Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(8):1029-1037. doi:10.7150/ijbs.19309 This issue Cite
Research Paper
FGFR3 deficient mice have accelerated fracture repair
Yangli Xie*, Fengtao Luo*, Wei Xu, Zuqiang Wang, Xianding Sun, Meng Xu, Junlan Huang, Dali Zhang, Qiaoyan Tan, Bo Chen, Wanling Jiang, Xiaolan Du ![]() , Lin Chen
, Lin Chen ![]()
Department of Rehabilitation Medicine, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China.
* Yangli Xie and Fengtao Luo contributed equally to this work.
Received 2017-1-23; Accepted 2017-3-29; Published 2017-7-18
Abstract

Bone fracture healing is processed through multiple biological stages that partly recapitulates the skeletal development process. FGFR3 is a negative regulator of chondrogenesis during embryonic stage and plays an important role in both chondrogenesis and osteogenesis. We have investigated the role of FGFR3 in fracture healing using unstabilized fracture model and found that gain-of-function mutation of FGFR3 inhibits the initiation of chondrogenesis during cartilage callus formation. Here, we created closed, stabilized proximal tibia fractures with an intramedullary pin in Fgfr3-/-mice and their littermate wild-type mice. Fracture healing was evaluated by radiography, micro-CT, histology, and real-time polymerase chain reaction (RT-PCR) analysis. The fractured Fgfr3-/- mice had increased formation of cartilaginous callus, more fracture callus, and more rapid endochondral ossification in fracture sites with up-regulated expressions of chondrogenesis related gene. The fractures of Fgfr3-/- mice healed faster with accelerated fracture callus mineralization and up-regulated expression of osteoblastogenic genes. The healing of fractures in Fgfr3-/- mice was accelerated in the stage of formation of cartilage and endochondral ossification. Downregulation of FGFR3 activity can be considered as a potential bio-therapeutic strategy for fracture treatment.
Keywords: FGFR3, fracture treatment
Introduction
Bone fracture are very common, exerting a marked impact on the quality of life and economy burden [1]. Although most fractures will be healed finally, there are still some of them experience delayed union or nonunion that is currently difficult to manage clinically. Understanding the mechanism of fracture healing will help clinicians to prevent the delayed union/nonunion, and promote the healing of fracture healing.
Fracture healing is a complex process involving a cascade of cellular events that include the initial bleeding and inflammation, recruitment and proliferation of mesenchymal cells, subsequent formation of cartilaginous callus and its gradual replacement by bony callus [2, 3]. Determined by their similar cellular processes, the molecular mechanisms underlying skeletal development and fracture healing are also comparable. It is well known that a variety of growth factors/cytokines regulating skeletal development and homeostasis can also regulate the fracture healing [4-6].
FGFs/FGFR3 signaling has been shown to play important roles in skeletal development and homeostasis. Expression of FGFR3 is observed in chondrocytes during early limb development and later in reserve and proliferating chondrocytes of the epiphyseal growth plates [7, 8]. Gain-of-function mutations in FGFR3 cause several types of the human skeletal dysplasias [9]. In contrast, loss-of-function mutation in FGFR3 leads to camptodactyly, tall stature, and hearing loss syndrome [10, 11]. Further studies found that FGFR3 negatively regulates chondrogenesis of long bone by inhibiting the proliferation and differentiation of chondrocytes, and negatively regulates autophagy and homeostasis of extracellular matrix (ECM) [12-15]. FGFR3 is also an important regulator of osteogenesis. FGFR3 has been found expressed in mature osteoblasts, osteocytes, and osteoclasts [16]. Both loss-of-function and gain-of-function mutations of FGFR3 result in osteopenia, suggesting the complex mechanisms underlying the role of FGFR3 in osteogenesis. Fgfr3 deficiency in osteoclast lineage cells leads to impaired bone resorption activity of osteoclasts [17]. Chondrocyte FGFR3 is involved in the regulation of bone formation and bone remodeling by a paracrine mechanism [18]. Above all, all these studies demonstrate that FGFR3 is an important regulator of multiple types of skeleton related cells including mesenchyme cells, chondrocytes, osteoblasts and osteoclasts, suggesting the important role of FGFR3 in the complex fracture healing processes.
Consistently, FGFR3 is strongly expressed in prehypertrophic chondrocytes, osteoblasts and periosteum during fracture repair [20, 21], indicating that FGFR3 may regulate the cartilaginous callus formation and replacement by bone. We have investigated the role of FGFR3 in fracture healing using unstabilized fracture model of mice carrying gain-of-function mutation of FGFR3 [22], and found that FGFR3 inhibits the initiation of chondrogenesis and hypertrophic differentiation during cartilage formation in the soft callus. Since rigid fixation is the major currently used fixing method in orthopaedic surgeries including fracture fixation and limb lengthening that is commonly used in achondroplasia patients [23]. So it is very important to investigate the role of FGFR3 in fracture healing using stabilized fracture model. Furthermore, application of stable fracture model, in which fracture is mainly healed through intramembranous ossification, will help us to further explore the role of FGFR3 in osteogenesis. In our previous study, mice with gain-of-function mutation of FGFR3 were used, the results from which may not accurately reflect the real role of endogenous FGFR3 in fracture healing of majority of patients that have no FGFR3 mutations.
In this study, we used FGFR3 deficient mice with stabilized fracture model. We observed that Fgfr3-/- mice exhibit more rapid fracture healing with accelerated fracture callus formation, mineralization and bone remodeling. Our results will deepen our understanding about the role of FGFR3 in fracture healing, subsequently provide an experiment basis for developing FGFR3-based biological therapies for bone fracture.
Materials and Methods
Mice
Fgfr3 knock out (Fgfr3-/-) mice were maintained on C3H/HeJ background and the genotyping was conducted as described previously [24]. All experiments were performed in accordance with protocols approved by the Institutional Animals Care and Use Committee of Daping Hospital (Chongqing, China).
Tibial Shaft Fracture Surgery
8 week old male mice were underwent a standardized, unilateral tibial shaft fracture, adapted from that described by Bonnarens [25], with intramedullary fixation. Briefly, a 5-mm vertical incision in the skin was made to expose the proximal 1/2 of tibia, then a 30-gauge needle was inserted vertically through the tibial plateau to the intramedullary canal. After blunt dissection the soft tissue, gradual pressure on the tibia was applied until a fracture was induced by microscissors, then skin was sutured. Buprenorphine was used for control of post-operative pain. Animals were monitored for general postsurgical health and function of the fractured limb.
Histology and analysis
A CO2 overdose was used for the euthanasia for all mice. The fractured tibias were dissected and collected on post fracture days (PFD) 3, 5, 7, 10, 14, 21 and 28 with excess muscle and soft tissue excised. The intramedullary needle was removed carefully from the fractured tibia. Four specimens from each group were fixed in 4% paraformaldehyde. The specimens were decalcified for 21 days in 15% EDTA (pH 7.4), embedded in paraffin, and sectioned at a thickness of 5μm. The sections were stained using Safranin O/Fast Green and Tartrate Resistant Acid Phosphatase (TRAP, Sigma) staining. Total callus area, total cartilage area, and total woven bone area were quantified. Histomorphometric analyses were performed with the OsteoMeasure (OsteoMetrics).
X-ray imaging and Micro-computed tomography
Bone radiographs were obtained using a Faxitron MX20.After X-ray detection, imaging of the architecture of the fracture callus was scanned by SCANCO vivo CT40 (70KV,114μA).
Bone marrow cell culture
Bone marrow cells were derived from 6- to 8-week-old mice as described previously [26]. Briefly, the bone marrows were flushed from the tibia and femora under aseptic conditions using α-MEM (Gibco) containing 10% FBS (Gibco). The second generation of cells was seeded at 1×105 cells/well in 24-multiwell plates. One day after plating, the medium was exchanged for fresh medium supplemented with 50 mg/ml ascorbic acid, 10 mM β-glycerophosphate and 10nM dexamethasone (Sigma). Medium was changed every 3 days. Osteogenic differentiation of bone marrow cells was assessed on day 7 and 14 using ALP activity assay and staining. Mineralization was detected on day 14 and 21 by alizarin red staining.
RNA isolation and Real-time PCR
Total RNA was extracted from the fracture calluses using Trizol reagent (Invitrogen) according to the manufacturer's instruction. The expressions of genes of interest were measured using a Mx3000P PCR machine (Stratagene) and SYBR Premix Ex TaqTM kit (Takara) at least 3 times. Expression levels for each gene of interest were normalized to their corresponding values of endogenous control gene Cyclophilin A. The primers for the genes of interest will be provided upon request.
Biomechanical Testing
The biomechanical properties of the fracture calluses were examined by a three-point bending test according to the protocol described previously [27]. Briefly, a downward bending load was applied to the shaft of the posterior aspect of the fractured tibia (with the loading nose directly over the fracture site (Instron). A load-displacement curve was generated to determine stiffness (N/mm), ultimate force (N), work to failure (mJ) and ultimate displacement (μm).
Statistical analysis
The data were presented as Means ± SD. Statistical significance was ascertained by two-way ANOVA. When significant levels (P<0.05) were achieved, Tukey's Post Hoc test was performed using PASW 18.0. The results were considered significantly different at P<0.05.
Results
Effects of FGFR3 deficiency on bone fracture healing
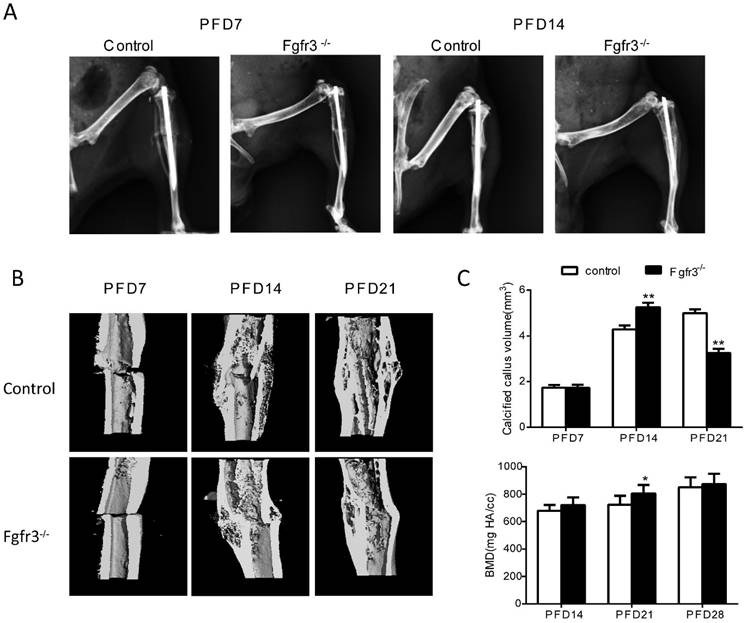
To determine the effects of FGFR3 on the process of fracture healing, we generated closed stabilized tibial fractures in WT and Fgfr3-/- mice and examined the healing process at different stages. There was no callus formation at PFD3 in either genotype (Data not shown). As shown by X-ray radiographs (Fig. 1A), at PFD7 there was radiographic evidence of a bony bridge in Fgfr3-/- mice compared with control mice (Fig. 1A). At PFD14 and PFD21, X-ray (Fig. 1A) and Micro-CT (Fig. 1B) showed that the fracture calluses of tibia from Fgfr3-/- mice were much larger than that observed in tibia from control mice. The mineralized calluses were examined by Micro-CT analysis (Fig. 1B). Consistent with the results from radiographic analysis, at PFD14 tibias were completely bridged by a calcified callus in Fgfr3-/- mice, whereas wild-type mice still exhibited a gap in the fracture site (Fig. 1 B). Longitudinal sections (Fig. 1B) of micro-CT 3-dimensional reconstructions and quantitative analysis (Fig. 1C) demonstrated that the size of callus and calcified callus volume at 14 days post-fracture were increased in the Fgfr3-/- mice and showed accelerated reduction at PFD21. The mineral density was also determined by Micro-CT. The data showed a trend of increased mineral density in Fgfr3-/- mice at PFD14 and 21, but this increase was blunted at PFD28. Our data suggest accelerated formation and remodeling of the fracture callus.
FGFR3 deficiency mice exhibit accelerated fracture healing. Tibia fractures were created in 8-week-old Fgfr3-/- mice and wild-type (WT) controls. Tibias with fractures were harvested at 7, 14, 21 and 28 days after fracture. (A) Representative radiographs of fractured tibias. (B) Longitudinal sections of micro-CT 3-dimensional reconstructed calluses from WT and Fgfr3-/- mice. (C) The quantitative analysis for calcified callus volume and BMD. Data are presented as Means ± SD (N=6-8/group, *p<0.05, **p<0.01).
Effects of FGFR3 deficiency on mechanical properties of fractured tibia
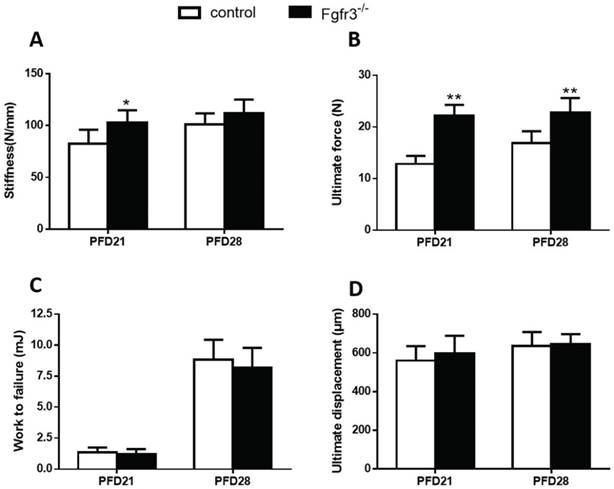
To assess the effects of FGFR3 deficiency on the mechanical properties of fractured tibia, biomechanical properties were examined by three-point bending in tibias at 3 and 4 weeks after fracture wild-type and Fgfr3-/- mice. At PFD 21, the Fgfr3-/- group showed greater stiffness than the control group, but had no significant difference at PFD28 (Fig. 2A). The Fgfr3-/- group showed an increase in the ultimate force, but no difference in work to failure required compared with the control groups at 21 and 28 days (Fig.2B, C). There were no differences between WT and Fgfr3-/- mice in the ultimate displacement throughout the times (Fig. 2D).
Biomechanical properties of the fracture calluses. (A) Stiffness, (B) ultimate force to failure, (C) work to failure, and (D) ultimate displacement results were assessed by three-point bending in tibias from 21 and 28 days post-fracture in Fgfr3-/- mice and wild-type (WT) controls. Data are presented as Means±SD (N=6/group, *p<0.05, **p<0.01).
Fractures in Fgfr3-/-mice undergo accelerated endochondral bone formation
Since FGFR3 is a negative regulator of endochondral bone formation during development, we wonder whether this function also works in the fracture healing. To determine whether Fgfr3 deficiency affect cartilaginous callus formation and its transformation into bony callus, callus tissues from control and Fgfr3-/- mice were analyzed by histology and computer-assisted image analysis. Compared to those of control mice, the total callus areas and cartilaginous callus areas were increased in Fgfr3-/- mice at PFD7 and PFD10. At PFD14, the cartilage tissue in fracture site of Fgfr3-/- mice was dramatically reduced with a relative increase of bonny tissue (Fig. 3A -D). In contrast, cartilage tissue is apparent in the central area of callus in the control mice at that time (Fig. 3A). In addition to the accelerated mineralization observed above, the callus of Fgfr3-/-mice underwent more rapid remodeling. Total callus area peaked on day 10 in Fgfr3-/-mice and progressively decreased with time, while the bony callus showed the similar trend. In contrast, peak callus area is present at PFD10 to 14 in control mice. Control mice had a larger callus than Fgfr3-/- mice from PFD14, which persisted during the observation period.
Fractures in Fgfr3-/-mice undergo accelerated endochondral bone formation. (A) Representative histology of fractured tibias stained with Safranin O/Fast Green show cartilage (red) and bone (green) formation. (B-D) Histomorphometric measurements (total callus area, cartilage area, bony callus area) were made from 20 sections for each group (N=5-6 mice/group). (E) Real-time RT-PCR mRNA expression analyses were performed for evaluating expressions of genes involved in chondrogenesis in callus from WT and Fgfr3-/- mice after fracture and results were expressed as fold changes relative to expression level of WT callus at PFD3. The analysis was repeated for three times. Data are presented as Means±SD ( N=5-6 mice/group, *p<0.05, **p<0.01).
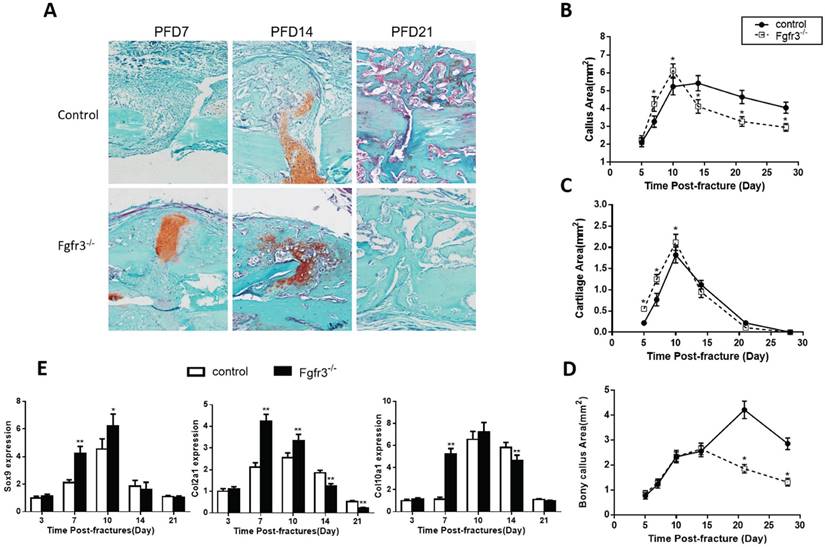
To determine whether the alterations of the callus areas were associated with the changes of the expressions of genes associated with endochondral bone formation, quantitative real-time PCR analysis was performed. Both Sox 9, collagen type II (Col2a1) and collagen type X (Col10a1) expression reached at peak value earlier during fracture repair in the Fgfr3-/- mice, the expression levels of these genes were significantly higher at PFD7 and 10 in the Fgfr3-/- mice (Fig. 3E). In contrast, the expressions of Col2a1 and Col10a1 peaked in wild-type fractures at PFD14. The earlier appearance and disappearance of these chondrogenic genes in the fracture callus are consistent with the accelerated endochondral bone formation in the Fgfr3-/- mice.
Osteoblast differentiation is accelerated in fractures in Fgfr3-/-mice
The bony tissue in callus was present and peaked earlier in the Fgfr3-/-mice. Despite the accelerated chondrogenesis, we checked the expressions of genes involved in osteoblast differentiation in the callus tissue of fractures site in control and FGFR3 deficiency mice. Expression levels of Runx2 were accelerated in fracture calluses of Fgfr3-/-mice (Fig. 4A). Runx2 had the maximal expression on PFD14 in the Fgfr3-/-mice, whereas it peaked at PFD21 in wild-type callus tissue (Fig. 4A). The osteoblast differentiation markers, including collagen type I (Col1a1), alkaline phosphatase (ALP), and osteocalcin (OCN) also had their maximal expression earlier in fracture callus from Fgfr3-/-mice, consistent with the accelerated bony tissue formation in fracture site (Fig.4B-D).
The expression of genes involved in osteogenesis is altered in callus from Fgfr3-/ -mice. Total RNA was extracted from callus in Fgfr3-/- mice and controls (N=5 mice/group) at various time points after fracture. The following primer sets were used: RUNX2 (A), Col1a1 (B), ALP (C), OCN (D), FGFR1 (E), and FGFR2 (F). Results were expressed as fold changes relative to expression level of WT callus at PFD3. The analysis was repeated for three times. Data are presented as Means±SD ( N=5-6 mice/group, *p<0.05, **p<0.01).
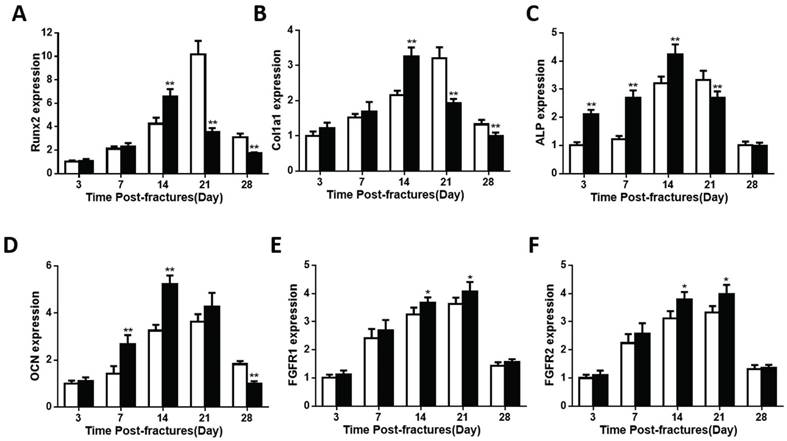
Furthermore, the expression levels of both ALP and OCN were higher in fractures in the Fgfr3-/- mice. Both the mRNA expressions of FGFR1 and FGFR2 showed a little increase at PFD14 and PFD21 (Fig.4E, F). These findings suggest that osteoblasts undergo a more rapid differentiation in Fgfr3-/- mice during fracture healing. To determine whether mesenchymal stromal cells from Fgfr3-/-mice have enhanced osteogenic potential, bone marrow progenitor cells were collected and analyzed. After culture in osteogenic medium for 7 or 14 days, alkaline phosphatase staining revealed more colonies formed with increased alkaline phosphatase in the Fgfr3-/- bone marrow stem cell cultures (Fig.S1). Similarly, alizarin red staining was stronger in the FGFR3 deficiency bone marrow progenitor cell cultures compared with control cultures at D14 (Fig.S1). These data indicate that the accelerated process of bony callus formation in Fgfr3-/- mice may be associated with the accelerated initiation of endochondral ossification and osteogenic differentiation.
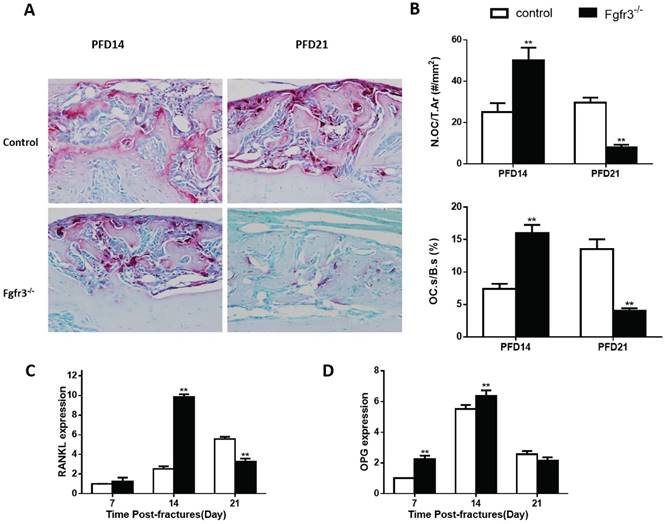
Effects of FGFR3 deficiency on osteoclastic bone resorption in calluses
Since the above radiographic and histologic analysis suggested that he fractures of Fgfr3-/- mice underwent more rapid remodeling (Fig.1), the fracture calluses were further stained by TRAP to determine whether the osteoclastic bone resorption is disturbed during fracture healing. The number of TRAP positive osteoclasts was increased by 100% in PFD14 calluses from Fgfr3-/- mice compared with that in control wild-type mice, and the TRAP positive osteoclast surface was also increased, consistent with accelerated bone remodeling (Fig. 5B). At PFD21, the number of osteoclasts in the callus from control mice was further increased. The expression levels of RANKL and OPG, two critical molecules regulating osteoclastogenesis, were significantly and early up-regulated in the calluses from Fgfr3-/- mice. We further calculated the ratio of RANKL/OPG, a major determinant of osteoclastogenesis in osteoblasts, and found that their ratio was significantly elevated (Fig.5C, D). The peak levels of RANKL in fracture site tissue occurred at PFD14 in Fgfr3-/- mice, and at PFD21 in control mice. These data are consistent with the observed differences in osteoclast numbers by bone histomorphometry.
Fgfr3-/- mice have accelerated bone remodeling in fracture healing. Representative micrographs of sections of calluses from control and Fgfr3-/- mice at 14 and 21days post-fracture stained histochemically for TRAP. (B) Number of TRAP-positive osteoclasts related to tissue area (N.Oc/T.Ar, #/mm2) and osteoclast surface relative to bone surface (Oc.S/BS, %) were assessed by computer-assisted image analysis. RANKL (C) and OPG (D) RNA levels in the fracture callus were examined by RT-PCR. Results were expressed as fold changes relative to expression level of WT callus at PFD7. Data are presented as Means±SD ( N=5-6 mice/group, *p<0.05, **p<0.01).
Discussion
Bone fracture is common in clinic and has fatally consequences on both economy and patient morbidity. Although bone has the ability to regenerate after injuries, some patients still experience serious complications including nonunion or delayed healing. Present therapies, such as surgery, bone grafting, growth factors and stem cell therapies, etc, have limited effect for these complications. Thus, deeper understanding of the underlying mechanisms is urgently needed for developing effective biological measures for accelerating fracture healing and preventing or promoting the healing of nonunion or delayed healing.
Skeleton development and fracture healing are similarly exerted by both chondrogenesis and osteogenesis. Both gain-of-function and loss-of-function mutations in FGFR3 lead to maldeveloped skeleton with abnormal growth plates [28], suggesting the potential important role of FGFR3 in fracture healing. We and other researchers have revealed that FGFR3 not only regulate chondrogenesis, but also affect osteogenesis by directly regulating osteogenic differentiation of bone marrow cells and indirectly by regulating chondrogenesis [18] and osteoclastogenesis [17, 29, 30]. FGFR3 is strongly expressed in prehypertrophic chondrocytes, osteoblasts and periosteum during fracture repair [20, 21]. Our previous data showed that FGFR3 is expressed during whole fracture healing processes by in situ hybridization and RT-PCR. The temporal and spatial expression patterns and roles of FGFR3 strongly indicate that FGFR3 may regulate fracture healing by affecting both chondrogenesis and osteogenesis. Indeed, our present study revealed that Fgfr3 deficient mice exhibited both dysregulated chondrogenesis and osteogenesis during fracture healing.
We found that, although stable fracture model was used, the total callus and cartilaginous callus areas were still increased in Fgfr3-/- mice at the early stage, indicating in Fgfr3-/- mice the chondrogenesis is enhanced in the healing of stabilized fracture. These data are consistent with the well-known negative role of FGFR3 in chondrogenesis during bone development, and also consistent with our previous finding that FGFR3 inhibits chondrogenesis during the healing process of unstabilized fracture of ACH mice. Besides its inhibition on chondrocyte proliferation and hypertrophic differentiation [31-33], FGFR3 also decreases the ECM content, the major component of cartilaginous callus, by reducing the ECM synthesis and promoting its degradation [14, 15]. In agreement with these findings, here we also found an upregulation in Col II levels (reflecting an increase in the ECM content) and a decrease in levels of MMP9 and MMP13 (two key enzymes for degradations of ECM) in our Fgfr3 -/- mice, indicating that FGFR3 plays a similar negative regulation mechanism in both bone development and bone repairing.
FGFR3, a major regulator of chondrogenesis, also modulate osteogenesis. The role of FGFR3, however, in osteogenesis is still controversial, as we have showed that both Fgfr3 -/- mice and mice with gain-of-function mutations of FGFR3 display decreased bone mass [30, 34, 35] In our previous study using unstable fracture model of ACH mice to study the role of FGFR3 in fracture healing, we had not analyzed the osteogenesis in detail. So in this study, we used stabilized fracture model to explore the role of FGFR3 in fracture healing related osteogenesis. We found enhanced osteogenesis as evidenced by accelerated and larger bony callus, indicating that FGFR3 inhibits osteogenesis during fracture healing.
The mechanism for the increased bony callus in FGFR3 deficient mice is not clarified. The increased expression of Runx2, an important transcription factor promoting osteoblastogenesis, in the fracture callus of Fgfr3-/- mice suggests the enhanced osteogenic differentiation during fracture healing, which is further supported by the elevated osteogenic differentiation of Fgfr3-/- bone marrow stromal cells.
Considering the coordination between chondrogenesis and osteogenesis during skeletal development and fracture healing, the enhanced chondrogenesis may also be involved in the augmented bony callus formation in Fgfr3-/- mice. In endochondral ossification during skeleton development and fracture, hypertrophic chondrocytes (cartilage) serve as a scaffold for bone formation as the mineralized cartilaginous templates will finally be replaced by bone following the invasion of blood vessels. The increased size of cartilaginous callus may be partially responsible for the subsequently increased bony callus in fracture site of Fgfr3-/- mice. In addition, recent studies showed that hypertrophic chondrocytes can directly transdifferentiate into osteoblasts during endochondral bone formation [36-38]. In Fgfr3-/-mice, the chondrocyte hypertrophy is enhanced resulting in more hypertrophic chondrocytes, we do not know at this moment whether their transdifferentiation into osteoblasts in involved in the strengthened formation of bony callus. Osteogenesis can also be indirectly affected by chondrogenesis. We recently found that FGFR3 can indirectly influence osteogenesis through its regulation of the secretion of several osteoblastogenesis related molecules by chondrocytes, such as Ihh, Bmp2, Bmp4 [18]. Whether the augmented cartilaginous callus in FGFR3 deficient mice indirectly affects the subsequent bony callus formation through the mechanisms described above need to be studied.
It is important to note the limitations of this study. Fracture healing is an integrated process in which multiple types of cells are involved. FGFR3 is expressed in several cell lineages involved in fracture healing. The unique role of FGFR3 in individual cell types can be dissected by using mice with conditional deletion of FGFR3 in future. For example, cell lineage specific knock out approach may help to dissect the direct and chondrocyte-dependent indirect role of FGFR3 in bony callus formation as discussed above. Osteoclast-specific modulation of FGFR3 will help to clarify the role of FGFR3-modulated osteoclastogenesis in callus formation and remodeling. In addition, the resources of progenitor cells for callus formation are not fully clear presently, which needs to be studied by approaches including linage tracing and in vivo dynamic imaging, etc.
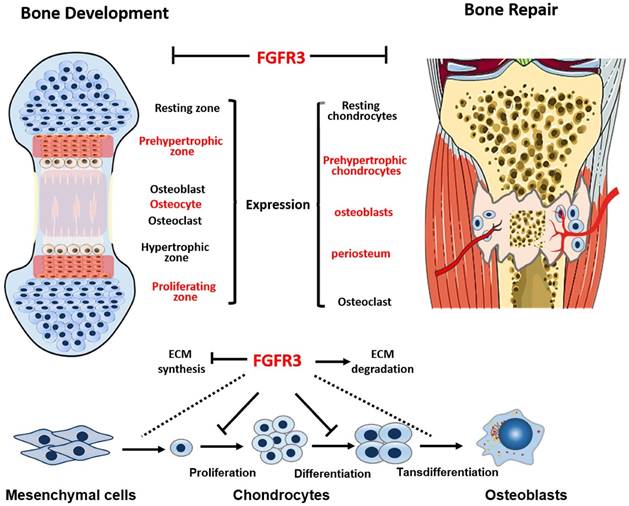
In summary, this study, by using the stabilized fracture model in Fgfr3 deficient mice, we demonstrate that deletion of Fgfr3 significantly accelerates fracture healing by an increase in both chondrogenesis and osteogenesis via a mechanism that enhances ECM productions and decreases degradations (Fig.6). Our studies suggest that FGFR3 plays a negative role in bone repairing. Thus, blocking FGFR3 functions by using a neutralizing antibody against FGFR3 or FGFR3 binding peptide may enhance bone repairing in patients with un-union or delayed fracture healing.
Schematic illustration of the role of FGFR3 in bone repairing. FGFR3 is strongly expressed in prehypertrophic chondrocytes, osteoblasts and periosteum during fracture repair. FGFR3 plays a negative role in bone repairing which is similar with its role in bone development.
Supplementary Material
Supplementary figure.
Acknowledgements
National Natural Science Foundation of China (81220108020, 81530071), Special Funds for Major State Basic Research Program of China (2014CB 942904), Foundation of army (16CXZ016) and Basic and Advanced Research Project in Chongqing (CSTC2013jcyjC0009).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Loi F, Cordova LA, Pajarinen J, Lin TH, Yao Z, Goodman SB. Inflammation, fracture and bone repair. Bone. 2016;86:119-30
2. Shapiro F. Bone development and its relation to fracture repair. The role of mesenchymal osteoblasts and surface osteoblasts. Eur Cell Mater. 2008;15:53-76
3. Hadjiargyrou M, O'Keefe RJ. The convergence of fracture repair and stem cells: interplay of genes, aging, environmental factors and disease. J Bone Miner Res. 2014;29:2307-22
4. Vortkamp A, Pathi S, Peretti GM, Caruso EM, Zaleske DJ, Tabin CJ. Recapitulation of signals regulating embryonic bone formation during postnatal growth and in fracture repair. Mechanisms of development. 1998;71:65-76
5. Chen Y, Alman BA. Wnt pathway, an essential role in bone regeneration. Journal of cellular biochemistry. 2009;106:353-62
6. Deschaseaux F, Sensebe L, Heymann D. Mechanisms of bone repair and regeneration. Trends Mol Med. 2009;15:417-29
7. Szebenyi G, Savage MP, Olwin BB, Fallon JF. Changes in the expression of fibroblast growth factor receptors mark distinct stages of chondrogenesis in vitro and during chick limb skeletal patterning. Dev Dyn. 1995;204:446-56
8. Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nature genetics. 1996;12:390-7
9. Ornitz DM, Marie PJ. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002;16:1446-65
10. Escobar LF, Tucker M, Bamshad M. A second family with CATSHL syndrome: Confirmatory report of another unique FGFR3 syndrome. American journal of medical genetics Part A. 2016;170:1908-11
11. Toydemir RM, Brassington AE, Bayrak-Toydemir P, Krakowiak PA, Jorde LB, Whitby FG. et al. A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome. Am J Hum Genet. 2006;79:935-41
12. Xie Y, Zhou S, Chen H, Du X, Chen L. Recent research on the growth plate: Advances in fibroblast growth factor signaling in growth plate development and disorders. Journal of molecular endocrinology. 2014;53:T11-34
13. Wang X, Qi H, Wang Q, Zhu Y, Wang X, Jin M. et al. FGFR3/fibroblast growth factor receptor 3 inhibits autophagy through decreasing the ATG12-ATG5 conjugate, leading to the delay of cartilage development in achondroplasia. Autophagy. 2015;11:1998-2013
14. Foldynova-Trantirkova S, Wilcox WR, Krejci P. Sixteen years and counting: the current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum Mutat. 2012;33:29-41
15. Krejci P, Masri B, Fontaine V, Mekikian PB, Weis M, Prats H. et al. Interaction of fibroblast growth factor and C-natriuretic peptide signaling in regulation of chondrocyte proliferation and extracellular matrix homeostasis. Journal of cell science. 2005;118:5089-100
16. Xiao L, Naganawa T, Obugunde E, Gronowicz G, Ornitz DM, Coffin JD. et al. Stat1 controls postnatal bone formation by regulating fibroblast growth factor signaling in osteoblasts. J Biol Chem. 2004;279:27743-52
17. Su N, Li X, Tang Y, Yang J, Wen X, Guo J. et al. Deletion of FGFR3 in Osteoclast Lineage Cells Results in Increased Bone Mass in Mice by Inhibiting Osteoclastic Bone Resorption. J Bone Miner Res. 2016;31:1676-87
18. Wen X, Li X, Tang Y, Tang J, Zhou S, Xie Y. et al. Chondrocyte FGFR3 Regulates Bone Mass by Inhibiting Osteogenesis. J Biol Chem. 2016;291:24912-21
19. Marie PJ, Miraoui H, Severe N. FGF/FGFR signaling in bone formation: progress and perspectives. Growth Factors. 2012;30:117-23
20. Nakajima A, Shimizu S, Moriya H, Yamazaki M. Expression of fibroblast growth factor receptor-3 (FGFR3), signal transducer and activator of transcription-1, and cyclin-dependent kinase inhibitor p21 during endochondral ossification: differential role of FGFR3 in skeletal development and fracture repair. Endocrinology. 2003;144:4659-68
21. Rundle CH, Miyakoshi N, Ramirez E, Wergedal JE, Lau KH, Baylink DJ. Expression of the fibroblast growth factor receptor genes in fracture repair. Clinical orthopaedics and related research. 2002:253-63
22. Su N, Yang J, Xie Y, Du X, Lu X, Yin Z. et al. Gain-of-function mutation of FGFR3 results in impaired fracture healing due to inhibition of chondrocyte differentiation. Biochem Biophys Res Commun. 2008;376:454-9
23. Kitoh H, Mishima K, Matsushita M, Nishida Y, Ishiguro N. Early and late fracture following extensive limb lengthening in patients with achondroplasia and hypochondroplasia. Bone Joint J. 2014;96B:1269-73
24. Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911-21
25. Bonnarens F, Einhorn TA. Production of a standard closed fracture in laboratory animal bone. J Orthop Res. 1984;2:97-101
26. Su N, Sun Q, Li C, Lu X, Qi H, Chen S. et al. Gain-of-function mutation in FGFR3 in mice leads to decreased bone mass by affecting both osteoblastogenesis and osteoclastogenesis. Hum Mol Genet. 2010;19:1199-210
27. Weng T, Mao F, Wang Y, Sun Q, Li R, Yang G. et al. Osteoblastic molecular scaffold Gab1 is required for maintaining bone homeostasis. Journal of cell science. 2010;123:682-9
28. Su N, Jin M, Chen L. Role of FGF/FGFR signaling in skeletal development and homeostasis: learning from mouse models. Bone Research. 2014;3:9-32
29. Su N, Sun Q, Li C, Lu X, Qi H, Chen S. et al. Gain-of-function mutation in FGFR3 in mice leads to decreased bone mass by affecting both osteoblastogenesis and osteoclastogenesis. Hum Mol Genet. 2010;19:1199-210
30. Valverde-Franco G, Liu H, Davidson D, Chai S, Valderrama-Carvajal H, Goltzman D. et al. Defective bone mineralization and osteopenia in young adult FGFR3-/- mice. Hum Mol Genet. 2004;13:271-84
31. Ornitz DM. FGF signaling in the developing endochondral skeleton. Cytokine Growth Factor Rev. 2005;16:205-13
32. Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332-6
33. Krejci P. The paradox of FGFR3 signaling in skeletal dysplasia: why chondrocytes growth arrest while other cells over proliferate. Mutation research Reviews in mutation research. 2014;759:40-8
34. Eswarakumar VP, Schlessinger J. Skeletal overgrowth is mediated by deficiency in a specific isoform of fibroblast growth factor receptor 3. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3937-42
35. Mugniery E, Dacquin R, Marty C, Benoist-Lasselin C, de Vernejoul MC, Jurdic P. et al. An activating Fgfr3 mutation affects trabecular bone formation via a paracrine mechanism during growth. Hum Mol Genet. 2012;21:2503-13
36. Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B. Chondrocytes Transdifferentiate into Osteoblasts in Endochondral Bone during Development, Postnatal Growth and Fracture Healing in Mice. PLoS Genet. 2014;10:e1004820
37. Yang L, Tsang KY, Tang HC, Chan D, Cheah KS. Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation. Proc Natl Acad Sci U S A. 2014;111:12097-102
38. Yang G, Zhu L, Hou N, Lan Y, Wu XM, Zhou B. et al. Osteogenic fate of hypertrophic chondrocytes. Cell Res. 2014;24:1266-9
Author contact
![]() Corresponding author: Lin Chen, PhD & MD, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991; Fax: 86-23-68702991; E-mail: linchen70com. Xiaolan Du, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991; Fax: 86-23-68702991; E-mail: DXL_xiaolancom
Corresponding author: Lin Chen, PhD & MD, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991; Fax: 86-23-68702991; E-mail: linchen70com. Xiaolan Du, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing 400042, China. Tel: 86-23-68702991; Fax: 86-23-68702991; E-mail: DXL_xiaolancom