Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Heterogeneity of breast cancer
Single cell DNA sequencing in...
Technical challenges in single...
Single cell DNA sequencing...
Conclusion and Future Aspects
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(8):949-960. doi:10.7150/ijbs.19627 This issue Cite
Review
Current Progresses of Single Cell DNA Sequencing in Breast Cancer Research
Jianlin Liu, Ragini Adhav, Xiaoling Xu ![]()
Faculty of Health Science, University of Macau, Macau SAR, China
Received 2017-2-13; Accepted 2017-5-8; Published 2017-7-18
Abstract

Breast cancers display striking genetic and phenotypic diversities. To date, several hypotheses are raised to explain and understand the heterogeneity, including theories for cancer stem cell (CSC) and clonal evolution. According to the CSC theory, the most tumorigenic cells, while maintaining themselves through symmetric division, divide asymmetrically to generate non-CSCs with less tumorigenic and metastatic potential, although they can also dedifferentiate back to CSCs. Clonal evolution theory recapitulates that a tumor initially arises from a single cell, which then undergoes clonal expansion to a population of cancer cells. During tumorigenesis and evolution process, cancer cells undergo different degrees of genetic instability and consequently obtain varied genetic aberrations. Yet the heterogeneity in breast cancers is very complex, poorly understood and subjected to further investigation. In recent years, single cell sequencing (SCS) technology developed rapidly, providing a powerful new way to better understand the heterogeneity, which may lay foundations to some new strategies for breast cancer therapies. In this review, we will summarize development of SCS technologies and recent advances of SCS in breast cancer.
Keywords: Cancer stem cells, Intratumor heterogeneity, Intertumor heterogeneity, Breast cancer, Single cell sequencing.
Introduction
Breast cancer is the most common malignancy in women worldwide. Drug resistance, cancer recurrence and metastasis are the main causes of mortality of breast cancer patients [1, 2]. According to the cancer stem cell (CSC) hypothesis, CSCs are responsible for phenotypic and functional heterogeneity within the tumor in some cancers [3-6], and for disease recurrence and metastasis [7-11], which make cancer hard to treat. Within tumor microenvironment, CSCs are differentially originated in different area within a tumor, leading to cancer heterogeneity [12]. On the other hand, clonal evolution theory suggests that heritable genetic and epigenetic changes contribute to tumor heterogeneity. Similar to hematologic malignancies or other cancers, breast cancers also have high heterogeneity in pathology and molecular profiles [13]. It is essential to reveal the initiating events in molecular nature of breast cancer and understand the nature of heterogeneity, and then to identify putative predictive biomarkers for aberrant oncogene, thereby to develop molecularly targeted therapies for breast cancer patients. Emerging technologies, such as massive parallel genomic sequencing, multiplexed somatic mutation genotyping, are commonly used to classify cancers into molecular subsets [14]. In the past 5-10 years, single cell sequencing undergoes a rapid progress, which greatly advanced our understanding of heterogeneity in cancers.
Heterogeneity of breast cancer
Breast cancer is a heterogeneous malignancy and there are highly diversities among different cancers (Intertumor heterogeneity), and within a single tumor (Intratumor heterogeneity).
Intertumor heterogeneity
Human breast cancers are categorized into 18 subtypes by the histological features of primary tumors, such as cellular arrangement, cellular features, lesion size, the presence of necrosis, etc. [15]. However, this kind of classification based on histological criteria is confusing due to a number of factors, including regions of different morphologies within one tumor, and different scoring subjectivity, etc. [13]. Recently, microarray analysis provides a new way to categorize human breast cancers into at least 6 subtypes by gene expression profiling, including luminal A, luminal B, ERBB2, claudin-low, normal-like, and basal-like [16-19]. Definitely, this pattern of gene expression in luminal and basal cells is useful for defining the original molecular portraits of breast tumors, and facilitating patient care and treatment [16, 20]. By using a sophisticated lineage tracing approach in vivo, a pool of Axin2+ cells are considered to contribute to the origin of basal and luminal cell populations [21]. Luminal progenitor cells with an Flf5-driven Cre transgene only give rise to the luminal population of cells [22]. ERα-/PR- luminal progenitors only generate ERα-/PR- differentiated luminal alveolar cells [23]. Subtype-specific tumor cell-of-origin and transforming events have been proposed to be responsible for intertumor heterogeneity [24]. It has been proposed that HER2+ tumors correlate with enrichment in the fetal mammary stem cell signature and luminal progenitors serve as precursors for basal-like cases [25-28]. Other studies provided evidence illustrating that the luminal progenitors are original cells for breast cancer associated gene-1 (BRCA1) mutant carriers [25, 28]. Basal mammary stem cells can develop into both basal and luminal cells in varied stages of development [22, 29]. In these cases, genetic lineage tracing helps us to understand the mammary epithelial hierarchies and origin of cancers.
Intratumor heterogeneity
Besides the huge differences among different tumors, the cancer cells within one tumor are also highly diverse with cellular genetic alternations [30]. A tumor might be a complexity of different subtypes of cell, understanding the origin and evolution of the tumor is helpful for understanding of either intratumor or intertumor heterogeneity of cancer, facilitating cancer therapy, especially the precision medicine for cancer. Two main theories have been proposed to explain intratumor heterogeneity, including cancer stem cell theory [31] and clonal evolution model [32], which might be complementary during cancer development.
Cancer stem cells (CSCs)
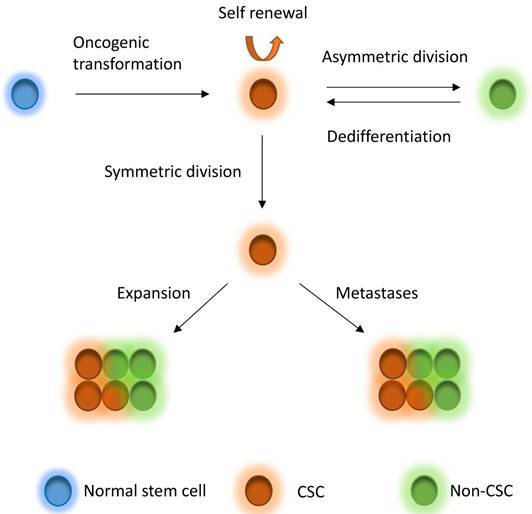
CSCs, which are a very small pool of tumorigenic cells that have self-renewal, proliferative and differentiation abilities, have been used to explain intratumor heterogeneity. Breast CSCs were first isolated and characterized from primary and metastatic human breast tumor in 2003 [33]. The identification of breast CSCs depends on the recognition of the cell surface marker expression, such as CD44+CD24-/low, CD55+, CD61+, and ALDH1 [33-36]. CSCs derive from several fashions, including oncogenic transformation of a normal stem cell, non-CSC-to-CSC plasticity, etc. [31]. According to the CSC theory, CSCs are on the top of the hierarchy of cells and drive the growth and progression of the tumor mass and seed metastases [31]. The CSCs hierarchy is bidirectional conversions between CSC and non-CSC states (Figure 1). CSCs undergo symmetric division to produce more CSCs. On the other hand, the most tumorigenic cells undergo asymmetric division to generate non-CSCs with less tumorigenic and metastatic potential [31]. In addition, non-CSCs can also undergo a dedifferentiation process and revert to the CSC phenotype under certain conditions [10, 31, 37-40]. CSCs may comprise a group of heterogeneous and functionally varied population of cells that may undergo different genetic and epigenetic changes and non-CSCs can convert to CSCs with various degrees of efficiency depending on the stimuli from the microenvironment, probably giving rise a vast degree of diversity within tumors [31]. Increasing studies also demonstrated that CSC model is applicable to many other solid tumors including breast tumor [33], glioblastoma [7, 41], colorectal tumor [42, 43], pancreatic tumor [44] and ovarian tumor [45-47]. However, the CSCs might be dynamically variable within tumors under distinct microenvironmental cues [31, 48] and the markers for CSCs might also be variable in different subtypes or stages of diseases [33, 46, 49-54], although some cases might not follow the CSC model [55-58]. The mammary gland is a special organ, of which development occurs through different stages throughout embryonic and pubertal development and reproductive life. Most recently, evidences show that the cancer risk is attributed to random mutation arising in normal stem cells divisions [59, 60], which suggests that mammary gland might be an organ has relative high cancer risk, whereas another study suggests that the mutation accumulation is tissue specific in adult stem cells [61]. Although these studies differ in the pattern of mutation occurring, they indeed support the CSC theory to some extent.
CSC model. Normal stem cells can undergo oncogenic transformation to give rise to cancer stem cells (CSCs). CSCs can generate CSCs and non-CSC through symmetric division and asymmetric division, respectively, driving tumor growth and seeding metastases. On the other hand, non-CSCs can also dedifferentiate back to CSCs under stimuli from microenvironment. CSCs to non-CSCs is bidirectional and dynamic conversion, leading to great diversity within tumors.
Clonal evolution
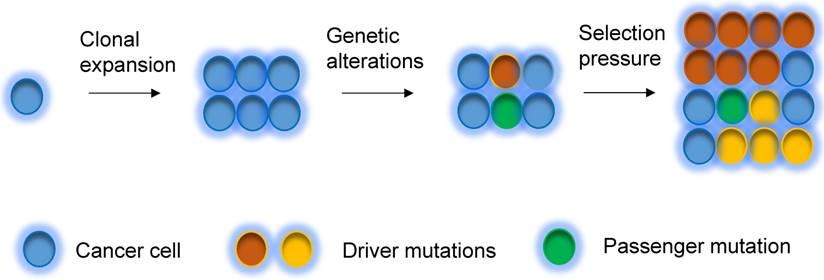
A tumor initially arises from a single cell, which then undergoes clonal expansion to a population of cancer cells. During tumorigenesis and evolution process, cancer cells undergo different degrees of genetic instability and consequently obtain varied genetic aberrations. Clonal expansions driven by the acquisition of different mutations are main portraits for clonal evolution [32, 62] (Figure 2), but not all expansions are induced by genetic events. Driver mutations (i.e. mutations that allow cells gain growth advantages) are the key mutational events that drive clonal expansions in a given microenvironment [63]. Under a certain set of selective pressure, clones acquire driver mutations, and are also accompanied by passenger alterations, which may change into driver aberrations if the selective pressures change [63]. During the tumor progression, the mutational rate also changes, subsequently the clones acquire new mutations, which lead to genetic heterogeneity within the tumor [63, 64]. The clonal evolution has been revealed in breast cancer. Several somatic coding mutations that vary between primary and metastatic breast tumor have been discovered by using next generation sequencing in patients, suggesting that evolution occurs in breast cancer progression [65]. In addition, some other events, such as epigenetic modification during tumorigenesis could also substantially contribute to heterogeneity of cancers [66, 67].
Clonal evolution model. Tumor can evolve through clonal expansion of cancer cell, giving rise to heterogeneity within tumors, which is created by genetic changes. Under selection pressure, earlier diver mutations (red color), and new driver mutations (yellow color) obtain advantage for outgrowth of clones and drive tumors grow.
Single cell DNA sequencing in breast cancer
Because the quantity of CSCs is very low compared with the total cells of a tumor, the genetic alterations of the CSCs probably are masked when tumors are sequenced as a whole. This problem can be overcome by using single cell sequencing (SCS), which is a powerful technology to study the evolution and heterogeneity in tumor and to understand the role of rare cells in cancer progression [68]. Compare to mixed cell sequencing, SCS could unravel key points much clearer in cancer biology that is difficult to address with bulk tumor sequencing. Thus, this method will be greatly helpful for our understanding of initiation, progression, invasion, metastasis, resistance and recurrence in cancer, subsequently guiding a more efficient early detection and targeted therapy in cancer therapies in clinic.
Technical challenges in single cell DNA sequencing
For the single cell DNA research, following several main procedures are involved in a sequential order: 1) Single cell isolation; 2) Amplification (including whole genome amplification, library construction); 3) Sequencing; and 4) Bioinformatics analysis. To date, the single cell sequencing is still technologically challenging due to the bias and errors caused during the whole single cell DNA sequencing workflow.
Single cell isolation
The first step of single cell sequencing is to capture the single cell of interest. Several approaches have been developed for capturing the single cell from abundant pools, including serial dilution [69], micropipetting [70], microwell dilution [71], optical tweezers [72], microfluidic platforms [73-75], and fluorescence-activated cell sorting (FACS) [76] (Table 1). Basically, serial dilution is conducted by diluting a pool of cells for a number of times in a constant dilution factor to obtain one single cell per microliter [69]. Micropipetting uses a special glass micropipette under the microscope to pick the cell of interest into individual PCR tubes for downstream amplification [70], which is laborious and low-throughput. Microwell dilution is a method that distributes single cells into wells by using a microwell array, followed by genetic material amplification simultaneously [71]. Most recently, QIAscout (QIAGEN) has been developed for isolating the single cell effectively and fast by microrafts (https://www.qiagen.com/mo/). Optical tweezers employ highly focused laser beam in the combination of imaging-based cell selection to capture individual cells [72, 77, 78]. Microfluidic platforms usually use reconfigurable flow-routing capabilities of integrated microvalve technology, which can deposit single cells into nanoliter-volume storage chambers of microfluidic chips, followed by amplification in droplets containing reagents and cell [73, 74]. C1 Single-Cell Auto Pre System is popular one of microfluidic devices that uses pneumatic components to control the microfluidic integrated fluidic circuit (IFC) and uses thermal components for preparatory chemistry, which has 96-capture sites per IFC [75]. Drop-seq [79] and the Chromium™ Controller from 10x genomics (https://www.10xgenomics.com/single-cell/) can also be used for single cell capture, but the present designs are only applicable for downstream RNA-seq not for DNA-seq. FACS can isolate thousands of single cells in microdroplets by electric charge at much high pressure. Antibodies against cell markers or dye can be used for labeling live cell or cell nuclei to isolate sub-groups of cell or cell nuclei from frozen or formalin-fixed paraffin-embedded (FFPE) samples [76, 80]. However, isolating rare single cell is much more challenging. In this regards, laser capture microdissection (LCM) is used for the isolation of cells within certain context without contamination from surrounding cells from fresh or archival specimens by using infrared (IR) capture system [81, 82] or ultraviolet (UV) cutting system [83-86] (Table 2). Circulating tumor cells (CTCs) are rare cells present in peripheral blood from cancer patients, which occur at much low frequency [87]. So far, several platforms have been developed for capturing CTCs (Table 2). CTC-chip is a unique microfluidic platform that can be used to separate viable CTCs efficiently via antibody EpCAM-coated microposts [88]. So far, FDA (Food and Drug Administration) has approved the CellSearch system, which uses ferrofluid particles conjugated with anti-EpCAM and anti-CD45 antibodies [89, 90], for the capture of CTCs.
Methods for capturing single cell from abundant cell population
| Methods | Descriptions | Advantages | Disadvantages |
|---|---|---|---|
| Serial dilution | Serial dilution to single cell per microliter | Low cost | Time-consuming; high possibility of capturing multiple cells |
| Micropipetting | Capture single cell using special micropipette | Low cost; higher possibility for capturing rare population cells | Time-consuming; low throughput |
| Microwell dilution | Isolate single cell using microwell array | High throughput; low contamination (nanolitre volumes reaction); less reagent cost | Expensive consumables and equipment; time-consuming |
| Optical tweezers | Trap single cell using focused laser beam | Low contamination (nanolitre volumes reaction); fluorescent cells can be captured | Highly dependent both on the size and shape of the cell; low throughput |
| Microfluidic devices | Capture single cell into flow chambers using microfluidic chips | Low contamination (nanolitre volumes reaction); less reagent cost | Expensive consumables and equipment; hard to avoid cell doublets or empty well |
| FACS sorting | Sort single cell by electric charge at high pressure | High throughput; fluorescent cell surface markers can be used for capturing specific population of cells; dye can be used for sorting nuclei from broken cell of frozen or FFPE samples | Expensive equipment |
Methods for capturing single cell from rare cell population
| Methods | Descriptions | Advantages | Disadvantages |
|---|---|---|---|
| Laser capture microdissection | Isolate single cell from tissue section using a laser | Context of cell can be identified | Expensive equipment; DNA materials may be damaged by UV |
| CTC-chip | Separate CTC via antibodies-coated micropost | High-throughput | Expensive consumables |
| CellSearch | Capture CTC using ferrofluid particles conjugated with EpCAM antibody | High-throughput | CTC with mesenchymal-like phenotype can be difficult to isolate |
Amplification of genome
Since there is only one copy of DNA within one single cell, whole genome amplification (WGA) is required for next generation sequencing. However, artefacts can be introduced during WGA, such as amplification bias (decreased coverage uniformity, allelic imbalance), genome loss, mutations and chimaeras [91]. There are several conventional approaches used for WGA, such as degenerate oligonucleotide primed PCR (DOP-PCR) [92], multiple displacement amplification (MDA) [93, 94], PicoPLEX [95], multiple annealing and looping-based amplification cycles (MALBAC) [70] (Table 3). DOP-PCR is a special polymerase chain reaction (PCR) that employs oligonucleotides of partially degenerated sequence with a low annealing temperature, which enables the priming at multiple evenly dispersed sites along a genome, resulting in DNA amplification at a rapid, efficient manner [92]. Only 10% of physical coverage can be achieved along a single-cell genome but levels of copy number can be accurately retained using DOP-PCR [68, 76, 96], which makes it suitable for copy number variants (CNVs) detection not for single nucleotide variants (SNVs) measurement. MDA amplifies the randomly primed regions of genome by using random hexamers and Phi29 DNA polymerase in an isothermal condition, which has a low error rate due to the high proof-reading capability of Phi29 DNA polymerase [93, 94, 97]. High physical coverage (>90%) can be achieved along a single-cell genome using MDA, however the coverage is not uniform, making it a good method to detect the SNVs but a poor method for CNVs detection [70, 98-100]. PicoPLEX and MALBAC follow a very similar protocol, which use random degenerate primers to add a common sequence in the first step, followed by priming for subsequent PCR amplification [95]. MALBAC modifies this protocol by using a new common sequences and temperature cycling, which results in more uniform amplification [70]. This method has high false positive error rates in SNVs, which suggests it more suitable for CNVs detection [70]. The comparison of these three methods has been done. One report find that coverage generated by MDA is better than MALBAC, leading to higher detection rates of SNVs [101]. Another report reveals that coverage breath of MDA is better than MALBAC and DOP-PCR [102]. But the uniformity of MALBAC and DOP-PCR is greater, which results in better detection of CNVs [101, 102]. So far several groups improve the WGA method by performing MDA for single cell DNA in microfluidic emulsions. Microwell displacement amplification system (MIDAS) performs the MDA for single cell in nanoliter wells, which can both reduce contamination and improve amplification uniformity [71]. Droplet MDA can minimize bias and amplification of contaminants by using microfluidic-generated picoliter droplets for WGA reactions [103]. Emulsion WGA (eWGA) also divides DNA into a large number of picoliter aqueous droplets in oil for DNA amplification, which can improve the uniformity and accuracy of the amplification markedly [104]. Usually the WGA induces a lot of bias and errors and library construction follows the WGA, amplifying the bias. However, a new robust, scalable, and high-fidelity method called direct library preparation (DLP) is developed for library preparation using nanoliter-volume transposition reactions without preamplification, which results in greater coverage uniformity and more reliable detection of copy-number alterations compared with existing methods [105]. Most recently, a novel method, linear amplification via transposon insertion (LIANTI), is developed for single-cell WGA [106]. Unlike the exponential amplification of genomic DNA in conventional WGA methods, this method combines Tn5 transposition and T7 promoter to linearly amplify the genomic DNA into thousands of copies of RNAs, followed by reverse transcription and second-strand synthesis into double-stranded amplicons for DNA library construction, which outperforms existing approaches by greatly reducing amplification bias and errors, enabling micro-CNV detection with kilobase resolution by digital-counting analysis of inferred fragment number [106].
Methods for whole genome amplification
| Methods | Description | Advantages | Disadvantages |
|---|---|---|---|
| Conventional WGA methods | |||
| Degenerate oligonucleotide primed PCR (DOP-PCR) | PCR-based amplification using degenerate oligonucleotides and thermostable polymerase | High uniformity (better for calling CNVs) | High error rate; low coverage |
| Multiple displacement amplification (MDA) | Isothermal amplification of randomly primed regions of genome using random hexamers and Phi29 polymerase | Low error rate; great coverage (better for calling SNVs) | Lack of uniformity |
| Multiple annealing and looping-based amplification cycles (MALBAC) or PicoPLEX | Limited isothermal amplification using degenerate primers followed by PCR amplification | High uniformity (better for calling CNVs) | Intermediate error rate |
| Modified MDA methods | |||
| Microwell displacement amplification system (MIDAS) | Perform MDA in microwell of nanoliter | Low contamination; improved uniformity than conventional MDA | Limited efficiency of amplification |
| Droplet MDA | Perform MDA in microfluidic-generated picoliter droplets | Low contamination; improved uniformity than conventional MDA | - |
| Emulsion WGA (eWGA) | Perform MDA in picoliter aqueous droplets in oil | Higher coverage; higher accuracy and finer resolution in simultaneous detection of SNVs and CNVs | - |
| Direct library preparation (DLP) | Directly construct single-cell whole-genome library using nanoliter-volume transposition reactions without preamplification | High uniformity; reliable for detection of CNVs | - |
| Novel WGA methods | |||
| Linear amplification via transposon insertion (LIANTI) | Combine Tn5 transposition and T7 promoter in vitro transcription to linearly amplify the genomic DNA | Lowest amplification bias and errors | - |
Next generation sequencing
There are several commonly used high-throughput sequencing platforms, including Roche 454 sequencing [107], Ion torrent [108, 109], Illumina sequencing [110], the Complete Genomics platform [111], and the Real-time Sequencer (RS) of Pacific Biosciences [112], although with different errors rate (Table 4). These approaches may generate some common errors: nucleotide substitutions, insertions and deletions (indels), and coverage biases. It is reported that indels errors in Illumina platform are much less than substitution errors, in addition, overall error rate of Illumina is the lowest comparing with other platforms [113]. The error profiles of individual platforms have been discussed in this review [113]. For the Illumina platform, some errors come up as the lower quality scores in the specific positions such as some sequence motifs, especially the indels error in GC-rich sequence and around inverted repeats. These types of errors can be removed by normalizing with the quality score (Q>20) [114-117]. However, some other errors introduced in library or sample preparation could not be reflected in the sequence quality score, such as the errors owing to PCR, which could be reduced by using a PCR-free library construction [114].
Error rate of high-throughput sequencing platforms
| Platforms | Substitution | Indels |
|---|---|---|
| Roche 454 sequencing | Low | Intermediate |
| Ion torrent | Low | Intermediate |
| Illumina sequencing | Low | Low |
| The Complete Genomics platform | High | Low |
| The Real-time Sequencer (RS) by Pacific Biosciences | High | High |
Bioinformatics analysis
Analysis of high-throughput sequencing data is a very complicated and time-consuming process. In general, analyses of bulk sequencing and single cell sequencing follow a similar pipeline as briefly summarized below. 1) Pre-processing raw sequence data, including alignment to a reference genome and some data cleanup. 2) Variant discovery, including identification of genomic variation in each sample and filter the data using the appropriate methods based on experiment design. 3) Callset refinement, including using meta-data to increase genotyping accuracy and determine the overall quality of the callset. However, due to much more bias (amplification bias and chimeric DNA rearrangements) introduced during WGA, the computational methods for analysis of single cell data should be different. So far, many methods or algorithms are developed for bulk DNA sequence data, the tools originally designed for analyzing single cell data are relatively lacking. Several tools developed for bulk sequence alignment are used for single cell genome alignment currently, including BWA [70, 100, 118], Bowtie [80, 119], SOAPaligner [98, 99, 120] etc. Single nucleotide variants (SNVs) caller for bulk samples also have been used for single cell sequencing, such as SNVdetector [121], SOAPsnp [98, 99, 122], VarScan [123, 124], GATK [70, 100, 125-127], MuTech [128], etc.. Current variant callers basically do not include allelic dropout (ADO), allelic imbalance, coverage non-uniformity and false-positive errors, so they are not exactly applicable for single cell DNA sequencing. Monovar is a novel method designed for detecting and genotyping SNVs in single cell data, which outperforms standard methods commonly used to identify driver mutations and delineating clonal substructure [129]. Filtering the data using the appropriate methods or criteria based on experiment design is important for single cell analysis. Due to bias and errors introduced during the previous steps, all the possibilities are required to be taken into account. It is crucial to compare the variant alleles found in the single cells to those identified in the bulk to avoid selection bias for single cell capture [74]. In addition, the errors introduced in WGA and sequencing, which results in SNVs false positive calls, are required to be considered. To overcome this problem, firstly, it requires that the coverage of the mutant alleles should be 10X or more reads, which leads to very low false positive rate [99]. Secondly, the criterion that a mutation occurs in bulk sample or in at least two cells simultaneously is required [70, 74]. However, this strategy may lead to loss of some extremely rare mutations that only occur in one single cell. This can be partially overcome by validation of these mutations using PCR-Sanger sequencing in the original single cell DNA, but still depends on how early the errors are introduced. Loss of genome and non-uniform amplification can cause false positive or false negative in calling copy number aberrations (CNAs) detection. Improvement of uniformity of WGA is essential for calling CNAs. Usually CNAs calling also mainly relies on algorithms, including circular binary segmentation, rank segmentation, and hidden Markov models, which can normalize the bias during WGA to identify the regions that are really biologically over- or under- amplified [70, 96, 130]. Ginkgo is also a web platform designed for single cell CNAs analysis, which can automatically construct copy-number profiles of cells [131]. Determining genetic relationships between single cells, consequently understanding the mutational heterogeneity within tumors and inferring of a tumor evolutionary lineage trees are important task for single cell DNA sequencing. OncoNEM is a method for reconstruction of tumor clonal lineage tress from somatic SNVs of single cells, which accounts for genotyping errors and tests for unobserved subpopulations [132]. Ginkon can construct phylogenetic trees of cells by using CNAs [131]. Single Cell Genotyper (SCG) infers clonal genotype and population structure by inputting a cell-target matrix, which simultaneously addresses the technical noise in single cell data by taking doublets into account and predicting genotypes accordingly [133]. SCITE is a tool for the identification of the evolutionary history of a tumor by analyzing single cells, which is able to calculate the maximum-likelihood mutation history by using a flexible Markov chain Monte Carlo sampling scheme [134]. SiFit is another novel method for tumor phylogenies from noisy single cell data by using a finite-sites model, which could improve inference of tumor phylogenies (bioRxiv: http://dx.doi.org/10.1101/091595).
Single cell DNA sequencing application in breast cancer
Single cell DNA sequencing is mainly used for calling SNVs, CNAs, and structure variants (SVs) in breast cancer research. So far studies mainly focus on CNAs and SNVs, however, few study on SVs is published.
CNAs
The current single cell genome sequencing in breast cancer mainly focus on CNAs by whole genome sequencing (WGS), revealing the heterogeneity within the tumor and the clonal structure and evolution of tumors. Calling for CNAs does not require high coverage of sequencing. The first single cell DNA sequencing research in cancer was published by Navin etc. in 2011 [76], which called CNAs using bins of different length with uniform expected unique counts for correcting the bias in WGA. Eventually, they sequenced 100 single cells of a polygenomic triple negative (ER-/PR-/Her2-) breast cancer and 100 single cells of a monogenomic triple negative breast primary cancer and its liver metastasis using single nucleus sequencing with low coverage (~6%), revealing three different subclones that might represent sequential clonal expansion. The CNAs analysis also revealed a clonal expansion leading to the formation of original tumor and the metastasis respectively. In addition, their findings suggested a new pattern of tumor growth called “punctuated clonal evolution” with few persistent intermediates, which is contradict to gradual models. This evolution model was supported by another report from the same group three years later [100]. To increase coverage and reduce the ADO and false positive rate induced in WGA, they sorted nuclei from cells at G2/M phase for sequence. Fifty single nuclei of an oestrogen-receptor positive (ER+/PR+/Her2-) (ERBC) breast tumor were sequenced, which showed the tumor shared highly similar CNAs, suggesting a monoclonal population. Meanwhile, fifty single nuclei from triple-negative breast cancer were sequenced, and the CNAs profiles revealed highly similar aneuploid and hypodiploid within each subpopulation. These findings indicated that the tumor evolve by early rearrangements in chromosome, followed by stable clonal expansions. This model was further confirmed by sequencing 1,000 single cells of triple negative breast tumors from 12 patients [135]. Eventually, this model established by single cell CNAs suggested important implications for tumor evolution, diagnostics and therapy in breast cancer, especially in TNBC. In addition, single cell CNAs also has been used for tracing the origin of disseminated tumor cell in breast cancer [136]. In this study, genomes of 63 single cells from 6 non-metastatic breast cancer patients were sequenced. Fifty-three percent of cells were defined as disseminated tumor cells (DTCs) and remaining cells were non-aberrant 'normal' cells or 'aberrant cells of unknown origin' based on CNAs. Further, the prevalence of the aberrant cells of unknown origin was age-dependent and one subset of them was hematopoietic in origin, in addition, data also revealed that the DTCs were originated from the main tumor clone, primary tumor subpopulation, or subpopulation in lymph node metastasis. Unraveling the origin of DTCs facilitated our understanding for metastasis of breast cancer and further prevention in future. Most recently, single cell CNAs analysis from FFPE samples also have been reported in synchronously diagnosed ductal carcinoma in situ (DCIS) and invasive breast cancer [80], which showed similar CNAs profiles to those of frozen tissue and concordant with CNAs profiles of bulk tissue. They identified six different but highly related subclones, implying that either invasion was unrelated to the CNAs or invade occurred in early stage of disease followed by genome instability and that multiple diverse DCIS subclones developed in parallel then progressed to invasive disease in one case. Mover, they revealed two major subpopulations in another case, suggesting that intratumor genetic heterogeneity occurred in early stage of disease and progression from DCIS to invasive disease occurred via clonal selection.
SNVs
SNVs calling usually requires high coverage depth (>10X), which is highly cost for WGS due to a 3 Gb human genome. Thus, researchers so far primarily focused on SNVs calling mainly on protein coding region (the exome; 30-60 Mb) using single cell whole exome sequencing (WES). Two reports applied single cell WES research to myeloproliferative neoplasm and kidney tumor [98, 99]. In these studies, they established a routine workflow and criteria for WES and SNVs calling, which are very important for single cell WES. The quantity of 25 of single cells were considered sufficient for calling most of mutations in this myeloproliferative cancer case, and another study also claimed that 20-40 single cells were necessary to detect the major subpopulations with 95% power [98, 135]. Of the routine, they developed a reliable way to verify the called somatic mutations, which use PCR-Sanger sequencing by randomly choosing 30 somatic mutations and examining their status in 52 randomly selected cells. Finally, they identified some essential thrombocythemia related mutant genes, including SESN2 and NTRK1, revealed a monoclonal evolution in JAK2-negative myeloproliferative neoplasm and delineated the intra-tumor genetic heterogeneity, and identified some important gene such as AHNAK in kidney tumor. The first single cell WES research in breast cancer was reported by Yong Wang, et al. in 2014 [100]. In this study, a new approach was developed for verifying the called somatic mutations, which is single-molecule targeted deep sequencing (more than 110,000X) in the bulk tissue. They firstly sequenced 4 single tumor nuclei of ERBC from G2/M phase at high coverage breadth (80.79±3.31%) and depth (46.75X±5.06) using WGS, and found 12 clonal non-synonymous mutations (also present in bulk tissue sequencing) and 32 subclonal non-synonymous mutations. In addition, they sequenced 59 nuclei of ERBC from G2/M phase (47 tumor cells and 12 normal cells) with 92.77% coverage breadth and 46.78X coverage depth using WES, identifying 17 clonal mutations, 19 new subclonal mutations, and 26 de novo mutations that were present in only one tumor cell, such as MARCH11, CABP2. On the other hand, they sequenced 16 single tumor nuclei of TNBC from the G2/M phase and 16 single normal nuclei and identified 374 clonal non-synonymous mutations present in bulk tissue, 145 subclonal non-synonymous mutations, and 152 de novo mutations, including AURKA, SYNE2, TGFB2, etc. This data suggested that the point mutations evolved gradually, leading to extensively clonal diversity, and that the TNBC had more mutation rate (13.3), whereas the ERBC did not. This work identified some mutant genes, including some rare novel mutations that might be involved in breast cancer. Meanwhile it also raised questions, such as what roles these mutations play in breast cancer, which genes are real drivers, and which genes are passengers? It could be expected that more single cell WES on breast cancer will be reported in the coming years, which will accelerate our understanding of origin, progression and metastasis of breast cancer, facilitating prevention and therapy of this disease.
Conclusion and Future Aspects
Heterogeneity in genetics and pathologies of breast cancer casts difficulties in cancer treatment and patient care. Recently developed SCS technology makes it possible for providing a better understanding about heterogeneity of breast cancer. Although this technology develops rapidly with increasing efficiency and accuracy, some problems remain in the whole procedure of single cell preparation, whole genome amplification, library construction, sequencing or data analysis, such as low coverage, bias, errors, depending on different work platform or method. More efficient approaches for capturing single cell, a better method for WGA, a better platform for sequencing, some better tools or algorithms for data analysis still need to be developed in the future.
Using single cell sequencing for deciphering breast cancer heterogeneity is still in the early stages for providing a comprehensive understanding of cancer initiation and progression, although enormous efforts have been delivered to sequence cancer cell lines, primary and metastatic cancers [76, 100, 135-137]. Moreover, the present studies only showed how the tumor evolved by CNAs and how many genes got mutated in some ER and TNBC cases, yet many basic biological questions are still unknown. This includes 1) which genes are the real driver for cancer initiation, and progression; 2) could such drivers change their roles during the process of tumorigenesis; 3) what are the differences among different CSCs within a single cancer; and 4) which cell or group of cells is the origin for breast tumor. The further development of SCS technology should keep up with demand for the precise answers. The mutation spectrum will be more intact and detailed along the whole genome as well as SVs by sequencing more cells using WGS if the cost can be reduced in the future, especially the more mutations in intron region, which have important roles in breast cancer [138].
Currently, the single-cell genomic approaches are limited to freshly isolated or recently frozen cancer samples due to the chemical alterations caused by formalin fixation. This technical barrier is overcome recently. Martelotte et al. (2017) developed a powerful methodology for profiling whole-genome copy-number of single nuclei isolated from FFPE breast cancer samples [80]. We can foresee that future efforts may also be emphasized on the application of SCS technology to a broad spectrum in breast cancer research, such as cancer metastasis, recurrence, drug resistance, and phylogenetic, etc., which will lead better understandings of breast cancer origin, therapeutic treatment, and patient care.
Acknowledgements
We thank members of the Xu laboratory for critical reading and discussion. This work is supported by the Startup Research Grant (SRG2015-00042-FHS)) of University of Macau, FDCT grant (027/2015/A1) and MYRG grant (MYRG2016-00138-FHS).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Weigelt B, Peterse JL, van 't Veer LJ. Breast cancer metastasis: markers and models. Nature reviews Cancer. 2005;5:591-602
2. Cheng X, Chen H. Tumor heterogeneity and resistance to EGFR-targeted therapy in advanced nonsmall cell lung cancer: challenges and perspectives. OncoTargets and therapy. 2014;7:1689-704
3. Dick JE. Stem cell concepts renew cancer research. Blood. 2008;112:4793-807
4. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105-11
5. Shackleton M, Quintana E, Fearon ER, Morrison SJ. Heterogeneity in cancer: cancer stem cells versus clonal evolution. Cell. 2009;138:822-9
6. Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313-9
7. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756-60
8. Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780-3
9. Oravecz-Wilson KI, Philips ST, Yilmaz OH, Ames HM, Li L, Crawford BD. et al. Persistence of leukemia-initiating cells in a conditional knockin model of an imatinib-responsive myeloproliferative disorder. Cancer Cell. 2009;16:137-48
10. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY. et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704-15
11. Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G. et al. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin Cancer Res. 2006;12:5615-21
12. Albini A, Bruno A, Gallo C, Pajardi G, Noonan DM, Dallaglio K. Cancer stem cells and the tumor microenvironment: interplay in tumor heterogeneity. Connective tissue research. 2015;56:414-25
13. Stingl J, Caldas C. Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nature reviews Cancer. 2007;7:791-9
14. Sleijfer S, Bogaerts J, Siu LL. Designing transformative clinical trials in the cancer genome era. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:1834-41
15. Tavassoli FA, Devilee P. Pathology and genetics of tumours of the breast and female genital organs: Iarc; 2003.
16. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA. et al. Molecular portraits of human breast tumours. Nature. 2000;406:747-52
17. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H. et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10869-74
18. Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A. et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8418-23
19. Sotiriou C, Neo SY, McShane LM, Korn EL, Long PM, Jazaeri A. et al. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10393-8
20. Jones C, Mackay A, Grigoriadis A, Cossu A, Reis-Filho JS, Fulford L. et al. Expression profiling of purified normal human luminal and myoepithelial breast cells: identification of novel prognostic markers for breast cancer. Cancer research. 2004;64:3037-45
21. van Amerongen R, Bowman AN, Nusse R. Developmental stage and time dictate the fate of Wnt/beta-catenin-responsive stem cells in the mammary gland. Cell stem cell. 2012;11:387-400
22. Rios AC, Fu NY, Lindeman GJ, Visvader JE. In situ identification of bipotent stem cells in the mammary gland. Nature. 2014;506:322-7
23. Rodilla V, Dasti A, Huyghe M, Lafkas D, Laurent C, Reyal F. et al. Luminal progenitors restrict their lineage potential during mammary gland development. PLoS biology. 2015;13:e1002069
24. Polyak K. Breast cancer: origins and evolution. J Clin Invest. 2007;117:3155-63
25. Molyneux G, Geyer FC, Magnay FA, McCarthy A, Kendrick H, Natrajan R. et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell. 2010;7:403-17
26. Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO. et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci U S A. 2011;108:7950-5
27. Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M. et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell. 2011;8:149-63
28. Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH. et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med. 2009;15:907-13
29. Wang D, Cai C, Dong X, Yu QC, Zhang XO, Yang L. et al. Identification of multipotent mammary stem cells by protein C receptor expression. Nature. 2015;517:81-4
30. Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochim Biophys Acta. 2010;1805:105-17
31. Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem. 2013;59:168-79
32. Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6:924-35
33. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983-8
34. Ikeda J, Morii E, Liu Y, Qiu Y, Nakamichi N, Jokoji R. et al. Prognostic significance of CD55 expression in breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008;14:4780-6
35. Vaillant F, Asselin-Labat ML, Shackleton M, Forrest NC, Lindeman GJ, Visvader JE. The mammary progenitor marker CD61/beta3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer research. 2008;68:7711-7
36. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M. et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell stem cell. 2007;1:555-67
37. Yu CC, Lo WL, Chen YW, Huang PI, Hsu HS, Tseng LM. et al. Bmi-1 Regulates Snail Expression and Promotes Metastasis Ability in Head and Neck Squamous Cancer-Derived ALDH1 Positive Cells. Journal of oncology. 2011:2011
38. Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N. et al. MOZ-TIF2, but not BCR-ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer cell. 2004;6:587-96
39. Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML, Weissman IL. Similar MLL-associated leukemias arising from self-renewing stem cells and short-lived myeloid progenitors. Genes & development. 2003;17:3029-35
40. Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PloS one. 2008;3:e2888
41. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T. et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396-401
42. Dalerba P, Dylla SJ, Park IK, Liu R, Wang X, Cho RW. et al. Phenotypic characterization of human colorectal cancer stem cells. Proc Natl Acad Sci U S A. 2007;104:10158-63
43. O'Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106-10
44. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V. et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030-7
45. Zhang S, Balch C, Chan MW, Lai HC, Matei D, Schilder JM. et al. Identification and characterization of ovarian cancer-initiating cells from primary human tumors. Cancer Res. 2008;68:4311-20
46. Stewart JM, Shaw PA, Gedye C, Bernardini MQ, Neel BG, Ailles LE. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. Proc Natl Acad Sci U S A. 2011;108:6468-73
47. Curley MD, Therrien VA, Cummings CL, Sergent PA, Koulouris CR, Friel AM. et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells. 2009;27:2875-83
48. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328-37
49. Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM. et al. A hierarchy of self-renewing tumor-initiating cell types in glioblastoma. Cancer Cell. 2010;17:362-75
50. Joo KM, Kim SY, Jin X, Song SY, Kong DS, Lee JI. et al. Clinical and biological implications of CD133-positive and CD133-negative cells in glioblastomas. Lab Invest. 2008;88:808-15
51. Wang J, Sakariassen PO, Tsinkalovsky O, Immervoll H, Boe SO, Svendsen A. et al. CD133 negative glioma cells form tumors in nude rats and give rise to CD133 positive cells. Int J Cancer. 2008;122:761-8
52. Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ. et al. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010-5
53. Taussig DC, Vargaftig J, Miraki-Moud F, Griessinger E, Sharrock K, Luke T. et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34(-) fraction. Blood. 2010;115:1976-84
54. Curtis SJ, Sinkevicius KW, Li D, Lau AN, Roach RR, Zamponi R. et al. Primary tumor genotype is an important determinant in identification of lung cancer propagating cells. Cell Stem Cell. 2010;7:127-33
55. Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature. 2012;488:527-30
56. Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M. et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337:730-5
57. Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK. et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 2013;152:25-38
58. Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M. et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature. 2007;449:1003-7
59. Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347:78-81
60. Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017;355:1330-4
61. Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N. et al. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260-4
62. Marusyk A, Polyak K. Tumor heterogeneity: causes and consequences. Biochimica et Biophysica Acta (BBA)-Reviews on Cancer. 2010;1805:105-17
63. Greaves M, Maley CC. Clonal evolution in cancer. Nature. 2012;481:306-13
64. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
65. Shah SP, Morin RD, Khattra J, Prentice L, Pugh T, Burleigh A. et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809-13
66. Dominguez PM, Teater M, Shaknovich R. The new frontier of epigenetic heterogeneity in B-cell neoplasms. Curr Opin Hematol. 2017 (Epub ahead of print)
67. Beca F, Polyak K. Intratumor Heterogeneity in Breast Cancer. Adv Exp Med Biol. 2016;882:169-89
68. Wang Y, Navin NE. Advances and applications of single-cell sequencing technologies. Mol Cell. 2015;58:598-609
69. Ham RG. Clonal Growth of Mammalian Cells in a Chemically Defined, Synthetic Medium. Proc Natl Acad Sci U S A. 1965;53:288-93
70. Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012;338:1622-6
71. Gole J, Gore A, Richards A, Chiu YJ, Fung HL, Bushman D. et al. Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells. Nat Biotechnol. 2013;31:1126-32
72. Landry ZC, Giovanonni SJ, Quake SR, Blainey PC. Optofluidic cell selection from complex microbial communities for single-genome analysis. Methods Enzymol. 2013;531:61-90
73. Leung K, Zahn H, Leaver T, Konwar KM, Hanson NW, Page AP. et al. A programmable droplet-based microfluidic device applied to multiparameter analysis of single microbes and microbial communities. Proc Natl Acad Sci U S A. 2012;109:7665-70
74. Gawad C, Koh W, Quake SR. Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics. Proc Natl Acad Sci U S A. 2014;111:17947-52
75. Pollen AA, Nowakowski TJ, Shuga J, Wang X, Leyrat AA, Lui JH. et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol. 2014;32:1053-8
76. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J. et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90-4
77. Yoshimoto N, Kida A, Jie X, Kurokawa M, Iijima M, Niimi T. et al. An automated system for high-throughput single cell-based breeding. Sci Rep. 2013;3:1191
78. Kovac JR, Voldman J. Intuitive, image-based cell sorting using optofluidic cell sorting. Anal Chem. 2007;79:9321-30
79. Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M. et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202-14
80. Martelotto LG, Baslan T, Kendall J, Geyer FC, Burke KA, Spraggon L. et al. Whole-genome single-cell copy number profiling from formalin-fixed paraffin-embedded samples. Nat Med. 2017;23:376-85
81. Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR. et al. Laser capture microdissection. Science. 1996;274:998-1001
82. Bonner RF, Emmert-Buck M, Cole K, Pohida T, Chuaqui R, Goldstein S. et al. Laser capture microdissection: molecular analysis of tissue. Science. 1997;278:1481 3
83. Kolble K. The LEICA microdissection system: design and applications. J Mol Med (Berl). 2000;78:B24-5
84. Micke P, Ostman A, Lundeberg J, Ponten F. Laser-assisted cell microdissection using the PALM system. Methods Mol Biol. 2005;293:151-66
85. Schutze K, Becker I, Becker KF, Thalhammer S, Stark R, Heckl WM. et al. Cut out or poke in-the key to the world of single genes: laser micromanipulation as a valuable tool on the look-out for the origin of disease. Genet Anal. 1997;14:1-8
86. Espina V, Wulfkuhle JD, Calvert VS, VanMeter A, Zhou W, Coukos G. et al. Laser-capture microdissection. Nat Protoc. 2006;1:586-603
87. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC. et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351:781-91
88. Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L. et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450:1235-9
89. Yu M, Stott S, Toner M, Maheswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J Cell Biol. 2011;192:373-82
90. Riethdorf S, Fritsche H, Muller V, Rau T, Schindlbeck C, Rack B. et al. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: a validation study of the CellSearch system. Clin Cancer Res. 2007;13:920-8
91. Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet. 2016;17:175-88
92. Telenius H, Carter NP, Bebb CE, Nordenskjold M, Ponder BA, Tunnacliffe A. Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics. 1992;13:718-25
93. Dean FB, Nelson JR, Giesler TL, Lasken RS. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001;11:1095-9
94. Zhang DY, Brandwein M, Hsuih T, Li HB. Ramification amplification: a novel isothermal DNA amplification method. Mol Diagn. 2001;6:141-50
95. Langmore JP. Rubicon Genomics, Inc. Pharmacogenomics. 2002;3:557-60
96. Baslan T, Kendall J, Rodgers L, Cox H, Riggs M, Stepansky A. et al. Genome-wide copy number analysis of single cells. Nat Protoc. 2012;7:1024-41
97. Dean FB, Hosono S, Fang L, Wu X, Faruqi AF, Bray-Ward P. et al. Comprehensive human genome amplification using multiple displacement amplification. Proc Natl Acad Sci U S A. 2002;99:5261-6
98. Hou Y, Song L, Zhu P, Zhang B, Tao Y, Xu X. et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell. 2012;148:873-85
99. Xu X, Hou Y, Yin X, Bao L, Tang A, Song L. et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012;148:886-95
100. Wang Y, Waters J, Leung ML, Unruh A, Roh W, Shi X. et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155-60
101. Hou Y, Wu K, Shi X, Li F, Song L, Wu H. et al. Comparison of variations detection between whole-genome amplification methods used in single-cell resequencing. Gigascience. 2015;4:37
102. Huang L, Ma F, Chapman A, Lu S, Xie XS. Single-Cell Whole-Genome Amplification and Sequencing: Methodology and Applications. Annu Rev Genomics Hum Genet. 2015;16:79-102
103. Nishikawa Y, Hosokawa M, Maruyama T, Yamagishi K, Mori T, Takeyama H. Monodisperse Picoliter Droplets for Low-Bias and Contamination-Free Reactions in Single-Cell Whole Genome Amplification. PLoS One. 2015;10:e0138733
104. Fu Y, Li C, Lu S, Zhou W, Tang F, Xie XS. et al. Uniform and accurate single-cell sequencing based on emulsion whole-genome amplification. Proc Natl Acad Sci U S A. 2015;112:11923-8
105. Zahn H, Steif A, Laks E, Eirew P, VanInsberghe M, Shah SP. et al. Scalable whole-genome single-cell library preparation without preamplification. Nat Methods. 2017;14:167-73
106. Chen C, Xing D, Tan L, Li H, Zhou G, Huang L. et al. Single-cell whole-genome analyses by Linear Amplification via Transposon Insertion (LIANTI). Science. 2017;356:189-94
107. Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376-80
108. Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, Davey M. et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475:348-52
109. Merriman B, Ion Torrent R, Team D, Rothberg JM. Progress in ion torrent semiconductor chip based sequencing. Electrophoresis. 2012;33:3397-417
110. Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG. et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53-9
111. Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG. et al. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78-81
112. Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G. et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133-8
113. Laehnemann D, Borkhardt A, McHardy AC. Denoising DNA deep sequencing data-high-throughput sequencing errors and their correction. Brief Bioinform. 2016;17:154-79
114. Ross MG, Russ C, Costello M, Hollinger A, Lennon NJ, Hegarty R. et al. Characterizing and measuring bias in sequence data. Genome Biol. 2013;14:R51
115. Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR. et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341
116. Minoche AE, Dohm JC, Himmelbauer H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biol. 2011;12:R112
117. Nakamura K, Oshima T, Morimoto T, Ikeda S, Yoshikawa H, Shiwa Y. et al. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 2011;39:e90
118. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754-60
119. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25
120. Gu S, Fang L, Xu X. Using SOAPaligner for Short Reads Alignment. Curr Protoc Bioinformatics. 2013;44:11 1-7
121. Zhang J, Wheeler DA, Yakub I, Wei S, Sood R, Rowe W. et al. SNPdetector: a software tool for sensitive and accurate SNP detection. PLoS Comput Biol. 2005;1:e53
122. Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K. et al. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19:1124-32
123. Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER. et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283-5
124. Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L. et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568-76
125. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297-303
126. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491-8
127. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11 0 1-33
128. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013;31:213-9
129. Zafar H, Wang Y, Nakhleh L, Navin N, Chen K. Monovar: single-nucleotide variant detection in single cells. Nat Methods. 2016;13:505-7
130. Zhang C, Zhang C, Chen S, Yin X, Pan X, Lin G. et al. A single cell level based method for copy number variation analysis by low coverage massively parallel sequencing. PLoS One. 2013;8:e54236
131. Garvin T, Aboukhalil R, Kendall J, Baslan T, Atwal GS, Hicks J. et al. Interactive analysis and assessment of single-cell copy-number variations. Nat Methods. 2015;12:1058-60
132. Ross EM, Markowetz F. OncoNEM: inferring tumor evolution from single-cell sequencing data. Genome Biol. 2016;17:69
133. Roth A, McPherson A, Laks E, Biele J, Yap D, Wan A. et al. Clonal genotype and population structure inference from single-cell tumor sequencing. Nat Methods. 2016;13:573-6
134. Jahn K, Kuipers J, Beerenwinkel N. Tree inference for single-cell data. Genome Biol. 2016;17:86
135. Gao R, Davis A, McDonald TO, Sei E, Shi X, Wang Y. et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet. 2016;48:1119-30
136. Demeulemeester J, Kumar P, Moller EK, Nord S, Wedge DC, Peterson A. et al. Tracing the origin of disseminated tumor cells in breast cancer using single-cell sequencing. Genome Biol. 2016;17:250
137. Eirew P, Steif A, Khattra J, Ha G, Yap D, Farahani H. et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature. 2015;518:422-6
138. Bailey SD, Desai K, Kron KJ, Mazrooei P, Sinnott-Armstrong NA, Treloar AE. et al. Noncoding somatic and inherited single-nucleotide variants converge to promote ESR1 expression in breast cancer. Nat Genet. 2016;48:1260-6
Author contact
![]() Corresponding author: Xiaoling Xu, Tel: +853 8822 4925 Fax: +853 8822 2314 E-mail: xiaolingxmo
Corresponding author: Xiaoling Xu, Tel: +853 8822 4925 Fax: +853 8822 2314 E-mail: xiaolingxmo