database (Oxford). 2016
47. Karagianni P, Talianidis I. Transcription factor networks regulating hepatic fatty acid metabolism. Biochim Biophys Acta. 2015;1851(1):2-8
48. Ninkovic J, Steiner-Mezzadri A, Jawerka M, Akinci U, Masserdotti G, Petricca S. et al. The BAF complex interacts with Pax6 in adult neural progenitors to establish a neurogenic cross-regulatory transcriptional network. Cell Stem Cell. 2013;13(4):403-18
49. Han H, Shim H, Shin D, Shim JE, Ko Y, Shin J. et al. TRRUST: a reference database of human transcriptional regulatory interactions. Sci Rep. 2015;5:11432
50. Takeda S, Takeuchi Y. The role of PPAR gamma for the osteoblastic differentiation. Nihon Rinsho. 2004;62(Suppl 2):789-93
51. Vasheghani F, Zhang Y, Li YH, Blati M, Fahmi H, Lussier B. et al. PPARgamma deficiency results in severe, accelerated osteoarthritis associated with aberrant mTOR signalling in the articular cartilage. Ann Rheum Dis. 2015;74(3):569-78
52. Jones D. Potential remains for PPAR-targeted drugs. Nat Rev Drug Discov. 2010;9(9):668-9
53. Shao YY, Wang L, Hicks DG, Tarr S, Ballock RT. Expression and activation of peroxisome proliferator-activated receptors in growth plate chondrocytes. J Orthop Res. 2005;23(5):1139-45
54. Kawai M, Rosen CJ. PPARgamma: a circadian transcription factor in adipogenesis and osteogenesis. Nat Rev Endocrinol. 2010;6(11):629-36
55. Scholtysek C, Katzenbeisser J, Fu H, Uderhardt S, Ipseiz N, Stoll C. et al. PPARβ/δ governs Wnt signaling and bone turnover. Nat Med. 2013;19(5):608-13
56. Ratneswaran A, LeBlanc EA, Walser E, Welch I, Mort JS, Borradaile N. et al. Peroxisome proliferator-activated receptor delta promotes the progression of posttraumatic osteoarthritis in a mouse model. Arthritis Rheumatol. 2015;67(2):454-64
57. Monemdjou R, Vasheghani F, Fahmi H, Perez G, Blati M, Taniguchi N. et al. Association of cartilage-specific deletion of peroxisome proliferator-activated receptor gamma with abnormal endochondral ossification and impaired cartilage growth and development in a murine model. Arthritis Rheum. 2012;64(5):1551-61
58. Clockaerts S, Bastiaansen-Jenniskens YM, Feijt C, Verhaar JA, Somville J, De Clerck LS. et al. Peroxisome proliferator activated receptor alpha activation decreases inflammatory and destructive responses in osteoarthritic cartilage. Osteoarthritis Cartilage. 2011;19(7):895-902
59. Sforza C, Grandi G, Binelli M, Tommasi DG, Rosati R, Ferrario VF. Age- and sex-related changes in the normal human ear. Forensic Sci Int. 2009;187(1-3):110 e1-7
60. Luquet S, Gaudel C, Holst D, Lopez-Soriano J, Jehl-Pietri C, Fredenrich A. et al. Roles of PPAR delta in lipid absorption and metabolism: a new target for the treatment of type 2 diabetes. Biochim Biophys Acta. 2005;1740(2):313-7
61. Mizuno M, Takebe T, Kobayashi S, Kimura S, Masutani M, Lee S. et al. Elastic cartilage reconstruction by transplantation of cultured hyaline cartilage-derived chondrocytes. Transplant Proc. 2014;46(4):1217-21
62. Hansen MK, Connolly TM. Nuclear receptors as drug targets in obesity, dyslipidemia and atherosclerosis. Curr Opin Investig Drugs. 2008;9(3):247-55
Author contact
![]() Corresponding author: Jun Ren, Tel: +86-791-83813080; Fax: +86-791-83805967. E-mail: renjunedu.cn or renjunjxaucom.
Corresponding author: Jun Ren, Tel: +86-791-83813080; Fax: +86-791-83805967. E-mail: renjunedu.cn or renjunjxaucom.