Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(6):701-711. doi:10.7150/ijbs.17534 This issue Cite
Research Paper
Eupafolin Exhibits Potent Anti-Angiogenic and Antitumor Activity in Hepatocellular Carcinoma
Honglei Jiang1, Dan Wu2, Dong Xu1, Hao Yu1, Zheming Zhao1, Dongyan Ma1, Junzhe Jin1 ![]()
1. General Surgery department, the fourth affiliated hospital of China medical university, Shenyang, China;
2. Infectious disease department, Shengjing hospital of China medical university, Shenyang, China.
Received 2016-9-11; Accepted 2017-4-20; Published 2017-5-16
Abstract

Eupafolin is a flavonoid extracted from the common sage herb which has been used in China as traditional medicine. Previous studies had reported that eupafolin had antioxidative, anti-inflammatory and antitumor effects. However, the function and the mechanism of eupafolin to exert its antitumor activity, especially its effect on tumor angiogenesis, have not been elucidated. Herein, we showed that eupafolin significantly inhibited vascular endothelial growth factor (VEGF)-induced cell proliferation, migration and tube formation of human umbilical vascular endothelial cells (HUVECs) in a dose-dependent manner. Meanwhile, the new blood microvessels induced by VEGF in the matrigel plug were also substantially suppressed by eupafolin. The results of HCC xenograft experiments demonstrated eupafolin remarkably inhibited tumor growth and tumor angiogenesis in vivo, suggesting the antitumor activity exerted by eupafolin was closely correlated with its potency on tumor angiogenesis. Mechanism investigations revealed that eupafolin significantly blocked VEGF-induced activation of VEGFR2 in HUVEC cells as well as its downstream signaling pathway. In addition to the effect on endothelial cells, through inhibiting Akt activity in tumor cells, VEGF secretion in HepG2 was dramatically decreased after eupafolin treatment. Our study was the first to report the activity of eupafolin against tumor angiogenesis as well as the underlying mechanism by which eupafolin to exert its anti-angiogenic activity.
Keywords: Eupafolin, tumor angiogenesis
Introduction
Angiogenesis, the outgrowth of new vessels from pre-existing vasculature, plays an important part in neoplastic transformation and cancer development as well as in tumor metastasis [1]. It is widely accepted that when tumor volume reached 1mm3, new vascular network is required to supply adequate nutrition and energy for its rapid growth [2, 3]. The newly generated blood vessels not only support the growth of tumor mass, also benefit for the initiation of metastasis, which leads to >90% of mortality in various cancers including hepatocelluar carcinoma [4]. Angiogenesis is a complex multiple-step process, which involves in endothelial cell proliferation, migration, tube formation, vascular network reorganization and stabilization [5]. Although multiple proangiogenic factors have been identified, among these factors, vascular endothelial growth factor (VEGF), which is expressed and secreted by tumor cells in hypoxia conditions, is considered to be the most potent angiogenic factor in the process of tumor angiogenesis [3]. VEGF exerts its biological effects via binding to its receptors expressed on endothelial cells, namely vascular endothelial growth factor receptor 1(VEGFR1) and 2(VEGFR2). Previous studies had verified the biologically relevant VEGF signaling events were mainly mediated through VEGFR2 [6, 7]. Activation of VEGFR2 gives rise to the activation of a series of downstream signaling pathways in endothelial cells, including extracellular signal-regulated kinases (ERK), protein kinase C, Src family kinase, focal adhesion kinase, and phosphoinositide 3-kinase/Akt/eNOS pathway [8, 9].
Hepatocellular carcinomas (HCCs) are characterized with abundant microvessel density and high levels of circulating VEGF, making anti-agiogenesis as an attractive therapeutic strategy. VEGF and its receptors have been found to be overexpressed in HCC in comparison with normal liver tissue, and overexpression of VEGF was correlated with the poor prognosis [10-12]. So far, numbers of angiogenic inhibitors, including small molecular kinase inhibitors and monoclonal antibodies, have been investigated in clinical [13, 14]. Sorafenib, which is approved by FDA for HCC therapy, is a potent inhibitor against multiple angiogenic tyrosine kinases [15]. The success of sorafenib implies that it is feasible to control HCC via targeting angiogenesis in tumors [16]. For most of the anti-angiogenesis therapies available in clinic, significant side effect limited their further development [17, 18]. So identification of novel anti-angiogenic inhibitor is an urgent demand both for HCC prevention and treatment.
In comparison with the compounds from chemical synthesis, small molecules derived from nature products also provide a great opportunity for novel drug identification [19]. Eupafolin is a flavonoid isolated from the common sage herb, which is widely used as traditional medicine in China and India. Extensive studies reported eupafolin had good pharmacological activity in anti-inflammation mainly by inhibition of transcription factor NF-κB [20, 21], whereas the activity of eupafolin against tumor was rarely available. In the present studies, we found that eupafolin exhibited substantial antitumor activity in hepatocellular carcinoma xenograft model. Mechanism investigations demonstrated its activity against hepatocellular carcinoma was closely related to its anti-angiogenic activity.
Material and Methods
Cell line and reagents
HUVEC cells were isolated as previously described [22] and the presence of von Willebrand factor was validated with immunofluorescence staining. HUVECs were maintained in M199 medium containing 20% fetal bovine serum (FBS), 30μg/ml endothelial cell growth supplement(ECGS), 10ng/ml epidermal growth factor (EGF), 100 units/ml penicillin and 100 units/ml streptomycin. HCC cell lines HepG2 and Hep3B were purchased from Cell Bank of Chinese Academy of Sciences (Shanghai) and cultured with RPMI 1640 medium supplemented with 10% FBS, 100 units/ml penicillin and 100 units/ml streptomycin. Eupafolin was obtained from ChromaDex, Inc. Anti-β-actin antibody was product of Sigma. Anti-p-VEGFR2 (Tyr1175), anti-p-Akt (Ser473), anti-p-Akt (Thr308), anti-pan-Akt, anti-Akt1, anti-p-GSK3β (Ser9), anti-ERK1/2, anti-p-ERK1/2(Thr202/Tyr204) and anti-p-S6 (Ser235/236) antibodies were products of Cell Signaling Technology, Inc. (Danvers, MA). Anti-Ki67 (ab66155) antibody was products of Abcam (Cambridge, UK). Anti-CD31 antibody and , anti-rabbit IgG-HRP were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Myr-Akt1 was product of Addgene (Cambridge, MA, USA). Lipofectamin was obtained from Invitrogen (Carlsbad, CA). The VEGF assay kit was product of R&D Systems (Minneapolis, MN)
Cell proliferation assay
HUVECs (2× 104 per well) or HCC cells (2× 104 per well) were treated with or without VEGF (20 ng/mL) and eupafolin for 24 h. Cell viability was measured by using the CellTiter-Glo Luminescent Cell Viability Assay kit (MTS) purchased from Promega Corp. (Madison, WI) according to the instructions of the manufacturer's protocol.
VEGF ELISA Assay
VEGF amount in cell culture medium was detected by the ELISA kit (R&D Systems, Minneapolis, MN) according to the manufacturer's instructions. Each sample was analyzed in triplicate.
Western blotting
Cells treated with eupafolin were digested with trypsin and cell suspension was collected by centrifugation, then the pellets were lysed with RIPA buffer containing with protease cocktail (Roche, Germany) on ice. Protein concentrations were determined with the Bradford assay (Bio-Rad, Philadelphia, PA, USA). The protein in the lysate was separated by SDS-PAGE and then electronically transferred to PVDF membrane (Millipore, Billerica, MA, USA). Nonspecific binding site on the membrane was blocked with 5% non-fat dry milk at room temperature for 1 h and the membranes were incubated with specific primary antibodies at 4°C overnight. After washing three times with TBS-Tween 20 solution, the membranes were incubated with secondary antibodies conjugated with HRP at room temperature for 1h. Washing three times with TBS-Tween20, the bands on membrane were detected by ECL chemiluminescence reagents (Pierce Chemical Co., Rockford, lllinois, USA).
HUVEC cell wound scratch assay
HUVEC cells were seeded in six-well plate pre-coated with 0.1% gelatin in complete medium, after cells reached 70-80% confluence as a monolayer, cells were treated with 10 μg/ml mitomycin for 2 h to block cell proliferation and then the monolayer was scratched with a new 10 μl pipette tip across the center of the well. After scratching, the detached cells were removed by gently washing the well with culture medium twice. ECGM containing 0.5% FBS was added with or without 20 ng/mL VEGF and different concentrations of eupafolin. 12 h later, randomly chosen fields were photographed and the migrated cells were quantified by manual counting. Inhibition percentage was calculated by taking VEGF-induced wells as 100%.
Transwell migration assay
The Transwell (Corning, Inc.) was pre-coated with 0.1% gelatin, serum-free medium containing VEGF (20 ng/ml) was added into the bottom chambers, HUVECs were trypsinized and suspended with serum-free medium, 1 × 105 cells per well incubated with different concentration of eupafolin were seeded into the top chambers. The Transwell plate was incubated in a 5% CO2 incubator at 37°C for about 8-10 h, the cells on the top surface of the membrane (non-migrated cells) were wiped with a cotton swab and the cells located on the bottom membrane (invasive cells) were fixed with cold 4% paraformaldehyde for 30 min and stained with crystal violet solution. Images were taken using Olympus inverted microscope and the invasive cells were determined by counting the cells.
Tube formation assay
Matrigel was thawed at 4°C and 50 μl per well was added into 96-well plate, then the plate was incubated at 37°C for 45min to solidify the matrigel. HUVECs (2× 104) treated with different concentrations of eupafolin were seeded into 96-well plated in 100μl culture medium with or without VEGF (20 ng/ml). 6-8 h later, tubular structures of endothelial cells were examined using an inverted microscope (Olympus, Tokyo, Japan). The number of the tubes was quantified from five random fields [23].
Matrigel plug assay
The matrigel plug assay was performed as previous described [24]. Briefly, Growth factor-reduced Matrigel (BD Biosciences) containing 70 ng VEGF and 20 units of heparin with or without eupafolin were implanted subcutaneously into 5-6 week-old C57/BL/6 mice. After 7 days, the mice were sacrificed and Matrigel plugs were removed, each group contains 4-5 plugs, then the plugs were fixed and H&E staining was performed. The functional microvessels were quantified manually using a microscope to identify the formation of new microvessels. The number of erythrocyte-filled blood vessels was counted.
In vivo efficacy studies
Nu/nu athymic nude mice were used for in vivo experiments. The mice were fed under specific pathogen free (SPF) conditions according to the regulations of Institutional Animal Care and Use Committee. HepG2 or Hep3B cells (5×106 cell/mice) were s.c. injected into the right flank of nude mice. After the tumor was formed and the volume was about 100 mm3, the mice were randomly assigned. The control group was dosed with 0.5% sodium carboxymethylcellulose, the treatment group was administrated with 60 mg/kg eupafolin by intraperitoneal injection three times per week, five mice per group. Tumors volume were measured twice per week with microcalipers and calculated as V= (length×width2)/2. After experiment completion, mice were killed and the tumor tissue was weighed and photographed.
Immunohistochemical (IHC) staining
The tumor tissue from in vivo experiment was embedded in paraffin and cut into 5μM sections. After dewaxing in xylene and hydration in ethanol, the slides was immersed in 3% H2O2 to block the endogenous peroxidase activity. The slides were placed into boiling citric acid solution to perform antigen retrieval with a microwave oven. The tissues were incubated with serum from the host of the secondary antibody to block nonspecific binding site and then incubated with primary antibodies of anti-CD31 (1:100) or anti-Ki67 (1:250) respectively at 4°C overnight. After washing with PBS three times, the slides was incubated with biotinylated secondary antibodies at a 1:100 dilution, and then followed by Vectastin ABC solution. Finally, 3, 3-diaminobenzidine (DAB) solution was used to detected the binding of the antibodies on tumor tissue. Tissues were counterstained with harris' hematoxylin, dehydrated in xylene, and then mounted. Slides were observed and photographed with a light microscope, and the results were analyzed with Image-Pro Plus software (version 6.2) program.
Statistical analysis
The statistical analysis was done with SPSS software (version 13.0). All experiments were conducted in duplicate or triplicate. The quantitative data were shown as Mean ± SD and the statistical differences between two groups was examined by a two-tailed Student's t test. p< 0.05 indicated the significant difference.
Results
The effect of eupafolin in HUVEC cells was more potent than in HCC cells
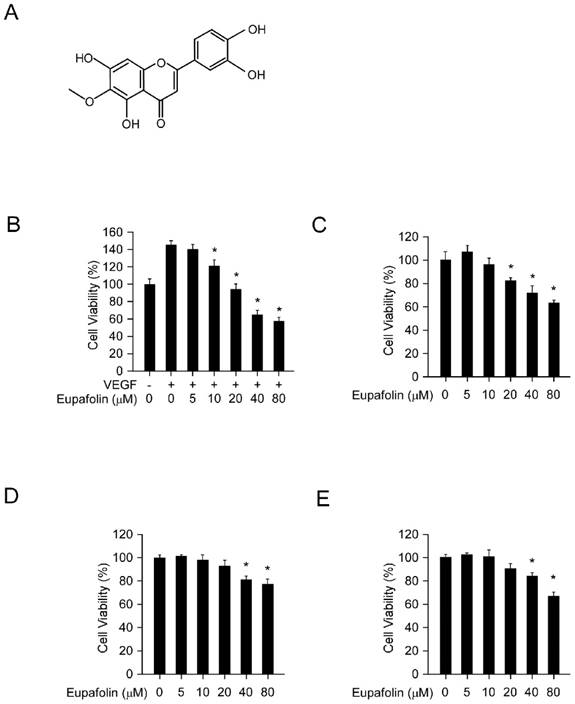
Eupafolin was a flavonoid with a molecular weight of 316.26 g/mol (Figure 1A). Firstly, we assessed the inhibitory effect of eupafolin on cell viability in HUVEC cells and HCC cell line HepG2 and Hep3B by MTS assay. As the results shown in Figure 1B, eupafolin substantially inhibited VEGF-induced endothelial cells proliferation from the concentration of 10μM and with an IC50 about 30 μM. However, under normal culture conditions (20% FBS), eupafolin inhibited cell proliferation at a higher concentration with an IC50 more than 80μM, implying that eupafolin was more effective in angiogenesis disease conditions. Moreover, we also examined the inhibitory effect of eupafolin on HCC cells, and the results showed that the IC50 of HepG2 and Hep3B cells was over 80 μM, suggesting that eupafolin was more effective in mediating the proliferation of endothelial cells than that of cancer cells.
Eupafolin inhibited cell viability in HUVECs and HCC cells. A, the chemical structure of eupafolin. B, eupafolin inhibited VEGF-induced HUVECs proliferation. HUVECs(2× 104/well) were seeded into 96-well plate and starved with 0.1% FBS medium overnight and then incubated with or without VEGF(20 ng/ml) and various concentrations of eupafolin for 24 h, cell viability was analyzed by MTS assay. C, inhibitory effect of eupafolin on HUVECs in normal conditions. HUVECs (2×104/well) were seeded into 96-well plate and treated with different concentrations of eupafolin for 24 h, cell viability was analyzed by MTS assay. D and E, eupafolin suppressed HCC cell lines HepG2(D) and Hep3B(E) proliferation (the conditions were the same as described in C). Columns, mean of three independent experiments ;bars, SD. The asterisk (*, p<0.05) indicated a significant decrease of cell viability after treated with eupafolin.
Eupafolin significantly inhibited VEGF-induced angiogenesis
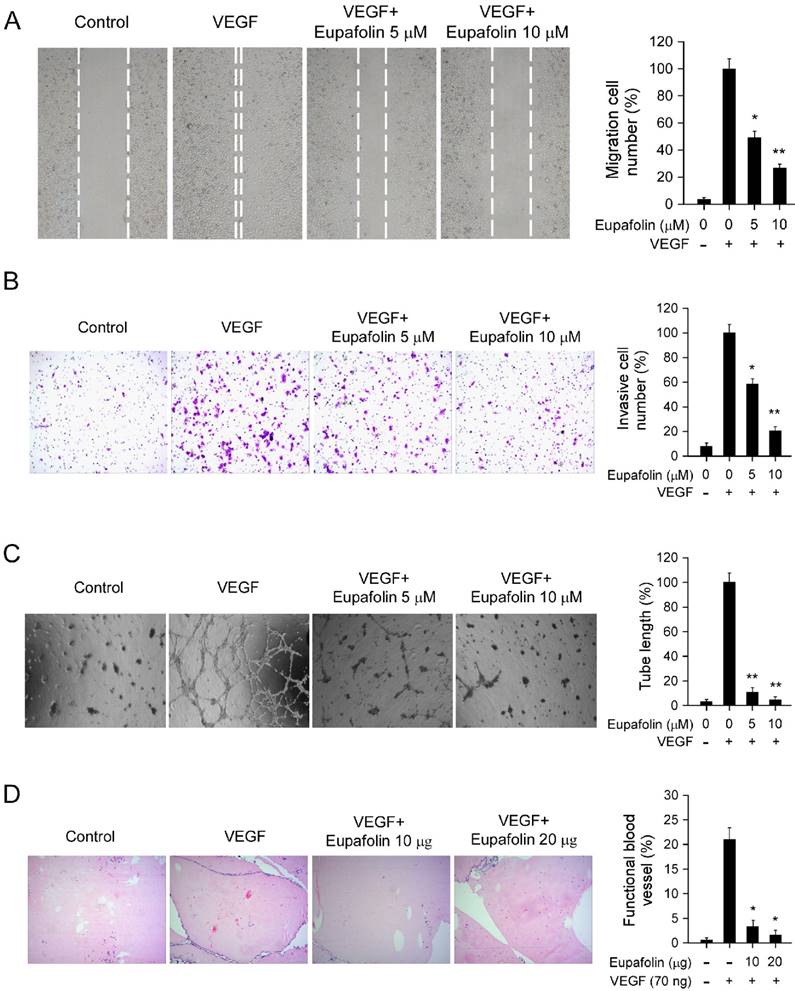
Endothelial cell migration is a critical step in the process of angiogenesis to form blood vessels. The activity of eupafolin against VEGF-induced chemotactic motility in HUVECs was assessed by wound-healing migration assay and transwell cell invasion assay. As shown in Figure 2, in contrast with VEGF-treated group, VEGF-induced HUVECs migration was inhibited by eupafolin in a dose-dependent manner. Eupafolin substantially inhibited HUVEC migration at 5 μM in the scratching cell assays (Figure 2A), and the compound also significantly suppressed HUVEC invasion at 5 μM in transwell assay (Figure 2B). In addition to the migration of endothelial cells, capillary-like tube formation is also a key step in the complex process of angiogenesis. Therefore, we examined how eupafolin affected HUVECs tube formation by two-dimensioned matrigel assay. As shown in Figure 2C, in VEGF-incubated group, elongated and robust tube-like structures were formed, but tube formation of HUVECs induced by VEGF was dramatically decreased after eupafolin treatment. 5 μM eupafolin inhibited 80% tube formation of HUVECs on matrigel and 10 μM eupafolin almost completely blocked the formation of tubular structure. Previous results shown in Figure 1B indicated that eupafolin at 5 μM had no obvious inhibitory effects on HUVECs viability, so the suppression of HUVEC migration and tube formation exerted by eupafolin was specific, not owing to the cytotoxicity. In order to further confirm the anti-angiogenic activity of eupafolin, we had used matrigel plug assay to evaluate the effect of eupafolin on VEGF-induced angiogenesis in vivo. As the results shown in Figure 2D, Matrigel plugs containing VEGF only were filled with intact RBCs, representing the functional vasculatures was developed inside the Matrigel through angiogenesis initiated by VEGF. In sharp contrast, vascular formation in matrigel plug containing eupafolin was dramatically decreased, and eupafolin (20 μg) totally blocked vasculature formation in the assays.
Eupafolin inhibited VEGF-induced agiogenesis in vitro and ex vivo. A, eupafolin inhibited HUVECs migration. HUVECs were grown into full confluence in six-well plate and treated with 10 μg/ml mitomycin C for 2 h, then cells were wounded with pipette and incubated with or without 20 ng/ml VEGF as well as various concentrations of eupafolin. 12 h later, the migrated cells were quantified by manual counting. B, eupafolin suppressed HUVECs invasion. Cells were seeded in the upper chamber of Transwell and incubated with various concentrations of eupafolin. The bottom chamber was added with culture medium with VEGF. About 8 to 10 h later, the invasive cells were fixed and stained with crystal violet solution, the invasive cells were quantified by manual counting. C, eupafolin inhibited VEGF-induced tube formation of HUVECs. HUVECs incubated with different concentrations of eupafolin were seeded into 96-well plate pre-coated with Matrigel. After 6 to 8 h, tubular structures were photographed and the number of the tubes was quantified. D, eupafolin inhibited VEGF-induced angiogenesis in Matrigel plug assay. The Matrigel plug assay was performed as described in “Materials and Methods”. The number of vessels was counted. Representative photographs of each experiment (left panels) were shown, and the graph (right panels) showed the data of at least three independent experiments. Columns, mean; bars, SE; The asterisk (*, p< 0.05; **, p< 0.01) indicated significant difference versus VEGF alone.
Eupafolin inhibited tumor angiogenesis and tumor growth in vivo
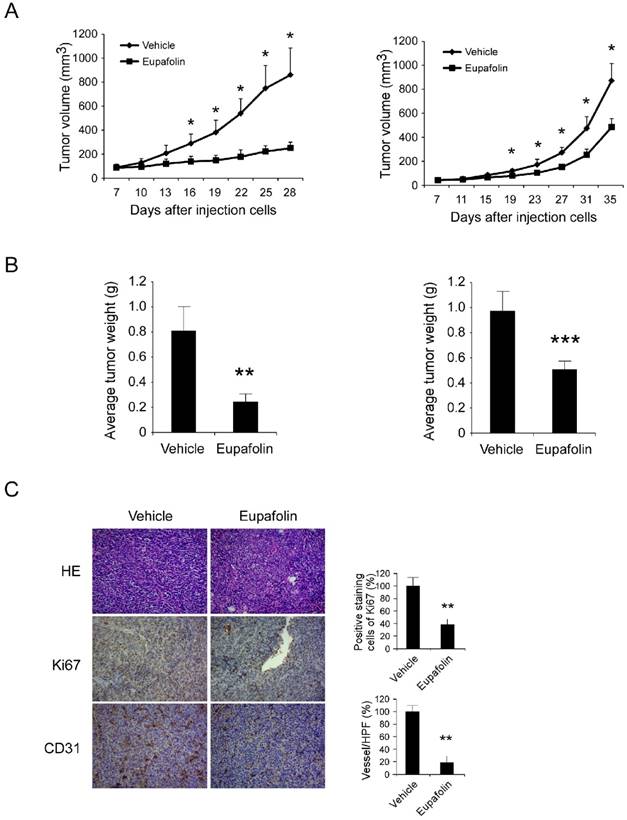
Tumor angiogenesis provided various resources such as oxygen, nutrients to support tumor growth, previous investigations had clarified eupafolin possessed excellent anti-angiogenic activity in vitro and in vivo. In order to further confirm the antitumor activity of eupafolin, the potency of eupafolin against HCC growth was determined in xenograft models. As the results shown (Figure 3A-B, supplementary1A), eupafolin had demonstrated substantial antitumor activity in HepG2 and Hep3B xenograft models. In HepG2 model, 21 days after eupafolin treatment, the tumor volume of vehicle group had reached about 800 mm3, whereas the volume in eupafolin group was around 200 mm3, and the tumor weight was 0.78g versus 0.23g. In Hep3B model, tumor growth was also substantially attenuated and about 50% growth inhibition was observed, the tumor volume was 900 mm3 versus 430 mm3 and tumor weight was 0.98g versus 0.51g respectively. No obvious toxicity was observed as evaluating the change of body weight of tumor-bearing mice between vehicle and eupafolin-treated group (supplementary 1B). To confirm the effect of eupafolin on tumor angiogenesis, tumor tissues were dissected and analyzed by using CD31 antibody to detect endothelial cells. In comparison with vehicle group, vessel density in xenograft from mice treated with eupafolin substantially reduced, with a decrease over 80%. Ki-67 is an important biomarker to indicate the potential of proliferation, as the result shown in Figure 3C, the expression of Ki67 in eupafolin group was decreased over 60% in contrast with vehicle group. With the inhibition of angiogenesis in tumor tissue, the supply of oxygen and nutrients to support tumor growth was blocked, and the proliferation ability of tumor cells was also weakened.
Eupafolin suppressed HCC cells growth and angiogenesis in vivo. Antitumor activity of eupafolin was investigated in HepG2 and Hep3B xenograft models, mice were randomly assigned into groups when tumor volume reached 100 mm3. The dosage of eupafolin was 60 mg/kg and was administrated three times per week by intraperitoneal injection. A, the growth curve of HepG2(left panel) and Hep3B (right panel) xenograft; B, the weight of tumors in vehicle and eupafolin-treated group, left: HepG2, right: Hep3B; C, tumor tissues were performed with immunohistochemistry analysis by staining with anti-Ki67, anti-CD31 antibody to detect the change of angiogenesis in tumor tissue. Left panel, representative photograph of tumor tissue per group (200×); right panel, the expression of indicated marker in per group was quantified, the asterisks (*, p<0.05, **, p<0.01, Student's t test) indicated significant difference.
Eupafolin inhibited the activation of VEGFR2 signaling pathway
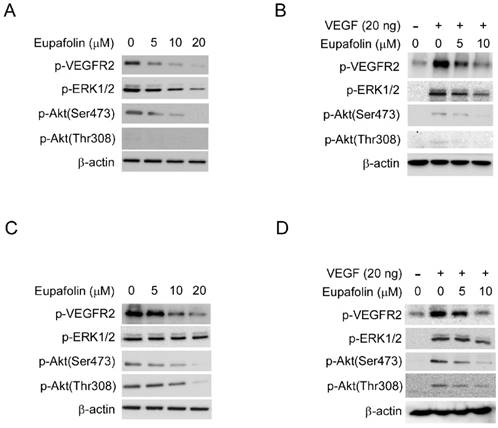
VEGFR2 was proved to play predominant roles for VEGF to exert its agiogenic activity. VEGFR2 activation leads to the activation of various downstream signaling pathway that are involved in regulating endothelial cell migration, proliferation, and survival. As shown in Figure 4A, exposure to eupafolin resulted in the inhibition of phosphorylation of VEGFR2 in a dose-dependent manner in HUVECs. With the suppression of VEGFR2 activation, the phosphorylation of ERK1/2 and Akt, which are the main downstream signaling pathways of VEGFR2, were both blocked. Moreover, we also examined the effect of eupafolin on VEGF-induced VEGFR2 activation. Likewise, VEGF-driven phosphorylation of VEGFR2 and the activation of its downstream signaling were also inhibited by eupafolin (Figure 4B). Additionally, we also investigated the effect of eupafolin on VEGFR2 phosphorylation in HepG2 cells. As the results shown in figure 4C and 4D, VEGFR2 phosphorylation in HCCs was also substantially inhibited by eupafolin. However, different from the effect on downstream signaling of VEGFR2 in HUVECs, with the inhibition of phosphorylation of VEGFR2, the activity of Akt was dramatically suppressed accordingly, but the activity of ERK had no obvious change in contrast with Akt, these results suggested that the role of VEGFR2 played in HepG2 cells maybe was different from its role in HUVEC cells.
Eupafolin inhibited VEGFR2 signaling pathway in HUVECs and HCC cells. A and C, eupafolin inhibited VEGFR2 phosphorylation in HUVECs and HepG2 cells. HUVECs(A) and HepG2 cells(C) were incubated with various concentrations of eupafolin for 24 h and the cell lysates were subjected to western blotting and probed with indicated antibodies, β-actin was used as loading control. B and D, eupafolin suppressed VEGF-induced VEGFR2 phosphorylation in HUVECs and HepG2 cells. HUVECs(B) and HepG2(D) cells were starved with 0.1% FBS overnight and then incubated with various eupafolin for 2 h, after stimulating with 20 ng/ml VEGF for 10 min, cell lysates were collected and western blotting was used to examine eupafolin's effect on the activity on VEGFR2, ERK1/2 and Akt, β-actin was used as loading control.
Eupafolin suppressed VEGF secretion in HCC cells via targeting Akt activity
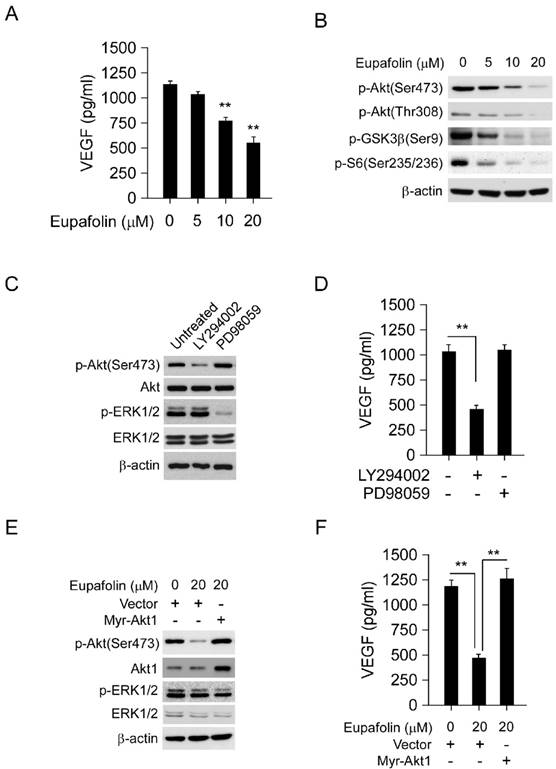
Generally, VEGF secreted by tumor cells was an important initiator to provoke angiogenesis in tumor tissue, therefore, blocking VEGF secretion by tumor cells was also very important for angiogenesis inhibition. As the results shown in Figure 5A, after eupafolin treatment, VEGF secreted by HepG2 cells was substantially decreased. Compared to the control, 50% decrease of VEGF production in HepG2 cell was observed after 20 μM eupafolin treatment. Previous studies had reported that Akt signaling pathway was involved in the mediation of VEGF secretion, so next step we investigated the effect of eupafolin on Akt signaling pathway. As expected, eupafolin had substantially inhibitory effect on Akt activity. The phosphorylation of Akt and downstream signaling pathway was dramatically inhibited by eupafolin in a dose dependent manner (Figure 5B). In order to confirm the important role of Akt signaling pathway played in the mediation of VEGF production in HepG2 cells, specific PI3K inhibitor (LY294002) and MEK inhibitor (PD98059) was adopted to treat HepG2 cells and to investigate the effect on VEGF secretion. As shown in Figure 5C and D, in contrast with PI3K inhibitor (LY294002), MEK inhibitor (PD98059) had little effect on VEGF production in HepG2 cell, suggesting that Akt signaling pathway was very important in mediating VEGF production. Moreover, In order to further elucidate the role of Akt played in eupafolin-mediated activity, HepG2 cells were transfected with Myr-Akt1 to overexpress Akt and then investigated eupafolin's effect on VEGF production. As shown in Figure 5E and F, after transfected with Myr-Akt1, the expression of Akt in HepG2 cells was significantly increased in contrast with the mock group, and the overexpression of Akt had no obvious effect on ERK signaling pathway. With the increase of Akt activation, eupafolin-mediated decrease of VEGF was almost completely rescued, which implied that VEGF decrease caused by eupafolin was mainly attributed to its effect on Akt signaling pathway.
Eupafolin suppressed VEGF secretion in HepG2 cells via inhibiting Akt signaling pathway. A, eupafolin inhibited VEGF secretion in HepG2 cells. HepG2 cells was treated with various concentrations of eupafolin for 24 h, the supernatant of cell culture medium was collected and the content of VEGF was examined with ELISA kit. B, eupafolin suppressed Akt signaling pathway in HepG2 cells. HepG2 cells were exposed to various concentrations of eupafolin for 24 h, and the cell lysates were analyzed by western blotting with indicated antibodies. C and D, Akt signaling pathway mediated VEGF secretion in HepG2 cells. HepG2 cells were treated with selective PI3K inhibitor(LY294002) or MEK inhibitor(PD98059) for 24 h respectively, cell lysates was subjected to western blotting with indicated antibodies (C), and the amount of VEGF in cell culture medium was analyzed with ELISA kit (D). E and F, exogenous overexpression of Akt attenuated eupafolin's effect on VEGF secretion. HepG2 cells were transfected with Myr-Akt1 for 24 h, and then treated with 20 µM eupafolin for 24 h, cell lysates were analyzed by western blot with indicated antibodies (E), VEGF secretion in cell culture medium was analyzed with ELISA kit (F). The graph showed the data of least three independent experiments and expressed as Mean±SD, the asterisks (*, p<0.05, Student's t test) indicated significant difference.
Discussion
Hepatocelluar carcinoma (HCC) was the fourth most common solid cancer and third leading cause of cancer-related death in China [25]. Sorafenib is the only targeted drug recommended by National Comprehensive Cancer Network (NCCN) as the standard first-line therapy for advanced HCC. Besides sorafenib, there are more than 50 reagents that are being investigated in clinical, however, no agent has yet proved superior to sorafenib [26]. Even so, the overall survival improvement of sorafenib is just near three months [27]. Therefore, substantial efforts are in great need to identify potential candidates with high potency and low toxicity. Previous studies reported that eupafolin, a flovonoid isolated from the common sage herb, had demonstrated antitumor activity in several cancers [28, 29], but littler information was available about its function in tumor angiogenesis and HCC. In the present study, we showed that eupafolin exerted potent antitumor activity in HCC xenograft models. Meanwhile, we also revealed eupafolin displayed its potency against HCC was closely correlated to its anti-agiogenesis activity. Multiple steps of VEGF-induced tumor angiogenesis, including endothelial cell proliferation, migration, invasion and tube formation, ex vivo and in vivo agiogenesis, were substantially inhibited by eupafolin. In addition to the effect on endothelial cells, with the inhibition of Akt signaling pathway in HCC cell, eupafolin also mediated tumor angiogenesis via blocking VEGF secretion by tumor cells.
As reported by Liu et al. [28], eupafolin at 20 μM had a direct effect against prostate cancer cells via targeting phosphatidylinositol 3-kinase-mediated Akt signaling pathway. Moreover, Chung KS et al. [29] showed that in cervical adenocarcinoma, eupafolin significantly induced tumor cells to undergo apoptosis at the concentration of 30 μM. However, in our tested HCC cell line HepG2 and Hep3B, eupafolin effectively induced growth inhibition of culture cells at the concentration over 40 μM. In contrast, eupafolin dramatically inhibited VEGF-induced angiogenic responses only at or <10 μM in all test angiogenesis assays. Due to VEGF played a predominant role in the formation of tumor vasculature, our results showed that tumor growth inhibition mediated by eupafolin in vivo was mainly through its antiangiogenic activity at a much lower concentration, rather than its direct cytotoxicity on tumor cells in HCC.
In HUVEC cells, VEGF-induced VEGFR2 phosphorylation was significantly inhibited by eupafolin in a dose-dependent manner. With the suppression of VEGFR2 activity, the activities of ERK and Akt in HUVECs, which were the downstream signaling of VEGFR2, also substantially inhibited. Given VEGFR2 was the major mediator of the angiogenic effects of VEGF, we thought the mechanism by which eupafolin to exert its activity against endothelial cell was closely related to its effects on VEGFR2 activity. Unlike most anti-angiogenesis inhibitors which displayed their activity through targeting endothelial cells in tumor tissue, in addition to the restraint of endothelial cell function, eupafolin also blocked tumor angiogenesis via suppressing VEGF production in tumor cells. In the process of tumor development, tumor-associated angiogenesis was thought to experience two different stages, which was separated by 'angiogenic switch' [30, 31]. The first was defined as an avascular stage, corresponding to the small lesions of no more than 1-2mm in diameter and most tumor cells stayed in dormancy. With small subset of dormant tumor cells enter into the vascular phase, exponential tumor growth ensued. Therefore, VEGF secreted by tumor cells was regarded as the initiator of tumor growth. In our studies, we showed that eupafolin substantially blocked VEGF secretion by tumor cells, which implied that eupafolin could eliminate the initiation step in tumor angiogenesis, and maybe delay the occurrence of resistance in contrast with other anti-angiogenesis therapies which only acted on endothelial cells.
Peng et al reported there was a VEGF-VEGFRs antocrine loop in hepatocellular carcinoma cells, VEGF promoted phosphorylation of VEGFR2 and internalization of p-VEGFR2, which was self-sustaining through elevating VEGF, VEGFR2 mRNA expression [32]. In our studies, our results demonstrated eupafolin was capable to block VEGF-induced VEGFR2 activation in HCC cells and inhibit downstream Akt phosphorylation, therefore, VEGF suppression by eupafolin in HCC cells maybe was related to the blockade of the autocrine loop. The Akt signaling pathway was a hub in the regulation of cancer progression and VEGF expression [33-36]. Liu et al and colleagues reported eupafolin could bind with PI3-K directly and suppress Akt activation [28]. Consistently, our results confirmed Akt downstream signaling pathway in HCC cells was significantly inhibited after eupafolin treatment. Recent studies demonstrated Akt played an important role in regulating normal vascularization and pathological angiogenesis and Akt activation was sufficient to mediate VEGF expression in human cancer cells [37, 38]. Activation of Akt induced VEGF expression through its two downstream molecules HDM2 and p70S6K1 [39, 40]. Moreover, Akt also transmitted the upstream signals from growth factors, cytokines, oncogenes to regulate the expression of proangiogenic factors such as VEGF, b-FGF by regulating hypoxia-inducible factor-1α expression at the translational level [41-44]. In present studies, as evidenced by PD98059, the selective MEK kinase inhibitor, had no effect on VEGF production in HCC cells, VEGF suppression caused by eupafolin was closely correlated with inhibition of Akt activity, rather than ERK pathway. Further investigations revealed that overexpression of Akt in HCC cells substantially rescued VEGF suppression caused by eupafolin, verifying the important role of Akt played in eupafolin- mediated VEGF suppression.
Potential limitation for eupafolin to be translated into clinical is the higher concentration for eupafolin activity. So far, no pharmacokinetic parameters of eupafolin have been available. In the future, development of appropriate delivery systems and chemical modification of eupafolin to enhance its efficacy are necessary. Moreover, in order to promote the potential of eupafolin to clinical use, conventional issues, such as bioavailability and toxicological profiles, have to be comprehensively investigated.
Taken together, our studies for the first time reported the anti-angiogenesis activity of eupafolin and showed eupafolin suppressed HCC xenografts growth in vivo via its anti-angiogenic effect. Moreover, mechanism investigations demonstrated eupafolin exerted its anti-angigogenic activity not only through targeting endothelial cells to restrain their functions in tumor angiogenesis, also targeting tumor cells to decrease VEGF secretion and block angiogenesis initiation.
Supplementary Material
Figure S1.
Acknowledgements
This work was supported by Natural Science Foundation of China (Grant No.81402473).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873-87
2. Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1-18
3. Ferrara N. VEGF and the quest for tumour angiogenesis factors. Nat Rev Cancer. 2002;2:795-803
4. Bishayee A, Darvesh AS. Angiogenesis in hepatocellular carcinoma: a potential target for chemoprevention and therapy. Curr Cancer Drug Targets. 2012;12:1095-118
5. Domigan CK, Iruela-Arispe ML. Recent advances in vascular development. Curr Opin Hematol. 2012;19:176-83
6. Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669-76
7. Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932-6
8. Shibuya M. Vascular endothelial growth factor (VEGF)-Receptor2: its biological functions, major signaling pathway, and specific ligand VEGF-E. Endothelium. 2006;13:63-9
9. Sia D, Alsinet C, Newell P, Villanueva A. VEGF signaling in cancer treatment. Curr Pharm Des. 2014;20:2834-42
10. Hu TH, Huang CC, Wu CL, Lin PR, Liu SY, Lin JW. et al. Increased endostatin/collagen XVIII expression correlates with elevated VEGF level and poor prognosis in hepatocellular carcinoma. Mod Pathol. 2005;18:663-72
11. Huang J, Zhang X, Tang Q, Zhang F, Li Y, Feng Z. et al. Prognostic significance and potential therapeutic target of VEGFR2 in hepatocellular carcinoma. J Clin Pathol. 2011;64:343-8
12. Cui J, Dong BW, Liang P, Yu XL, Yu DJ. Effect of c-myc, Ki-67, MMP-2 and VEGF expression on prognosis of hepatocellular carcinoma patients undergoing tumor resection. World J Gastroenterol. 2004;10:1533-6
13. Kim HY, Park JW. Clinical trials of combined molecular targeted therapy and locoregional therapy in hepatocellular carcinoma: past, present, and future. Liver Cancer. 2014;3:9-17
14. Germano D, Daniele B. Systemic therapy of hepatocellular carcinoma: current status and future perspectives. World J Gastroenterol. 2014;20:3087-99
15. Ge S, Huang D. Systemic therapies for hepatocellular carcinoma. Drug Discov Ther. 2015;9:352-62
16. Zhu AX, Duda DG, Sahani DV, Jain RK. HCC and angiogenesis: possible targets and future directions. Nat Rev Clin Oncol. 2011;8:292-301
17. Tanaka S, Arii S. Molecular targeted therapies in hepatocellular carcinoma. Semin Oncol. 2012;39:486-92
18. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417-27
19. Newman DJ. Natural products as leads to potential drugs: an old process or the new hope for drug discovery? J Med Chem. 2008;51:2589-99
20. Clavin M, Gorzalczany S, Macho A, Munoz E, Ferraro G, Acevedo C. et al. Anti-inflammatory activity of flavonoids from Eupatorium arnottianum. J Ethnopharmacol. 2007;112:585-9
21. Lai ZR, Ho YL, Huang SC, Huang TH, Lai SC, Tsai JC. et al. Antioxidant, anti-inflammatory and antiproliferative activities of Kalanchoe gracilis (L.) DC stem. Am J Chin Med. 2011;39:1275-90
22. Zhou Y, Chen Y, Tong L, Xie H, Wen W, Zhang J. et al. AL3810, a multi-tyrosine kinase inhibitor, exhibits potent anti-angiogenic and anti-tumour activity via targeting VEGFR, FGFR and PDGFR. J Cell Mol Med. 2012;16:2321-30
23. Ashton AW, Yokota R, John G, Zhao S, Suadicani SO, Spray DC. et al. Inhibition of endothelial cell migration, intercellular communication, and vascular tube formation by thromboxane A(2). J Biol Chem. 1999;274:35562-70
24. Yi T, Cho SG, Yi Z, Pang X, Rodriguez M, Wang Y. et al. Thymoquinone inhibits tumor angiogenesis and tumor growth through suppressing AKT and extracellular signal-regulated kinase signaling pathways. Mol Cancer Ther. 2008;7:1789-96
25. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
26. Miyahara K, Nouso K, Yamamoto K. Chemotherapy for advanced hepatocellular carcinoma in the sorafenib age. World J Gastroenterol. 2014;20:4151-9
27. Mazzoccoli G, Miele L, Oben J, Grieco A, Vinciguerra M. Biology, Epidemiology, Clinical Aspects of Hepatocellular Carcinoma and the Role of Sorafenib. Curr Drug Targets. 2016;17:783-99
28. Liu K, Park C, Chen H, Hwang J, Thimmegowda NR, Bae EY. et al. Eupafolin suppresses prostate cancer by targeting phosphatidylinositol 3-kinase-mediated Akt signaling. Mol Carcinog. 2015;54:751-60
29. Chung KS, Choi JH, Back NI, Choi MS, Kang EK, Chung HG. et al. Eupafolin, a flavonoid isolated from Artemisia princeps, induced apoptosis in human cervical adenocarcinoma HeLa cells. Mol Nutr Food Res. 2010;54:1318-28
30. Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401-10
31. Moserle L, Amadori A, Indraccolo S. The angiogenic switch: implications in the regulation of tumor dormancy. Curr Mol Med. 2009;9:935-41
32. Peng S, Wang Y, Peng H, Chen D, Shen S, Peng B. et al. Autocrine vascular endothelial growth factor signaling promotes cell proliferation and modulates sorafenib treatment efficacy in hepatocellular carcinoma. Hepatology. 2014;60:1264-77
33. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550-62
34. Li W, Peng C, Lee MH, Lim D, Zhu F, Fu Y. et al. TRAF4 is a critical molecule for Akt activation in lung cancer. Cancer Res. 2013;73:6938-50
35. Pore N, Liu S, Shu HK, Li B, Haas-Kogan D, Stokoe D. et al. Sp1 is involved in Akt-mediated induction of VEGF expression through an HIF-1-independent mechanism. Mol Biol Cell. 2004;15:4841-53
36. Huang Y, Hua K, Zhou X, Jin H, Chen X, Lu X. et al. Activation of the PI3K/AKT pathway mediates FSH-stimulated VEGF expression in ovarian serous cystadenocarcinoma. Cell Res. 2008;18:780-91
37. Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem. 2001;276:38349-52
38. Yang ZZ, Tschopp O, Hemmings-Mieszczak M, Feng J, Brodbeck D, Perentes E. et al. Protein kinase B alpha/Akt1 regulates placental development and fetal growth. J Biol Chem. 2003;278:32124-31
39. Skinner HD, Zheng JZ, Fang J, Agani F, Jiang BH. Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J Biol Chem. 2004;279:45643-51
40. Testa JR, Bellacosa A. AKT plays a central role in tumorigenesis. Proc Natl Acad Sci U S A. 2001;98:10983-5
41. Ong SH, Hadari YR, Gotoh N, Guy GR, Schlessinger J, Lax I. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci U S A. 2001;98:6074-9
42. Dong G, Chen Z, Li ZY, Yeh NT, Bancroft CC, Van Waes C. Hepatocyte growth factor/scatter factor-induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin-8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res. 2001;61:5911-8
43. Kang S, Bader AG, Zhao L, Vogt PK. Mutated PI 3-kinases: cancer targets on a silver platter. Cell Cycle. 2005;4:578-81
44. Jiang BH, Liu LZ. AKT signaling in regulating angiogenesis. Curr Cancer Drug Targets. 2008;8:19-26
Author contact
![]() Corresponding author: Junzhe Jin Ph.D. M.D., Address: The No.4 Chongshan east road Huanggu district, Shenyang Liaoning, 110032 Tel: 024-62043469 Fax: 024-62255001 E-mail: jzhjinedu.cn
Corresponding author: Junzhe Jin Ph.D. M.D., Address: The No.4 Chongshan east road Huanggu district, Shenyang Liaoning, 110032 Tel: 024-62043469 Fax: 024-62255001 E-mail: jzhjinedu.cn