Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Conclusion
Materials and Methods
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(7):923-934. doi:10.7150/ijbs.17260 This issue Cite
Research Paper
Acetylation of hMOF Modulates H4K16ac to Regulate DNA Repair Genes in Response to Oxidative Stress
Jianing Zhong1,#, ![]() , Liying Ji2, #, Huiqian Chen2, Xianfeng Li3, 4, Jian'an Zhang2, Xingxing Wang2, Weilin Wu1, Ying Xu1, Fei Huang1, Wanshi Cai3,4, Zhong Sheng Sun2,3,4,
, Liying Ji2, #, Huiqian Chen2, Xianfeng Li3, 4, Jian'an Zhang2, Xingxing Wang2, Weilin Wu1, Ying Xu1, Fei Huang1, Wanshi Cai3,4, Zhong Sheng Sun2,3,4, ![]()
1. The Science Research Center, Gannan Medical University, Ganzhou 341000, China
2. Institute of Genomic Medicine, Wenzhou Medical University, Wenzhou 325035, China
3. Beijing Institutes of Life Science, Chinese Academy of Sciences, Beijing 100101, China
4. University of Chinese Academy of Sciences, Beijing 100049, China
# Co-first author
Received 2016-8-19; Accepted 2017-5-28; Published 2017-7-15
Abstract

Oxidative stress is considered to be a key risk state for a variety of human diseases. In response to oxidative stress, the regulation of transcriptional expression of DNA repair genes would be important to DNA repair and genomic stability. However, the overall pattern of transcriptional expression of DNA repair genes and the underlying molecular response mechanism to oxidative stress remain unclear. Here, by employing colorectal cancer cell lines following exposure to hydrogen peroxide, we generated expression profiles of DNA repair genes via RNA-seq and identified gene subsets that are induced or repressed following oxidative stress exposure. RRBS-seq analyses further indicated that transcriptional regulation of most of the DNA repair genes that were induced or repressed is independent of their DNA methylation status. Our analyses also indicate that hydrogen peroxide induces deacetylase SIRT1 which decreases chromatin affinity and the activity of histone acetyltransferase hMOF toward H4K16ac and results in decreased transcriptional expression of DNA repair genes. Taken together, our findings provide a potential mechanism by which oxidative stress suppresses DNA repair genes which is independent of the DNA methylation status of their promoters.
Keywords: hMOF, H4K16ac, Oxidative Stress
Introduction
Increased levels of oxidative stress in cellular microenvironments as well as the presence of reactive oxygen species such as superoxides, hydroxyl radicals, and hydrogen peroxide (H2O2) in the external cellular environment contribute to a key risk state for various human diseases. These diseases include age-associated neurodegenerative disorders, Alzheimer's disease, Parkinson's disease and various cancers [1-3]. It is well-established that oxidative stress-induced DNA damage activates cellular DNA repair mechanisms to maintain genomic stability by facilitating the recruitment of DNA damage and repair proteins [4-6]. Moreover, oxidative stress also occurs during chromatin-based processes in DNA damage repair, transcription, and DNA replication, which attenuate DNA repair efficiency via the epigenetic changes that include DNA methylation and histone modifications [7-9].
Previous studies have focused on the elevated transcriptional expression of DNA repair genes which are regulated by the activation of kinases and transcription factors. Breast cancer associated protein 1 (BRCA1), p53, NF-κB, and AP-1 are believed to be the most important transcription factors involved in the response to DNA damage [10-13]. For example, a subset of DNA repair genes involved in nuclear excision repair (NER; e.g. DDB2 and XPC via p53, ERCC1, XPF, and XPG via AP-1) are upregulated via p53 and/or AP-1 following exposure to UV light [14]. For example, two DNA repair genes, APEX1 and NEIL1 are involved in base excision repair (BER), and are activated via AP-1 in response to H2O2-induced oxidative stress [15]. However, most studies focus on elucidating the activation or inhibition of some particular gene, rather than the overall regulation pattern of all DNA repair genes. Therefore, a broad characterization of transcriptional regulation of all DNA repair genes is necessary.
Histone modification represents one of the major epigenetic regulatory mechanisms that function in response to environmental stimuli [16]. The spatial and temporal control of DNA damage repair may be critically dependent on histone and chromatin modifications [17]. In order to rapidly recruit DNA damage response and repair factors, the formation of open chromatin structures at double strand breaks (DSBs) is required. This dynamic process is tightly associated with the alteration of histone modifications, including phosphorylation and acetylation [17, 18]. γH2AX (pSer139), an early marker of DNA damage response, facilitates recruitment of DNA damage proteins to promote the DNA damage response [19]. Beyond aiding in DNA damage signaling, recruitment of chromatin modifiers to DNA damage sites could inhibit transcription in situ, preventing the repair process and/or DNA damage signaling from interference by transcription. For example, H4K16ac is a highly abundant activating modification, with ~80% of histone H4 molecules having an acetyl group on K16 [20]. Hypo-acetylation of H4K16 is a targeted epigenetic site for posttranslational modification in the DNA damage response, in addition to its established roles in modulating chromatin structure and in transcriptional activation [21]. hMOF (human MOF, also called KAT8), a member of the MYST (Moz-Ybf2/Sas3-Sas2-Tip60) family of HATs, is a critical HAT for acetylating histone H4 at K16 [22]. Several studies show that depletion of hMOF renders global reduction of H4K16ac and defects of DNA repair in budding yeast and mammal cells [23, 24]. Interestingly, overexpression of hMOF reverses gene silencing of certain tumor suppressor genes induced by H4K16 deacetylation [25]. On the other hand, histone deacetylase SIRT1, which was initially identified as a homologue of NAD+-dependent deacetylase Sir2 in budding yeast, has the ability to deacetylate H4K16ac as well as H3K9ac and H3K14ac [26]. It has been shown that Sir2/SIRT1 are required for DNA repair and maintenance of genomic stability in either yeast or mammals [27, 28]. Notably, the reduction of H4K16ac displays association with the deacetylation activity of SIRT1, which has been confirmed by impairing deacetylation of H4K16ac in Sirt1 deficient cells [29]. Furthermore, exogenous expression of SIRT1 directly counteracts the effect of hMOF on H4K16ac and sensitization to the topoisomerase II inhibitor in cancer cells [30]. Additionally, SIRT1 modulates deacetylation of hMOF and is required for chromatin accessibility and activity of hMOF. These observations suggest that the regulation of H4K16ac, as the substrate of hMOF and SIRT1, is complicated, especially in the response to DNA damage [31]. Therefore, the detailed mechanism by which hMOF and SIRT1 synergistically modulate the level of H4K16ac in response to DNA damage requires investigation.
Here, we employed RNA-seq to profile the expression of DNA repair genes which were either induced or repressed in mammalian colorectal cancer cells following exposure to H2O2. RRBS-seq analysis showed no significant alteration of DNA methylation in promoter of significantly downregulated DNA repair genes in comparing H2O2-treated cells with controls. Moreover, we provide a potential mechanism by which hMOF and SIRT1 synergistically modulate the level of H4K16ac, which consequently contributes to suppression of DNA repair genes in response to H2O2-induced oxidative stress. Collectively, these findings describe an overall pattern of DNA repair gene response in mammalian cells exposed to H2O2-induced oxidative stress, where histone acetylation primarily accounts for the transcriptional changes of DNA repair genes rather than DNA methylation.
Results
RNA-seq Analysis Reveal Transcriptional Alteration of DNA Repair Genes in Response to H2O2-induced Oxidative Stress
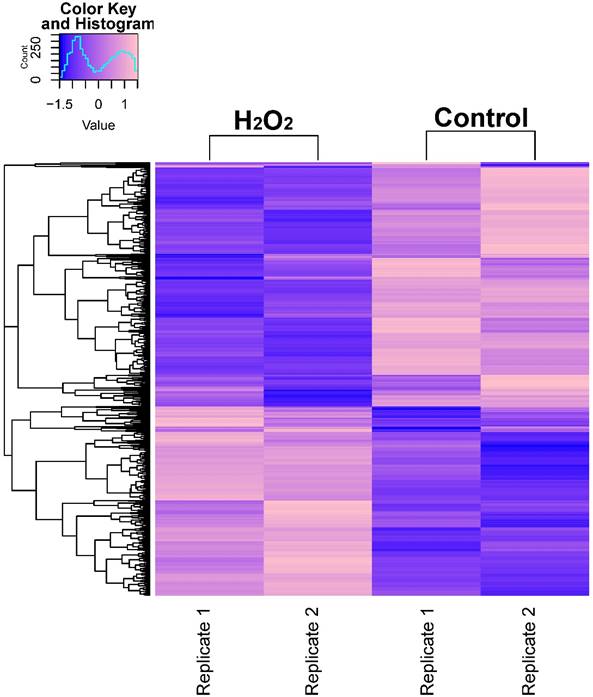
In order to describe the pattern of transcriptional expression of DNA repair genes in response to oxidative stress, we first performed RNA-seq on human colorectal cell samples (HCT116 cell line) treated with or without H2O2 in duplicate. Comparison of the transcriptional profiles identified 3,216 differentially expressed genes, among which 1,263 genes were elevated and 1,953 genes were repressed (Fig. 1A, Table S1). Gene ontology (GO) enrichment analysis was performed on these identified genes and z-scores were used to identify the proportion of upregulated genes in all differentially expressed genes for each term (Table S2). The data showed that significantly overrepresented biological processes were generally categorized into: cellular response to stress, DNA damage, apoptosis, signal transduction, cell death, and histone modification. Notably, the 24 differentially expressed DNA repair genes identified (8 increased and 16 decreased) were classified into different DNA repair pathways using the repairtoire database (http://repairtoire.genesilico.pl/, Table 1). The upregulated DNA repair genes were mainly involved in nucleotide excision repair (NER), DNA damage signaling (DDS), base excision repair (BER), and DNA damage reversal (DDR). Unexpectedly, we found a greater prevalence of significantly downregulated DNA repair genes. Downregulated genes were implicated in DDS, translesion synthesis (TLS), homologous recombination repair (HRR), non-homologous end joining (NHEJ), and BER. These results indicate that H2O2-induced oxidative stress may activate DNA damage signaling and single strand break repair pathways, while specifically repressing double strand break repair pathways via repression of HRR and NHEJ gene transcriptional expression.
RNA-seq Analysis Heatmap of Transcriptomes in H2O2-Treated or Untreated HCT116 Cells. Expression profiles of genes differentially expressed between H2O2-treated HCT116 cells and control cells. Hierarchical clustering of expression of these genes divides samples into the treated and control groups. The scale at top-left indicates the relative expression levels.
Significantly Differentially Expressed DNA Repair Genes in HCT116 Cells Involved in Different DNA Repair Pathways
| Pathways | Upregulated | Downregulated |
|---|---|---|
| MMR | N/A | POLE, MLH3 |
| NER | PCNA | POLE |
| DDS | RFC5, CDKN1A, DCLRE1A | FANCA, RAD50, ATM |
| TLS | N/A | POLM, REV3L, POLQ, REV1 |
| HRR | N/A | FANCA, BRCA2, EME2, MSH5 |
| NHEJ | N/A | POLM, PRKDC |
| BER | PCNA, APEX1, FEN1, UNG | NEIL1, POLE, PNKP |
| DRR | ALKBH2 | N/A |
Abbrevation: DDS: DNA damage signaling; DRR: DNA damage reversal; BER: base excision repair; NER: nucleotide excision repair; MMR: mismatch repair; HRR: homologous recombination repair; NHEJ: nonhomologous end-joining; TLS: translesion synthesis
RRBS-seq Analysis Reveal No Alteration in DNA Methylation in the Promoters of the DNA Repair Genes in Response to H2O2-induced Oxidative Stress
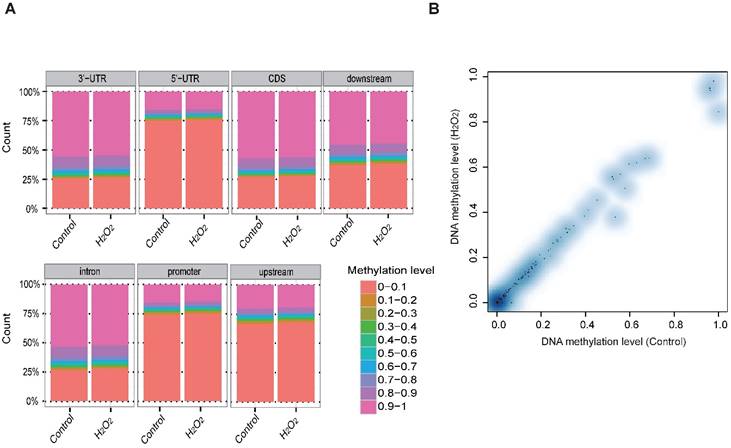
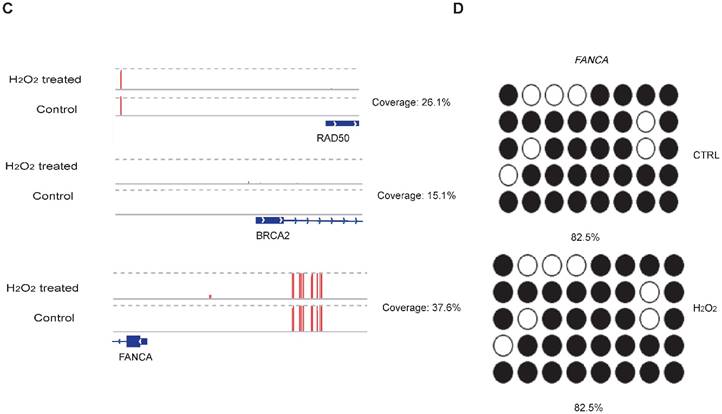
To examine whether DNA methylation affects the transcriptional alteration of DNA repair genes, we performed reduced representation bisulfite sequencing (RRBS-seq) in HCT116 cells after H2O2 treatment. We identified 785 differentially methylated regions (DMRs) using a sliding window approach [32]. The differential methylation levels in H2O2 -treated HCT116 were observed to occur on genomic features of 3' UTR, 5' UTR, CDS, intron, and promoter regions (Fig. 2A). The promoters of almost all the DNA repair genes displayed no significant methylation disparity between H2O2-treated and control cells (Fig. 2B). IGV mapping indicated that three DNA repair genes that were differentially transcriptionally repressed, RAD50, BRCA2, and FANCA, did not display differences in the methylation level between H2O2-treated and control cells (Fig. 2C). Further, no change in the DNA methylation of FANCA was observed via Bisulfite Sequencing (BS) (Fig. 2D). Therefore, we speculate that DNA methylation has a negligible effect on transcriptional regulation of DNA repair genes in response to H2O2-induced oxidative stress.
RRBS-seq Analysis Reveal No Alteration in DNA Methylation of the DNA Repair Genes on Their Promoters. (A) Density of different scales of methylation level on genomic features of H2O2 -treated HCT116 and control. The y-axis represents the accumulated percentage, and the stacked bars represent the proportion of different methylation levels indicated by different colors, as illustrated in the scales on the right. (B) DNA methylation of gene promoters in H2O2-treated HCT116 and control cells. The y-axis and x-axis represent DNA methylation levels in H2O2-treated HCT116 and control cells, respectively. The degree of blue color that surrounds the dots indicates the density of the indicated level of methylated regions. Lighter blue denotes fewer regions with the same methylation level as the surrounding dots, and vice versa. (C) Visualization of the methylation level on promoters of three representative DNA repair genes: RAD50, BRCA2, and FANCA. The length of the red bars denotes methylation levels on the indicated site. Deep blue blocks represent exons, and solid lines represent introns, on which the arrows indicate transcriptional direction. The promoter region is upstream of the first exon. CpG coverage was found to be 26.1%, 15.1%, and 37.6%, respectively. (D) Verification of DNA methylation of the FANCA gene via Bisulfite Sequencing.
H4K16ac Level is Significantly Decreased in Response to Acute H2O2-induced Oxidative Stress
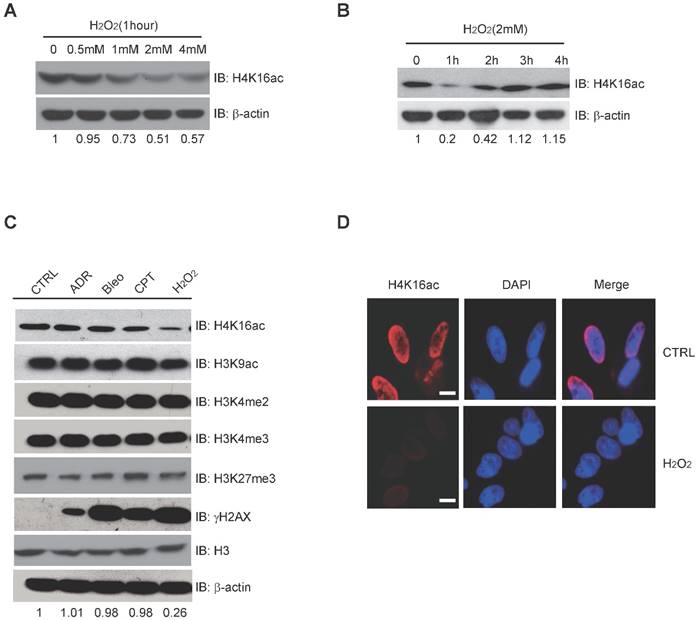
Histone modification may contribute to the expression of DNA repair genes. When treated with 0, 0.5 mM, 1 mM, 2 mM, and 4 mM H2O2, respectively, H4K16ac expression significantly decreases after 1-4 mM H2O2 (Fig. 3A). Following 2 mM H2O2 treatment for 0, 1 h, 2 h, 3 h, and 4 h, H4K16ac expression was observed to decreased initially but recover after 2 h of continuous H2O2 treatment (Fig. 3B). In order to determine the alterations of histone modifications in response to different DNA damage reagents, we treated HCT116 cells with adriamycin (ADR), bleomycin (Bleo), camptothecin (CPT), and hydrogen peroxide (H2O2). H2O2 treatment specifically resulted in significantly decreased H4K16ac, while the other histone markers, H3K9ac, H3K27m3, H3K4m2, and H3K4m3 exhibited no obvious changes (Fig. 3C). Fluorescent staining of H4K16ac in H2O2-treated HeLa cells was weakened, as determined via immunofluorescent staining (Fig. 3D). The result of decreased H4K16ac under H2O2 treatment prompts us to speculate that H4K16ac could play an important role in regulating gene expression in response to H2O2-induced oxidative stress.
H2O2-induced Oxidative Stress Leads to Hypo-acetylation of H4K16. (A) Western blot analysis of H4K16ac with incrementally increased concentrations of H2O2 treatment for 1 hour. The protein level of H4K16ac is normalized using Quantity One software. (B) Western blot analysis of H4K16ac abundance with 2 mM H2O2 treatment for various lengths of time. The protein level of H4K16ac is normalized using Quantity One software. (C) Western blot analysis of histone modifications levels, using specific antibodies, after treatment with 1 μM ADR, 1 μM Bleo, 1 μM CPT, and 2 mM H2O2 for 1 hour, respectively. The protein level of H4K16ac is normalized using Quantity One software. (D) Immunofluorescence staining of HeLa cells treated with or without H2O2 using H4K16ac antibody. Nuclei were stained with DAPI. The scale presents 10 μm.
hMOF and H4K16ac Bindings of DNA Repair Gene Promoters Both Decrease in Response to H2O2-induced Oxidative Stress
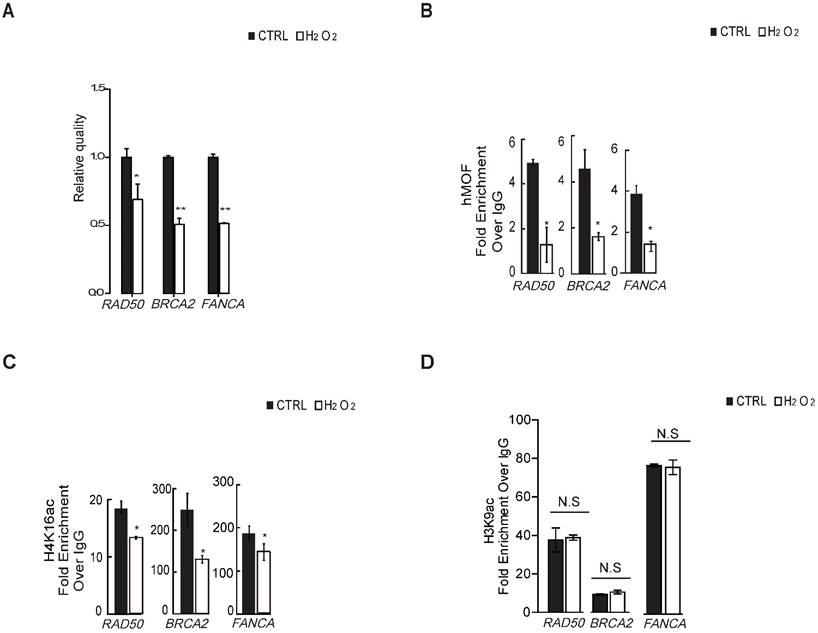
To verify whether H4K16ac contributes to the transcriptional expression of DNA repair genes in response to oxidative stress, we first determined that H2O2 treatment leads to transcriptional repression of DNA repair genes from significantly differentially downregulated DNA repair genes shown in Table 1 via RT-PCR (Fig. 4A), including BRCA2, RAD50, and FANCA, which are required for DNA double strand breaks repair. ChIP-qPCR assays revealed that hMOF and H4K16ac binding were co-enriched the similar promoter regions of these three DNA repair genes in control, both of which were decreased in cells exposed to H2O2 (Fig. 4B and 4C). Although H3K9ac also presented at similar regions of these DNA repair genes in control, no alteration in expression is observed after H2O2 treatment (Fig. 4D). These data indicated that the diminished binding of H4K16ac at promoters of DNA repair genes specifically contributes to H2O2-induced downregulation of these DNA repair genes.
Binding of hMOF and H4K16ac Both Decreases in HCT116 Cells Treated With or Without H2O2. (A) RT-PCR analysis of several significantly downregulated DNA repair genes, including RAD50, BRCA2, and FANCA (*p<0.05, **p<0.01). (B) ChIP-qPCR analysis of hMOF binding on the promoters of those genes (*p<0.05, **p<0.01). (C) ChIP-qPCR analysis of H4K16ac binding on the promoters of these genes (*p<0.05, **p<0.01). (D) ChIP-qPCR analysis of H3K9ac binding on the promoters of these genes (*p<0.05, **p<0.01).
SIRT1-Modulated Acetylation of hMOF Contributes to Downregulation of H4K16ac Levels in Response to Oxidative Stress
Previous studies have shown that chromatin affinity of hMOF is required for its acetylation activity toward H4K16ac [33]. Here, chromatin fractionation indicated that H2O2 treatment led to decreased chromatin affinity of hMOF (Fig. 5A). Further, we found that there was an increase of H4K16ac in HCT116 cells treated by an inhibitor of HDACs, Trichostatin A (TSA), but a decrease of H4K16ac in HA-hMOF overexpressing HCT116 cells (Fig. 5B). Thus, we speculate that some HDACs may regulate hMOF binding and activity toward H4K16ac.
SIRT1-Modulated Acetylation of hMOF Contributes to Hypo-acetylation of H4K16 Following Exposure to H2O2. (A) Chromatin fractionation analysis of hMOF binding on chromatin in HCT116 cells treated with or without H2O2. The detection of DNMT1 is treated as the positive control. The protein level of hMOF is normalized using Quantity One software. (B) Western blot analysis of H4K16ac in HA-hMOF overexpressing HCT116 cells and control cells treated with or without TSA, respectively. The protein level of H4K16ac is normalized using Quantity One software. (C) Western blot analysis of H4K16ac in HA-hMOF overexpressing or HA-hMOF (K274A) overexpressing HCT116 cells, with inhibition of SIRT1 using shRNA, respectively. The protein level of H4K16ac is normalized using Quantity One software. (D) Western blot analysis of H4K16ac in SIRT1-inhibited HCT116 cells treated with or without H2O2. The protein level of H4K16ac is normalized using Quantity One software. (E) Western blot analysis of H4K16ac in HA-hMOF overexpressing HCT116 cells and HA-hMOF (K274A) overexpressing HCT116 cells treated with or without H2O2, respectively. The protein level of H4K16ac is normalized using Quantity One software. (F) Western blot analysis of the DNA repair protein abundance in HA-hMOF overexpressing HCT116 cells and HA-hMOF (K274A) overexpressing HCT116 cells treated with or without H2O2, respectively. The protein levels of RAD50, BRCA2 and FANCA are normalized using Quantity One software.
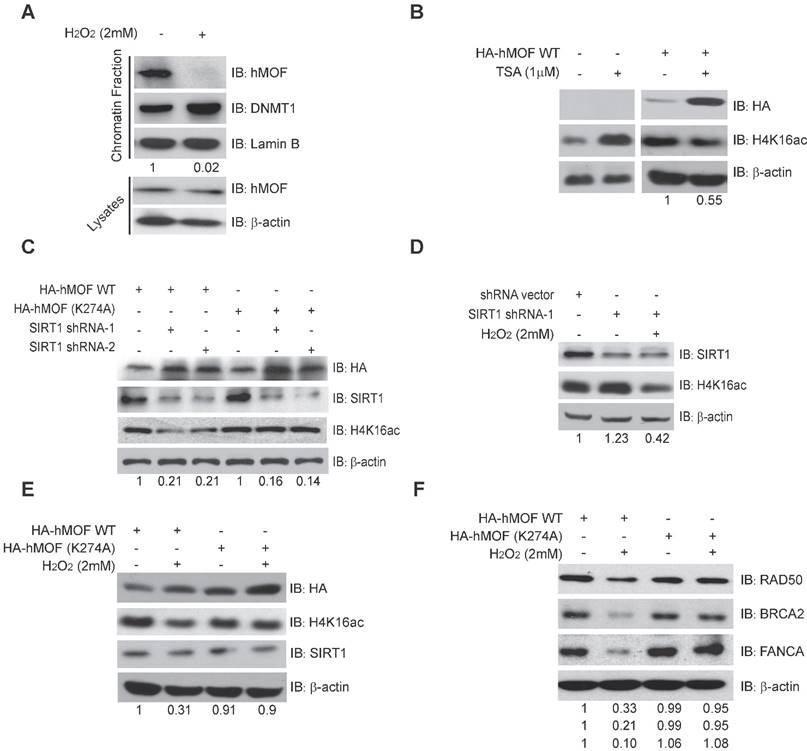
Given SIRT1 is a HDAC which has been shown to affect acetylation of lysine 274 of hMOF to modulate the H4K16ac [31, 33], we then constructed an unacetylated HMOF by mutagenesis of hMOF lysine 274(K274A). Our results demonstrated that the level of H4K16ac were increased after inhibition of SIRT1 via shRNA in HA-hMOF overexpressing HCT116 cells, whereas no alteration of H4K16ac occurred in HA-hMOF (K274A) overexpressing cells (Fig. 5C). In addition, H2O2 treatment could resulted in the decrease of H4K16ac in SIRT1-knockdown cells (Fig. 5D). These results suggest that SIRT1-mediated hMOF activity may contribute to the regulation of H4K16ac. Importantly, we found that H2O2 treatment induces decreased H4K16ac in HA-hMOF overexpressing cells, but not in HA-hMOF (K274A) overexpressing cells, while no effect of SIRT1 was observed (Fig. 5E). Moreover, H2O2 treatment resulted in downregulation of DNA repair proteins level, (including RAD50, BRCA2, and FANCA) in HA-hMOF overexpressing cells, but not in HA-hMOF (K274A) overexpressing cells (Fig. 5F). Collectively, these results conclude that acetylation of hMOF is required for the decrease of H4K16ac, which further leads to downregulation of DNA repair genes in cells exposed to oxidative stress.
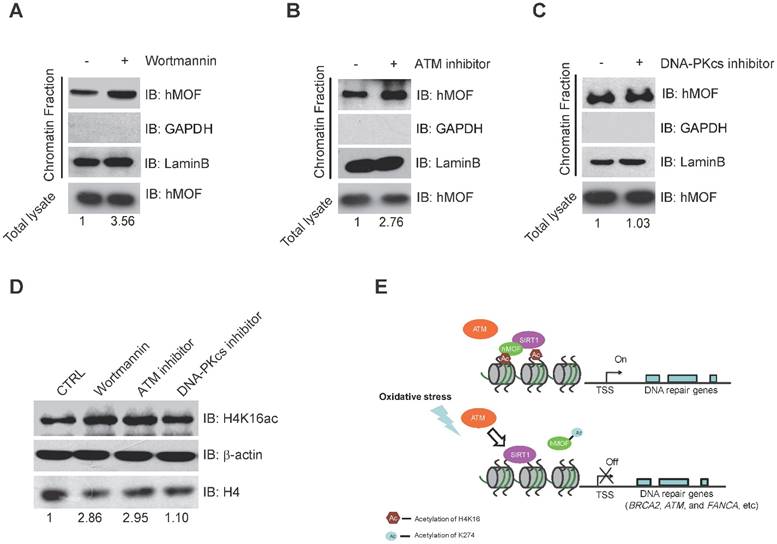
Binding of hMOF on Chromatin Is Dependent on ATM Kinase Activity
Previous studies have indicated that ATM is considered as one of the early response proteins in DNA oxidative damage signaling [6, 34]. And the binding of hMOF on chromatin is required for hMOF HAT activity toward H4K16 [31]. We therefore speculated that hMOF binding may be regulated via the signaling pathway dependent on ATM. We observed an increase in hMOF binding on chromatin after Wortmannin treatment (Fig. 6A). Wortmannin is an inhibitor of early DNA damage response proteins in the PIKK family (phosphatidylinositol 3-kinase-related protein kinases) which includes ATM, ATR, and DNA-PK, this result indicated that PIKK family kinases impede hMOF binding. Further analyses indicated that there was increased binding of hMOF after ATM inhibition when using its specific inhibitor, CGK733 (Fig. 6B). The change of hMOF binding was not observed in DNA-PK inhibitor (NU7026) treated cells (Fig. 6C). H4K16ac levels in cells treated with Wortmannin, CGK733 and NU7026 were also measured, respectively. Both Wortmannin and CGK733 treatments respectively resulted in a significant increase of H4K16ac. But the alteration of H4K16ac was not observed in NU7026 treated cells (Fig. 6D). These data suggest that the regulation of hMOF binding and H4K16ac is dependent on ATM kinase activity, but not DNA-PK kinase activity. Collectively, H2O2-induced oxidative damage can cause the alteration of hMOF binding dependent on ATM signaling pathway to lead to downregulation of DNA repair genes, therefore possibly feedback regulating DSBs repair.
Binding of hMOF on Chromatin is Dependent on ATM Kinase Activity. (A) Chromatin fractionation analysis of hMOF binding on chromatin in HCT116 cells with or without 1 μM Wortmaninn treatment. The protein level of hMOF is normalized using Quantity One software. (B) Chromatin fractionation analysis of hMOF binding on chromatin in HCT116 cells treated with or without 1 μM ATM inhibitor (CGK733) treatment. The protein level of hMOF is normalized using Quantity One software. (C) Chromatin fractionation analysis of hMOF binding on chromatin in HCT116 cells treated with or without 1 μM DNA-PK inhibitor (NU7026). The protein level of hMOF is normalized using Quantity One software. (D) Detection of H4K16ac before and after treatment of Wortmaninn, CGK733, and NU7026, respectively. H4 and β-actin were detected as loading control. The protein level of H4K16ac is normalized using Quantity One software.
Discussion
Here, we found that downregulation of DNA repair genes in response to H2O2-induced oxidative stress is independent of DNA methylation. Furthermore, our analyses revealed a potential mechanism of SIRT1-modulated hMOF acetylation, which affects hMOF recruitment and H4K16ac modification, thereby leading to downregulation of specific DNA repair genes. Our findings characterize transcriptional regulation of DNA repair genes upon oxidative stress and provide a molecular regulatory mechanism how some of the important DNA repair genes are suppressed (Fig. 6E).
Only 8 out of 87 detected DNA repair genes (PCNA, RFC5, DCLRE1A, APE1, CDKN1A, FEN1, UNG, and ALKBH2) in our RNA-seq data were activated in response to acute H2O2induced oxidative stress (Fig. 1 and Table 1). This result is mostly consistent with the previous studies [35-41]. However, in previous studies, H2DCF-DA (5-carboxy-2', 7'-dichlorodihydrofluorescein diacetate), is used to induce oxidative stress rather than H2O2. The discrepancy in results may be due to subtle differences in metabolism or rather, in molecular signaling. On the other hand, previous reports do not investigate the suppression of DNA repair genes in response to oxidative stress. Our findings indicate that 15 of 87 enriched DNA repair genes are repressed, and many of these are involved in HRR or NHEJ (Fig. 1 and Table 1), including FANCA, EME2, MSH5, POLM, PRKDC, and BRCA2. These results raise the possibility that oxidative stress may alternatively reduce the capacity of DNA double strand break repair via epigenetically repressing DNA repair genes involved in repair pathways.
DNA methylation is an important regulatory mechanism that contributes to transcriptional repression. Studies have examined the relationships between oxidative damage and epigenetic gene silencing using an engineered cell model in which an I-SceI restriction site is integrated into the CpG island of the E-cadherin promoter [42]. Based on these studies, a mechanism has been proposed in which H2O2-induced oxidative stress regulates epigenetic alteration of DNA methylation through the re-localization of DNMT1, EZH2 and SIRT1 on chromatin from non-CpG to CpG islands [43]. Here, our RRBS analyses indicate that the methylation level of promoters for almost all DNA repair genes remains unchanged (Fig. 2), which contrasts with the aforementioned reports. Our RRBS-seq data comprises 113 genes as opposed to the 124 previously identified DNA repair genes documented in repairtoir. However, the 11 genes not analyzed with RRBS-seq do not display transcriptional differences between H2O2-treated cells and controls. OGG1, a DNA repair gene implicated in the BER pathway, has been found to modulate the recruitment of members of the silencing complexes to chromatin [44]. However, OGG1 is not deregulated after H2O2 treatment in our study, which may help explain the discrepancy in gene subsets analyzed. Although the influence of DNA methylation in other genes cannot be ruled out, these findings suggest DNA methylation may not be the predominant regulatory mechanism in the transcriptional regulation of most DNA repair genes.
We speculate that chromatin modifications may play important roles in transcriptional regulation of DNA repair genes in response to oxidative stress. Previous studies have showed that H4K16ac is a highly abundant activating modification [20], is a target for posttranslational modification for double strand breaks (DSBs), and plays an important role in modulating chromatin structure and transcriptional activation. Our results further suggest that hypo-acetylation of H4K16 is required for suppression of DNA repair genes in response to oxidative stress (Fig. 3). Significant DNA damage may be required for activation of DNA repair via transcriptional upregulation of DNA repair genes at low genotoxic doses. In contrast, at high doses, transcriptional inhibition would be provoked by epigenetic alterations including DNA methylation and histone modification, which would counteract the effect of gene activation. Our results show that the H4K16ac level is significantly decreased after 1 mM or higher dosage of H2O2 treatment but no alteration occurred in treatments with 0.5 mM or lower H2O2 exposures (Fig. 3A, 3B). This result supports the hypothesis that a low dose of oxidative stress activates DNA damage signaling, but is not sufficient to alter histone modification.
hMOF is thought to be a major histone acetyltransferase for H4K16 and plays important roles in many biological processes, including chromatin de-condensation, the formation of chromatin boundaries and in DNA repair [25, 45, 46]. On the other hand, SIRT1 is a class III histone deacetylase that is able to deacetylate H4K16ac, and also functions in the modulation of chromatin structure [31]. SIRT1 also deacetylates chromatin modifying enzymes, including PCAF, p300, and hMOF [47] in addition to histone substrates and many other nuclear factors. Thus, SIRT1 may function in the modulation of H4K16ac via two mechanisms. First, SIRT1 may directly deacetylate H4K16ac in response to oxidative stress. Second, SIRT1 may regulate chromatin affinity and the activity of hMOF to indirectly promote hypo-acetylation of H4K16 [31]. In the present study, SIRT1 inhibition did not rescue the decrease of H4K16ac in response to H2O2 treatment (Fig. 5), which precluded the first putative mechanism. Previous study has determined the mechanism that hMOF lysine 274 acetylation is required for hMOF activity toward H4K16 via affecting chromatin affinity of hMOF [32]. Here, we found that this mutation of hMOF (K274A) blocked the decreased level of H4K16ac via SIRT1 inhibition in hMOF overexpressing cells (Fig. 5) supporting the second putative possibility that SIRT1-modulated hMOF acetylation was the predominant cause to downregulation of DNA repair genes. In summary, we determined a potential mechanism that chromatin affinity of the remodeler may be an important mechanism by which chromatin structure, and gene expression, is regulated in DNA repair pathways.
Conclusion
In this study, we provide evidence that the number of downregulated DNA repair genes is larger than that of upregulated DNA repair genes upon exposure to H2O2. Moreover, our data suggests that the reduction in gene expression is independent of DNA methylation of promoters in H2O2-treated cells. These determinations were made via RNA-seq and RRBS-seq analyses. Overall, we determine an underlying mechanism by which SIRT1-modulated hMOF acetylation causes a decrease of H4K16ac, and thereby represses important DNA repair genes in response to H2O2-induced oxidative stress. These results strengthen our understanding of DNA repair gene dysregulation in response to oxidative stress and in human diseases resultant from this dysregulation.
Materials and Methods
Cell culture, Plasmids, shRNA, Antibodies, Chemicals, and Primers
HCT116 cells and Hela cells were cultured in DMEM media (Hyclone, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone, USA) and penicillin-streptomycin (Invitrogen, USA). cDNA for hMOF (NM_032188) was generated from a cDNA library and confirmed by DNA sequencing. cDNA for SIRT1 (BC012499) was purchased from Genechem Corp. (Shanghai) and then subcloned into pcDNA3.0 vector, followed by sequencing validation. SIRT1 shRNA-1: GGGTCTTCCCTCAAAGTAAGA; SIRT1 shRNA-2: GCAGATTAGTAGGCGGCTTGA; The source of primary antibodies were as follows: anti-H4K16ac (Millipore, 07-329), anti-H3K27m3 (Millipore, 05-1951), anti-H3K4m2 (Millipore, 05-1338), anti-H3K4m3 (Millipore, 04-745), anti-γH2AX (Millipore, 16-193), anti-SIRT1 (Millipore, 05-1243), anti-hMOF (Abgent, AO1112a), anti-BRCA2 (Abcam, ab90541), anti-RAD50 (Abcam, ab204375), anti-FANCA (Abcam, ab201457), anti-GAPDH (Santa Cruz, sc-32233), anti-HA-tag (Convense, MMS-1019), and anti-β-actin (Sigma, A1978). The following secondary antibodies were used: anti-mouse IgG peroxidase labeled antibodies (Sigma, A4416), anti-rabbit IgG peroxidase labeled antibodies (Sigma, A6154), and anti-rabbit IgG. In addition, the following reagents were used: Adriamycin (Sigma, D4035), Bleomycin (Sigma, B5507), Camptothecin (Sigma, C9911), Hydrogen peroxide solution 30% (w/w) (Sigma, H1009), Trichostatin A (Sigma, T8552), Wortmannin (Sigma, W1628), CGK733 (Sigma, C9867), and NU7026 (Sigma, N1537). The primers information please see Supplementary Materials.
siRNA and Plasmids Transfection, RNA Isolation, and qRT-PCR
siRNA transfections were carried out with Lipofectamine 2000 (Invitrogen, USA) and all plasmid transfections were performed with Polyethylenimine (PEI, Bender, Austria) according to the manufacturer's instructions. Total RNA was isolated using the Trizol reagent kit (Invitrogen, USA). cDNA was reverse-transcribed from the total RNA using the First Strand Reverse Transcription Kit (Fermentas, K1691). qRT-PCR was performed on an ABI 7500 Real time PCR system using a SYBR Green Mixture (Roche, 4913914001).
Chromatin Fractionation
Cells were harvested and lysed in lysing buffer (10 mM HEPES pH 7.8, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.2% NP-40, 1X protease inhibitor, and 1X phosphatase inhibitor). After centrifuged at 1,500 X g for 10 min at 4°C and washed in ice-cold washing buffer (lysing buffer without NP-40), the pellets were then lysed in soluble nuclear buffer (3 mM EDTA, 0.2 mM EGTA, and 1X protein inhibitors). The suspension was centrifuged at 2,000 X g for 10 min at 4°C and washed in ice-cold soluble nuclear buffer. Finally, the pellets were added with buffer containing a high concentration of NaCl (50 mM Tris-HCl pH 8.0, 0.05% NP40, 0.45 M NaCl, and 1X protease inhibitor cocktail).
Immunofluorescence Staining
HeLa cells were fixed in 4% paraformaldehyde/PBS for 15 min and then washed with PBS containing 0.1% Triton X-100 (PBST) for 15 min at room temperature. Following blocked in PBS containing 5% BSA (Blocking buffer) for 30 min, cells were incubated with the primary antibodies (H4K16ac) which were diluted in blocking buffer for 1 h. Cells were washed 3X times with PBST and then incubated with Alexa Fluor 488 (Goat anti-rabbit IgG, 1: 200) in PBS/5% BSA for 1 h. After 3X times washed in PBST, cells were incubated with 1μg/ml DAPI and detected with a fluorescence microscope (Leica, Germany).
ChIP Assay
Cells were crosslinked with 1% formaldehyde for 15 min at RT and quenched in unreacted formaldehyde using 125 mM glycine. Cells were collected and re-suspended in ChIP dilution buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl pH 8.1, 150 mM NaCl and protease inhibitors [Roche, USA]). The lysates were then sonicated using a Sonics VibraCell sonicator resulting in average fragment sizes of approximately 200-500 bp which was confirmed by agarose gel electrophoresis. The sonicated lysates were then centrifuged at 4°C for 10 min, the supernatant was collected and then pre-cleaned with pre-washed protein A beads (Roche, USA) at 4°C for 60 min with rotation. Prewashed Dynabead®MyOne™ Streptavidin T1 beads (Invitrogen, USA) were incubated with the supernatant overnight at 4°C and precipitated by centrifugation. An aliquot of the pre-cleaned supernatant was preserved as a genomic control. The resultant DNA absorbed beads were washed with Buffer I (2% SDS) twice, Buffer II (0.1% Deoxycholate, 1% Triton X-100, 1 mM EDTA, 1 mM HEPES pH 7.5, 500 mM NaCl) once, Buffer III (250 mM LiCl, 0.5% NP-40, 0.5% Deoxycholate, 1 mM EDTA, 10 mM Tris-Cl pH 8.1) once, and TE (10 mM Tris pH 8.1, 1 mM EDTA) twice for 10 min at RT with agitation. The beads were precipitated using magnetic separation and SDS elution buffer (1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.1), added to the eluate and de-crosslinked overnight at 65°C. The eluate was treated with RNase A and Proteinase K and extracted with a PCR recycle Kit (Qiagen, Germany). The resultant DNA eluate was then used for qPCR analyses.
RRBS library preparation, Sequencing and Analysis
Before RRBS library preparation, total cellular DNA (treated with or without H2O2) was extracted using proteinase K/phenol. For each library, 500 ng of genomic DNA was digested overnight with MspI (20 units). After purification, the digested products were blunt-ended. dA was then added, followed by methylated-adapter (Illumina, USA) ligation. To obtain DNA fractions in the 40-120 bp and 120-220 bp size ranges of MspI-digested products, two ranges (160-240 bp and 240-340 bp) of adapter-ligated fractions were excised from a 2% agarose gel. The size-selected DNA was bisulfite-treated for two rounds using the EpiTect Bisulfite Kit (Qiagen, Venlo, Netherlands) following the manufacturer's standard protocol. Finally, libraries were generated by amplification with HiFi HotStart Uracil+ ReadyMix (KAPA, USA) and Illumina Multiplexing PCR Primers. An Agilent 2100 Bioanalyzer (Agilent Technologies, USA) and RT-PCR were used to assess the quality and quantity of the RRBS library. After RRBS library construction, Illumina HiSeq2000 was used to sequence the libraries according to the manufacturer's protocol. Paired-end short reads were generated from cells (with or without H2O2 treatment) via HiSeq2000 sequencing. Our published integrated software RRBS-Analyser was used to perform quality control, align the reads to the reference genome, identify methylated cytosines, and identify differentially methylated regions. Results indicated that the bisulfite conversion experiment was estimated to be more than 99% low non-CpG methylation levels.
Bisufite Sequencing (BS)
2 μg of cellular genomic DNA (before and after 2 mM H2O2 treatment) were then treated with bisulfite. Bisulfite treatment converts cytosine to uracil while 5-methyl cytosine is resistant to the conversion. After the treatment, genomic DNA was subject to subcloning and sequencing. The sequencing result was then compared to the original sequences, and any methylated/unmethylated cytosines are unambiguously determined.
Supplementary Material
Supplementary tables and methods.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (No. 81301778 and No. 31271266), the Zhejiang Provincial Natural Science Foundation (ZNSF) (No. LY13C060002 and LY16C060001), a Zhejiang Provincial Department of Education Research Project (No. 84612051), and a Startup Fund for scholars of Gannan Medical University (No. QD201605).
Author Contributions
J.N.Z., W.S.C., and Z.S.S. designed the experiments. J.A.Z. and J.L.J performed molecular biological assays. S.S.D., W.L.W and F.H. performed cell culture and cellular assays. W.S.C. aided with RNA-seq and RRBS. X.F.L. and H.Q.C performed RNA-seq and RRBS-seq data analysis. J.Y. helped in gene cloning. X.X.W. helped in cell culture. J.N.Z., Y.X., and Z.S.S. wrote the manuscript.
Competing Interests
The authors declare no competing financial interests.
References
1. Dizdaroglu M. Oxidatively induced DNA damage and its repair in cancer. Mutation research Reviews in mutation research. 2015;763:212-45
2. Srivastava KK, Kumar R. Stress, oxidative injury and disease. Indian journal of clinical biochemistry: IJCB. 2015;30:3-10
3. Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR. Oxidative stress and Parkinson's disease. Frontiers in neuroanatomy. 2015;9:91
4. Georgakilas AG. Oxidative stress, DNA damage and repair in carcinogenesis: have we established a connection? Cancer letters. 2012;327:3-4
5. Rusyn I, Asakura S, Pachkowski B, Bradford BU, Denissenko MF, Peters JM. et al. Expression of base excision DNA repair genes is a sensitive biomarker for in vivo detection of chemical-induced chronic oxidative stress: identification of the molecular source of radicals responsible for DNA damage by peroxisome proliferators. Cancer research. 2004;64:1050-7
6. Yan S, Sorrell M, Berman Z. Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cellular and molecular life sciences: CMLS. 2014;71:3951-67
7. Cencioni C, Spallotta F, Martelli F, Valente S, Mai A, Zeiher AM. et al. Oxidative stress and epigenetic regulation in ageing and age-related diseases. International journal of molecular sciences. 2013;14:17643-63
8. Gu X, Sun J, Li S, Wu X, Li L. Oxidative stress induces DNA demethylation and histone acetylation in SH-SY5Y cells: potential epigenetic mechanisms in gene transcription in Abeta production. Neurobiology of aging. 2013;34:1069-79
9. Nishida N, Kudo M. Oxidative stress and epigenetic instability in human hepatocarcinogenesis. Digestive diseases. 2013;31:447-53
10. Liu D, Xu Y. p53, oxidative stress, and aging. Antioxidants & redox signaling. 2011;15:1669-78
11. Okumura H, Ishii H, Pichiorri F, Croce CM, Mori M, Huebner K. Fragile gene product, Fhit, in oxidative and replicative stress responses. Cancer science. 2009;100:1145-50
12. Karin M, Takahashi T, Kapahi P, Delhase M, Chen Y, Makris C. et al. Oxidative stress and gene expression: the AP-1 and NF-kappaB connections. BioFactors. 2001;15:87-9
13. Nose K. [Transcriptional control of AP-1 by oxidative stress]. Tanpakushitsu kakusan koso Protein, nucleic acid, enzyme. 1999;44:2427-31
14. Lopez-Camarillo C, Ocampo EA, Casamichana ML, Perez-Plasencia C, Alvarez-Sanchez E, Marchat LA. Protein kinases and transcription factors activation in response to UV-radiation of skin: implications for carcinogenesis. International journal of molecular sciences. 2012;13:142-72
15. Pines A, Perrone L, Bivi N, Romanello M, Damante G, Gulisano M. et al. Activation of APE1/Ref-1 is dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. Nucleic acids research. 2005;33:4379-94
16. O'Hagan HM. Chromatin modifications during repair of environmental exposure-induced DNA damage: a potential mechanism for stable epigenetic alterations. Environmental and molecular mutagenesis. 2014;55:278-91
17. Kumar R, Horikoshi N, Singh M, Gupta A, Misra HS, Albuquerque K. et al. Chromatin modifications and the DNA damage response to ionizing radiation. Frontiers in oncology. 2012;2:214
18. Price BD, D'Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell. 2013;152:1344-54
19. Kinner A, Wu W, Staudt C, Iliakis G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic acids research. 2008;36:5678-94
20. Smith CM, Gafken PR, Zhang Z, Gottschling DE, Smith JB, Smith DL. Mass spectrometric quantification of acetylation at specific lysines within the amino-terminal tail of histone H4. Analytical biochemistry. 2003;316:23-33
21. Taylor GC, Eskeland R, Hekimoglu-Balkan B, Pradeepa MM, Bickmore WA. H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res. 2013;23:2053-65
22. Smith ER, Cayrou C, Huang R, Lane WS, Cote J, Lucchesi JC. A human protein complex homologous to the Drosophila MSL complex is responsible for the majority of histone H4 acetylation at lysine 16. Molecular and cellular biology. 2005;25:9175-88
23. Cao L, Zhu L, Yang J, Su J, Ni J, Du Y. et al. Correlation of low expression of hMOF with clinicopathological features of colorectal carcinoma, gastric cancer and renal cell carcinoma. International journal of oncology. 2014;44:1207-14
24. Sharma GG, So S, Gupta A, Kumar R, Cayrou C, Avvakumov N. et al. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Molecular and cellular biology. 2010;30:3582-95
25. Kapoor-Vazirani P, Kagey JD, Powell DR, Vertino PM. Role of hMOF-dependent histone H4 lysine 16 acetylation in the maintenance of TMS1/ASC gene activity. Cancer research. 2008;68:6810-21
26. Vaquero A, Sternglanz R, Reinberg D. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene. 2007;26:5505-20
27. Boulton SJ, Jackson SP. Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. The EMBO journal. 1998;17:1819-28
28. Uhl M, Csernok A, Aydin S, Kreienberg R, Wiesmuller L, Gatz SA. Role of SIRT1 in homologous recombination. DNA repair. 2010;9:383-93
29. Mishra BP, Zaffuto KM, Artinger EL, Org T, Mikkola HK, Cheng C. et al. The Histone Methyltransferase Activity of MLL1 Is Dispensable for Hematopoiesis and Leukemogenesis. Cell reports. 2014;7:1239-47
30. Hajji N, Wallenborg K, Vlachos P, Fullgrabe J, Hermanson O, Joseph B. Opposing effects of hMOF and SIRT1 on H4K16 acetylation and the sensitivity to the topoisomerase II inhibitor etoposide. Oncogene. 2010;29:2192-204
31. Lu L, Li L, Lv X, Wu XS, Liu DP, Liang CC. Modulations of hMOF autoacetylation by SIRT1 regulate hMOF recruitment and activities on the chromatin. Cell research. 2011;21:1182-95
32. Wang K, Li X, Dong S, Liang J, Mao F, Zeng C. et al. Q-RRBS: a quantitative reduced representation bisulfite sequencing method for single-cell methylome analyses. Epigenetics. 2015;10:775-83
33. Peng L, Ling H, Yuan Z, Fang B, Bloom G, Fukasawa K. et al. SIRT1 negatively regulates the activities, functions, and protein levels of hMOF and TIP60. Molecular and cellular biology. 2012;32:2823-36
34. Kansara K, Patel P, Shah D, Shukla RK, Singh S, Kumar A. et al. TiO2 nanoparticles induce DNA double strand breaks and cell cycle arrest in human alveolar cells. Environmental and molecular mutagenesis. 2015;56:204-17
35. Shan B, Morris G.F. Binding sequence-dependent regulation of the human proliferating cell nuclear antigen promoter by p53. Exp Cell Res. 2005;305(1):10-22
36. Grosch S, Kaina B. Transcriptional activation of apurinic/apyrimidinic endonuclease (Ape, Ref-1) by oxidative stress requires CREB. Biochem Biophys Res Commun. 1999;261(3):859-63
37. Christmann M. et al. Fen1 is induced p53 dependently and involved in the recovery from UV-light-induced replication inhibition. Oncogene. 2005;24(56):8304-13
38. Gomez V. et al. Regulation of DNA damage responses and cell cycle progression by hMOB2. Cell Signal. 2015;27(2):326-39
39. Sumual S. et al. Differential regulation of Snail by hypoxia and hyperglycemia in human proximal tubule cells. Int J Biochem Cell Biol. 2010;42(10):1689-97
40. Fachin A.L. et al. Gene expression profiles in human lymphocytes irradiated in vitro with low doses of gamma rays. Radiat Res. 2007;168(6):650-65
41. Lee S.Y. et al. TP53 regulates human AlkB homologue 2 expression in glioma resistance to Photofrin-mediated photodynamic therapy. Br J Cancer. 2010;103(3):362-9
42. O'Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS genetics. 2008;4:e1000155
43. O'Hagan HM, Wang W, Sen S, Destefano Shields C, Lee SS, Zhang YW. et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands. Cancer cell. 2011;20:606-19
44. Canugovi C, Maynard S, Bayne AC, Sykora P, Tian J, de Souza-Pinto NC, Croteau DL, Bohr VA. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair. 2010;9(10):1080-9
45. Fullgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q. et al. The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature. 2013;500:468-71
46. Gupta A, Guerin-Peyrou TG, Sharma GG, Park C, Agarwal M, Ganju RK. et al. The mammalian ortholog of Drosophila MOF that acetylates histone H4 lysine 16 is essential for embryogenesis and oncogenesis. Molecular and cellular biology. 2008;28:397-409
47. Vaquero A. The conserved role of sirtuins in chromatin regulation. The International journal of developmental biology. 2009;53:303-22
Author contact
![]() Corresponding author: Jianing Zhong: zhongning_003com; Zhong Sheng Sun: sunzsbiolscom
Corresponding author: Jianing Zhong: zhongning_003com; Zhong Sheng Sun: sunzsbiolscom