Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Correlation between alcohol...
Evidence of alcohol-mediated...
Future perspectives and...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(9):1100-1108. doi:10.7150/ijbs.21047 This issue Cite
Review
In Utero Alcohol Exposure and the Alteration of Histone Marks in the Developing Fetus: An Epigenetic Phenomenon of Maternal Drinking
Chanchal Mandal1, Debasish Halder1, Kyoung Hwa Jung1, 2, Young Gyu Chai1, 3 ![]()
1. Department of Molecular and Life Science, Hanyang University, Ansan, Republic of Korea;
2. Institute of Natural Science and Technology, Hanyang University, Ansan, Republic of Korea;
3. Department of Bionanotechnology, Hanyang University, Seoul, Republic of Korea.
Received 2017-5-16; Accepted 2017-7-7; Published 2017-9-5
Abstract

Ethanol is well known for its teratogenic effects during fetal development. Maternal alcohol consumption allows the developing fetus to experience the detrimental effects of alcohol exposure. Alcohol-mediated teratogenic effects can vary based on the dosage and the length of exposure. The specific mechanism of action behind this teratogenic effect is still unknown. Previous reports demonstrated that alcohol participates in epigenetic alterations, especially histone modifications during fetal development. Additional research is necessary to understand the correlation between major epigenetic events and alcohol-mediated teratogenesis such as that observed in fetal alcohol spectrum disorder (FASD). Here, we attempted to collect all the available information concerning alcohol-mediated histone modifications during gestational fetal development. We hope that this review will aid researchers to further examine the issues associated with ethanol exposure.
Keywords: Alcohol, Teratogenic effects, Epigenetics, FASD, FAS, Histone modifications.
Introduction
Epigenetics can induce changes in gene expression without altering the DNA sequence, thus affecting cellular ohenotype [1]. Living cells have tightly controlled and pre-programmed mechanisms to regulate epigenetic events. These events can also be altered by various factors, including environmental conditions, such as exposure to toxins, as well as by age, behavior, a specific disease, etc. Typically, these changes are not inherited, but they can be passed on to the next generation if the changes occur in a sperm or egg cell. Epigenetic regulation is thought to be primarily controlled through DNA methylation and histone modification and in part by non-coding RNAs (ncRNAs) [2-4]. In eukaryotic organisms, DNA winds around histone proteins, forming a complex called chromatin. The structural units of chromatin are nucleosomes, which consist of 147 base pairs of DNA wrapped around a octamer core of histone proteins [5]. The octamer core contains two of each of the histone proteins (H2A, H2B, H3, and H4). Two nucleosomes are in turn connected by a linker histone (H1), which contributes to the stability of the structure. Post-translational modifications at the N-terminus of the histone proteins are reffered to as histone modifications. These modifications include acetylation, methylation, phosphorylation, ubiquitinylation, ADP-ribosylation, and sumoylation of histone proteins [6-9]. Histone modifications can influence chromatin structure and the subsequent regulation of gene expression. Based on current knowledge, it is understood that histone acetylation is associated with active gene transcription, whereas histone methylation is associated with the suppression of transcription [10].
The exposure of cells to any toxin can result in harmful effects. The cells that are most vulnerable are those that are in the process of development and differentiation. Thus, the developing fetus is highly affected by alcohol exposure because the whole body is under development during the embryonic stage. It was well established that alcohol intake during pregnancy can result in abnormalities in infants. The types of abnormalities that result from ethanol exposure vary depending on the dose, duration, and frequency of consumption during the gestational period. Maternal genetics and metabolism are also important factors that can influence the developmental process [11]. Fetal alcohol spectrum disorder (FASD) is a term that is commonly used to refer to alcohol-related abnormalities in utero [12, 13]. Fetal alcohol syndrome (FAS) is the most severe form of FASD and includes growth retardation, craniofacial defects, and central nervous system (CNS) deficits [14, 15]. The developing fetus that is in the embryonic stage is known to be more severely affected by alcohol exposure compared to the later fetal stages [16]. Previous studies have suggested genetic susceptibilities to alcohol-mediated abnormalities. Microarray analysis and next-generation sequencing techniques have been applied to identify important developmental genes that are altered by maternal intake of alcohol during gestation [12-15, 17-20]. However, the specific genetic and epigenetic mechanisms behind alcohol-mediated abnormalities remain unknown. A combination of genetic and epigenetic approaches are needed to reveal the molecular mechanisms governing these disorders. In this review, we attempted to collect all the available information concerning exposure to ethanol in utero and the consequences of epigenetic alterations mediated by ethanol. We hope that this review will help researchers to better understand alcohol-directed epigenetic modifications, especially histone modifications, in teratogenicity.
Correlation between alcohol metabolism and histone modifications
Metabolites and metabolic enzymes are involved in epigenetic mechanisms. Thus, the transcriptional regulation of rate-limiting metabolic enzymes is important for controlling metabolic changes in cells. The levels of metabolites (e.g., NAD+, ATP, acetyl-CoA, and S-adenosyl methionine (SAM)) and metabolic hormones (e.g., insulin, leptin) contribute to the regulation of gene expression. The activities of major enzymes involved in epigenetic modification are in part regulated by the concentrations of available substrates and cofactors [21]. Thus, cellular metabolites are involved in the transcriptional regulation of genes as well as in epigenetic mechanisms.
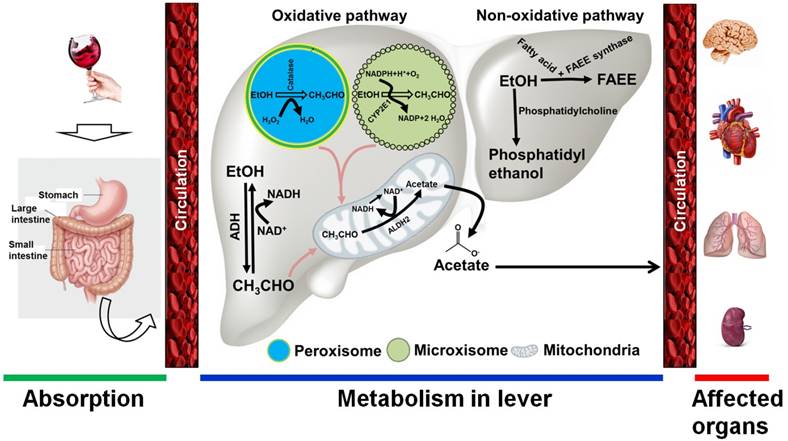
Alcohol is absorbed into the bloodstream by diffusion via small blood vessels in the walls of the stomach and the small intestine. Approximately 20% of ingested alcohol is absorbed through the stomach, while the remaining 80% is absorbed through the small intestine [22]. The small intestine has a large surface area that quickly allows for passage into the blood supply. The absorption of alcohol occurs more rapidly for beverages that have a higher alcohol content. A small proportion of ingested alcohol does not enter the systemic circulation and is oxidized in the stomach by alcohol dehydrogenase (ADH) enzymes, including class I and class III ADH molecules [23]. Following absorption, alcohol is transported to the liver, where it is enzymatically metabolized. The ability of the liver to metabolize alcohol is limited by time, and any excess is left to circulate throughout the body [23]. Thus, the intensity of alcohol's effects on the body is directly related to the amount consumed. Alcohol is also metabolized in tissues that do not contain ADH enzymes, such as in the brain, by the enzymes cytochrome P450 and catalase [24]. In general, alcohol metabolism is achieved through both oxidative and non-oxidative pathways. Alcohol oxidation occurs in the liver, where three major enzymes are responsible for completing the process. These enzymes are alcohol dehydrogenase, cytochrome P450 2E1 (CYP2E1), and catalase. ADHs, CYP2E1, and catalase are found in the cytosol, microsomes, and peroxisomes, respectively [25]. In the cytosol, ethanol is converted to acetaldehyde by the enzymatic action of ADH in the first step of the oxidative pathway. Nicotinamide adenine dinucleotide (NAD) accepts hydrogen atoms and electrons from alcohol to produce acetaldehyde. When alcohol intake is high, CYP2E1 is recruited to metabolize alcohol to acetaldehyde in the endoplasmic reticulum. Another minor pathway of alcohol oxidation is mediated by catalase in peroxisomes [26]. This pathway is capable of oxidizing ethanol in the presence of an H2O2-generating system. In the final step of this oxidative pathway, acetaldehyde is converted to acetate by mitochondrial acetaldehyde dehydrogenase. Much of the acetate produced through the oxidation of acetaldehyde leaves the liver and circulates to peripheral tissues [23, 25-27] (Figure 1). The non-oxidative pathway of ethanol metabolism involves the esterification of ethanol. The major enzyme involved in this pathway is fatty acid ethyl esters (FAEE) synthase, which forms esters from ethanol. Another product of the non-oxidative pathway is phosphatidylethanol, which is catalyzed by phosphatidylcholine-specific phospholipase D [23, 25, 27] (Figure 1).
Alcohol metabolism in the body. Following alcohol consumption, most is absorbed by the small intestine and the stomach. Through circulation, alcohol quickly reaches the liver, where it is broken down. Two major pathways are involved in ethanol metabolism: 1) the oxidative pathway, and 2) the non-oxidative pathway. 1) In the oxidative pathway, alcohol is metabolized to acetaldehyde by cytosolic alcohol dehydrogenase (ADH). If a large amount of ethanol is present, cytochrome P450 IIE1 (CYP2E1) in the endoplasmic reticulum may also be involved in alcohol metabolism. A minor oxidative reaction also occurs in the peroxisomes, where the catalase enzyme is responsible for alcohol metabolism. Consequently, the intermediate metabolite acetaldehyde is metabolized by aldehyde dehydrogenase 2 (ALDH2) to form acetate in the mitochondria. 2) Two enzymes are involved in the non-oxidative pathway. In the presence of fatty acids, ethanol is metabolized to fatty acid ethyl ester (FAEE) by FAEE synthase. A phosphatidylcholine-specific enzyme named phospholipase D is involved in producing phosphatidylethanol from ethanol. The products of both the oxidative and non-oxidative pathways are quickly released into circulation, through which ethanol metabolites affect the peripheral organs. EtOH, ethanol; CH3CHO, acetaldehyde; H2O2, hydrogen peroxide; NAD/NADH, nicotinamide adenine dinucleotide; ROS, reactive oxygen species.
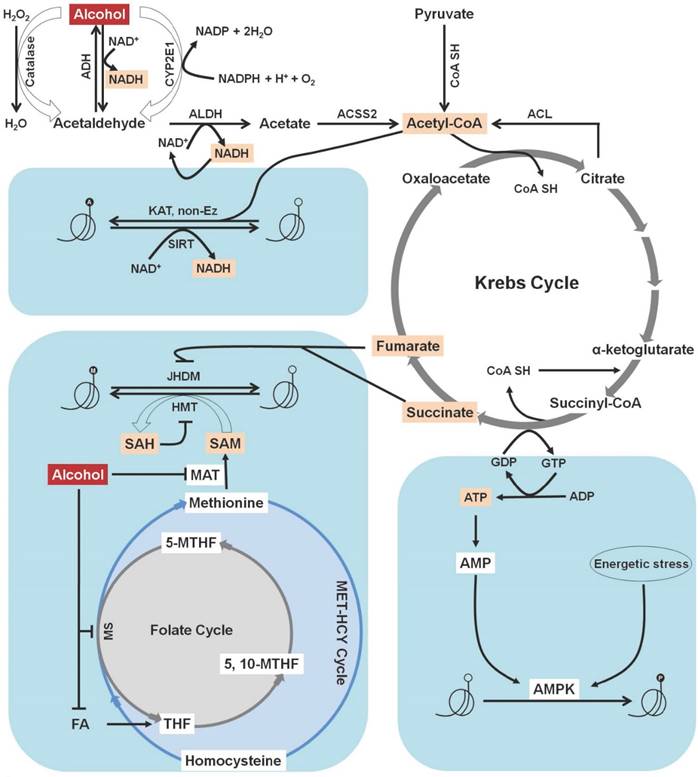
Increased levels of acetate quickly escape the liver and reach the blood stream and other parts of the body. In mitochondria, acetate is transformed to acetyl-CoA by the enzyme acyl-CoA synthetase short-chain family member 2 (ACSS2) [28]. There are two primary acetate-activating enzymes: acyl-CoA synthetase short-chain family members 1 and 2 (ACSS1 and ACSS2), which are regulated by sirtuins. NAD+-dependent HDACs are known as sirtuins (SIRTs 1-7) and are located in the nucleus, cytoplasm, and mitochondria [29]. SIRT3 is a deacetylase that activates long-chain acyl-CoA dehydrogenase within the mitochondrial matrix [30]. Acetylation inactivates ACSS1 and ACSS2, whereas deacetylation activates them. SIRT1 and SIRT3 deacetylate ACSS2 and ACSS1, respectively. Sirtuin-mediated deacetylation, in conjunction with ACSS enzyme-mediated acetyl-CoA synthesis, elevates the conversion rate of acetate to acetyl-CoA in all cellular compartments. Thus, any differences in cellular acetate levels may hamper total protein acetylation. The resulting acetyl CoA then enters the Krebs cycle and combines with oxaloacetate to form citrate. Citrate is either oxidized within the Krebs cycle or exported to the cytoplasm where it subjected to cleavage into acetyl-CoA by the enzyme ATP-citrate lyase (ACL) in an ATP-dependent manner [28]. Furthermore, acetyl-CoA can also acetylate histone proteins, either by influencing acetyltransferase catalytic activity or non-enzymatically (Figure 2). In eukaryotic cells, acetyl-CoA is regarded as the sole donor of acetyl groups to acetylating proteins [31].
Correlation between alcohol metabolites and histone modification. In the liver, ethanol is converted to acetate through the action of different metabolic enzymes. Increased levels of acetate quickly escape the liver and reach the blood stream and other parts of the body. In mitochondria, acetate is transformed to acetyl-CoA by the enzyme acyl-CoA synthetase short-chain family member 2 (ACSS2). The resulting acetyl CoA then enters the Krebs cycle and combines with oxaloacetate to form citrate. Citrate is either oxidized within the Krebs cycle or exported to the cytoplasm, where it subjected to cleavage into acetyl-CoA by the enzyme ATP-citrate lyase (ACL). Acetyl-CoA is the sole donor of acetyl groups to acetylate histone proteins, either by influencing acetyltransferase catalytic activity or non-enzymatically. An elevated level of acetate from alcohol metabolism also causes an increase in the NADH:NAD+ ratio. An increased NADH:NAD+ ratio causes functional impairment of NAD+-dependent enzymes and results in histone deacetylation. Alcohol-mediated folate deficiency reduces S-adenosylmethionine (SAM) level and the SAM to S-adenosyl homocysteine (SAH) ratio as well as increases the SAH concentration, which in turns inhibits histone methyl transferase (HMT). Additionally, the intermediate products of the Krebs cycle, fumarate and succinate, act as inhibitors of Jumonji-C domain-containing histone demethylases (JHDMs). AMP-activated protein kinase (AMPK) is involved in sensing ATP fluctuation in cells and becomes activated to phosphorylate histone proteins. Formation of energetic stress causes the activation of AMPK and leads to histone phosphorylation. H2O2: Hydrogen peroxide; H2O: Water; ADH: Alcohol dehydrogenase; NAD/NADH: Nicotinamide adenine dinucleotide; CYP2E1: Cytochrome P450 IIE1; ALDH: Aldehyde dehydrogenase; CoA-SH: CoA synthase; KAT: Lysine acetyltransferase; SIRT: Sirtuin; FA: Folic acid; THF: Tetrahydrofolate; MTHF: methylene tetrahydrofolate; MS: Methionine synthase; MAT: Methionine adenyltransferase; GDP: Guanosine diphosphate; GTP: Guanosine triphosphate; ATP: Adenosine triphosphate; ADP: Adenosine diphosphate; AMP: Adenosine monophosphate.
The NADH:NAD+ ratio affects transcriptional mechanisms through NAD+-dependent enzymatic activity. An NAD+-dependent enzyme, Sir2, exhibits histone deacetylase (HDAC) activity, which can induce gene silencing [32]. Gene silencing, circadian rhythm regulation, and other cellular functions require NAD+-dependent HDAC activity [33, 34]. As substrates of SIRT1, histone and p53 proteins are subject to conformational changes dictated by the ratio of NADH:NAD+. We have noticed that chronic alcohol exposure resulted in reduced NAD+ levels, required for sirtuin's deacetylase activity, and caused damage to the liver and other organs [35]. Metabolic reactions produce redox potential that plays an important role in circadian rhythm functioning. Previous studies have suggested that ethanol exposure (both acute and chronic) alters several aspects of the circadian rhythm [36-39].
SAM is the primary methyl group donor and is generated in the methionine cycle from methionine and ATP [11, 40]. Many studies have shown that folate deficiency reduces SAM levels and the SAM to S-adenosyl homocysteine (SAH) ratio, and increases SAH concentrations by inactivating methionine synthase (MS) and methionine adenyltransferase (MAT) [41, 42]. Elevated levels of SAH inhibit HMTs-mediated histone methylation [43, 44] (Figure 2). We already know that chronic alcohol consumption reduces the serum folate level [45, 46]. The Krebs cycle intermediates, succinate and fumarate, have been identified as inhibitors of Jumonji-C (JMJC) domain-containing histone demethylases (JHDMs) and promote methylation [47, 48].
ATP is a substrate for kinases in the phosphorylation process. Typically, kinases have a high affinity for ATP but its availability does not influence kinase activity. However, But AMP-activated protein kinase (AMPK) is involved in sensing the cellular energy level and becomes activated to phosphorylate histone proteins [49] (Figure 2). Like AMPK, many chromatin-modifying enzymes are also regulated by the availability of metabolites and can influence the capacity to write or erase post-translational modifications in the chromatin. Another aspect of alcohol metabolism is that it increases cellular ROS levels. Elevated ROS levels induce intracellular oxidative stress and can alter transcriptomic and epigenetic mechanisms [50, 51]. It was observed that superoxide plays an important role in regulating epigenetic events, including histone methylation and acetylation [52], in various normal and pathological processes.
Evidence of alcohol-mediated modification of histone marks
Epigenetic changes may be responsible for some of the detrimental effects of in utero alcohol exposure and may contribute to the abnormalities seen in FASD. The role of epigenetic mechanisms during prenatal alcohol exposure occurs in the early stages of development. Published studies (in vitro and in vivo) have shown that ethanol is responsible for protein modifications, particularly of the histones. Protein modifications may occur directly or indirectly, by disrupting epigenetic processes. Previous reports have suggested that ethanol can alter histone modifications, including acetylation, methylation, and phosphorylation, as discussed below.
Acetylation
In utero alcohol exposure is involved to cause both hyper- and hypoacetylation of histone protein in the fetus. The very first report of histone acetylation demonstrated that acute alcohol exposure caused acetylation of histone H3K9/18 and preceded apoptosis in fetal lung tissue [53]. Hyperacetylation of H3K9 was also reported in cardiac progenitor cells in vitro [54]. A similar result was also documented in the fetal heart, where ethanol induced H3K9 hyperacetylation and an alternation of gene expression related to heart development [55-57]. Extensive research recently showed that alcohol significantly enhanced the levels of phosphorylated ERK1/2 and induced hyperacetylation of histone3 in H9c2 cells [58]. Another report showed that ethanol exposure increased global HAT activity in the fetal hearts of mice without affecting HDAC activity. Thus, ethanol is involved in hyperacetylation in different parts of the developing fetus and can cause an elevation of the expression of developmental genes, including NKX2.5, β-MHC and Cx43 in the fetal heart [59]. Ethanol enhanced the specific acetylation of H3K14 at G9a exon 1 in the brain during synaptogenesis in a rodent model [60]. Hyperacetylation of H3K14 and induction of DHAND and EHAND genes was also reported in the developing fetal heart [61]. Ethanol-induced neonatal neurodegeneration enhanced the CB1R exon 1 acetylation signature at H4K8 [62]. It was also reported that ethanol elevated mRNA levels of histone modifying genes (HDAC2, HDAC4 and G9a) at the mRNA level in the fetal rat brain [63].
In utero alcohol exposure is also reported to cause hypoacetylation of histone protein in the developing fetus and reduced expression of related genes. Decreased levels of H3 and H4 acetylation were reported in the developing rat cerebellum due to the down-regulation of histone acetyl-transferase and CREB binding protein (CBP) [64]. They also reported hypoacetylation of H3K23 in the fetal brain. Another interesting finding showed that ethanol caused a reduction in the H3K9 acetylation level in the fetal brain [65, 66]. This contrasting phenomenon suggests that the ethanol-mediated alterations are context-dependent. A summary of alcohol-mediated effects on histone acetylation is presented in Table 1.
Summary of alcohol-mediated histone modifications in utero.
| Model organism | Pattern of alcohol exposure | Sampling | Status of histone signature | Reference |
|---|---|---|---|---|
| Mouse | 25% ethanol (3.75 g/kg) | E13.5 lung | Induction of H3K9/18ac | [53] |
| Cardiac progenitor cells | 200 mM in vitro | Cell culture exposed for 24 h | Induction of H3K9ac | [54] |
| Rat | 4 hr/day in the inhalation chamber (vapor of ethanol) | P8 brain (cerebellum) | Reduction of H3 and H4 acetylation. The level of H3K23ac is also reduced. | [64] |
| Cardiac progenitor cells | 200 mM in vitro | Cell culture exposed for 24 h | Elevation of H3K9ac and the expression of GATA4 and Mef2c genes. | [56] |
| Rat | ethanol in liquid diet | P60-P80 of each generation (F1, F2 and F3); brain | Reduction of H3K4me2, H3K4me3 and H3K9ace marks; induction of H3K9me2 mark. Reduction of mRNA levels of Set7/9; induction of G9a and HDAC2 mRNAs. | [65] |
| Rat | 6.7% v/v (in liquid diet) equivalent to ∼120-150 mg/dl in blood | P60-65 brain | Induction of H3K9me2 mark; deduction of H3K4me2, H3K4me3, H3K9ac, pH3S10 marks. Induction of mRNAs of G9a and Setdb1; reduction of Set7/9 mRNA level. | [66] |
| Mouse | 1.0 or 2.5 g/kg (subcutaneous injection) | P7 brain | Elevation of G9a mRNA and protein levels and G9a activity. Induction of H3K9me2 and H3K27me2. | [76] |
| Mouse (fetal cerebral cortical neuroepithelial stem cells) | 320 mg/dL (70mM) | Neurosphere Culture exposed for 5 days | Reduction of H3K4me3 mark at Nestin, Sox2, Dlx2, Pax6 and Vdr gene promoters. Reduction of H3K27me3 mark at Ascl1, Sox17, Twist1, Wnt5b, HoxA1, HoxA7, Msx2, Nanog, Nkx2.1 and Nkx2.2 gene promoters. Reduction of both H3K4me3 and H3K27me3 marks at Igf1, Smarca2, Dlx5 and Msx1 gene promoters. Global decline in the levels of H3K27me3 of the four transposable element families (LINE1, IAP, MusD and EtN). | [67] |
| Mouse | 10 μl/g/d (56% alcohol) by gavage | E11.5, E14.5, E17.5, and E18.5 fetal heart | Induction of HAT activity and H3K9ac signature. | [55] |
| Mouse | 5 ml/kg (56% v/v) intragastrically | E14.5 and E16.5 fetal heart; P0.5 and P7 neonatal heart | Induction of HAT activity and binding of P300, CBP, PCAF and SRC1 to the Gata4 promoter at E14.5. Induces hyperacetylation of H3K9. | [57] |
| Mouse | 1.0 g/kg (subcutaneous injection) | P7 brain | Induction of H3K14ac at G9a exon1. Elevation of H3K9me2 and H3K27me2 levels in the hippocampus and neocortex. | [60] |
| Mouse | 2.5 g/kg (subcutaneous injection) | P7 brain | Elevation of H4K8ac and reduction of H3K9me2 at exon1 of the CB1R gene. | [62] |
| Mouse | 6 g/kg/d (56%) intragastrically | E14.5 fetal heart | Elevation of H3K14ac mark and the expression of DHAND and EHAND genes. | [61] |
| Mouse | 2.5 g/kg (subcutaneous injection) | P7 brain | Reduction of H3K9me2 mark. | [77] |
| Rat (Fetal heart-derived cell line) | 200 mM in vitro | H9c2 cell culture | Induction of H3 acetylation level. | [58] |
| Mouse | 5 ml/kg (56% v/v) intragastrically | E14.5 and E16.5 fetal heart; P0.5 and P7 neonatal heart | Hyperacetylation of histone H3K9 on the NKX2.5 promoter. | [59] |
| Rat | 6.7% v/v (in liquid diet) equivalent to 120-150 mg/dl in blood | P60-P90 pituitary gland | Elevation of pituitary mRNA levels of histone modifying genes (HDAC2, HDAC4, G9a). | [63] |
| Mouse | 10% v/v in drinking water | P87 hippocampus | Enrichment of histone H3K4me3 mark at the Slc17a6 promoter. Induction of chromatin modifying enzymes (Dnmt1, Uhrf1, Ehmt1, Ash2l, Wdr5, and Kdm1b). | [68] |
| Mouse | 2.9 g/kg (intraperitoneal injection) | E17 forebrain | Elevation of H3K9me2, H3K9ace marks and reduction of H3K27me3 mark. Reduction of G9a, Setdb1, Kdm1a, Kdm4c, Uhrf1, Ezh2 and Dnmt1 mRNA levels. | [70] |
| Human dental pulp stem cells (DPSCs) | 20 or 50 mM (both acute and chronic exposure) | DPSC cell culture | Suppression of KDM6B mRNA level. | [69] |
Methylation
The ability of ethanol to influence histone methylation marks has also been reported. We have observed ethanol-mediated methylation as well as demethylation of histone protein. To the best of our knowledge, the first report concerning ethanol-mediated histone methylation was published by Govorko and colleagues [65]. They identified a reduction in H3K4me2, H3K4me3 and an increase in H3K9me2 marks in the arcuate nucleus of the hypothalamus in rat neonates. They also found a correlation between alcohol exposure and the reduced expression of Set7/9 and an induction of G9a and Setdb1 at the mRNA level. Set7/9, G9a and Setdb1 are responsible for the mono-methylation of H3k4, demethylation of H3k9me2 and mono-, di-, and trimethylation of H3K9, respectively. Recently, several reports on histone methylation have strengthened the notion that prenatal alcohol exposure can alter histone epigenetic marks in the developing fetus. Ethanol exposure caused a significant reduction in H3K4 trimethylation and H3K27 trimethylation in fetal cerebral cortical neuroepithelial stem cells [67]. Ethanol increased the specific methylation mark, H3K9me2, during the synaptogenesis period in a rodent model [60]. In utero ethanol exposure was reported to induce the H3K4me3 signature in the adult hippocampus [68]. The expression of the epigenetic modulator, KDM6B, was suppressed by ethanol during odontogenic/osteogenic differentiation in the DPSC model [69]. Osteopenia/osteoporosis is the hallmark feature of fetal alcohol spectrum disorders. A recent finding has shown that acute alcohol exposure on day 7 of gestation caused a significant alteration in the chromatin structure on day 17 [70]. A low dose of ethanol elevated the H3K4me3 and H3K27me3 marks, whereas a higher dose depleted these histone marks. The depletion of H3K9me2 was correlated with higher doses of ethanol. It was also described that the majority of epigenetic changes were associated with a repressed chromatin structure that correlated with the decreased expression of Dnmt1, Eed, G9a, EzH2, Kdm1a, Kdm4c, Setdb1, Sod3, Tet1 and Uhrf1 [70]. We noticed that most researches focused on reporting epigenetic alterations in the fetal brain, compared to other parts of the fetus. Thus, much remains to be elucidated concerning alcohol-mediated histone modifications in fetal development. A summary of ethanol-mediated alterations of histone methylation is presented in Table 1.
Phosphorylation
Recent studies have shown that the MAPK signaling pathway plays a pivotal role in the actions of ethanol in the cell. The exposure to the neural crest cells to ethanol resulted in the induction of p38 MAPK phosphorylation [71]. The phosphorylation of histone H3 at serine 10 and serine 28 has been associated with gene expression and regulation [72]. However, there is little direct evidence of prenatal alcohol exposure affecting the phosphorylation of histone protein in literature reports. The only available evidence is that prenatal alcohol exposure reduces the phosphorylation of histone H3 at serine 10 (pH3S10) [66]. Similar reports describe the alteration of histone H3 phosphorylation at the serine 10 and serine 28 residues in the rat liver [73, 74]. Acute EtOH exposure altered the intensity of histone H3 phosphorylation at the serine 10 in the rat hippocampus [75]. Further investigation is needed to uncover direct evidence for prenatal alcohol exposure and the subsequent phosphorylation of histone protein. It was recently reported that the phosphorylation of histone H3 at serine 10 and serine 28 is associated with histone acetylation, in particular, H3K9ac and H3K14ac, in EGF-stimulating cells [72]. Thus, it will be interesting to elucidate whether alcohol-mediated phosphorylation of histone H3 phosphorylation at the serine 10 and serine 28 is also associated with other histone modifications. A summary of alcohol-mediated phosphorylation of histones in utero is presented in Table 1.
Future perspectives and concluding remarks
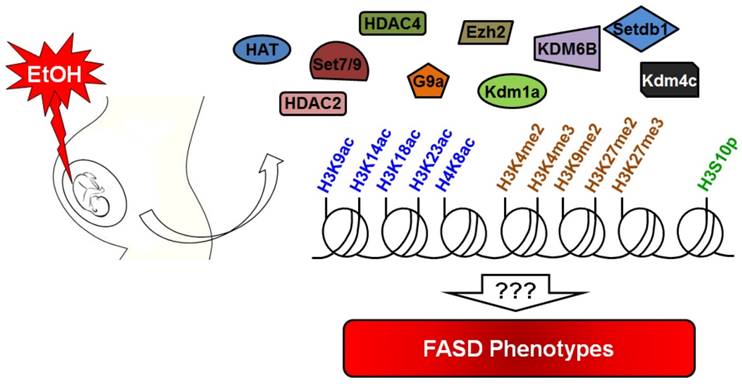
Prenatal alcohol exposure causes damage to the developing fetus through direct and indirect mechanisms. Excessive alcohol exposure can results in postnatal FASD or FAS. Various signaling pathways, cell types, and underlying molecular mechanisms are known to be induced or inhibited by ethanol, but the epigenomic and epigenetic mechanisms affected by ethanol exposure are less well known. Recent research has reported on the effects of alcohol on histone modifications. However, we still do not know the relationship between specific histone modifications and FASD phenotypes (Figure 3). Another important avenue for future research is to identify drugs that can inhibit ethanol-mediated teratogenicity. Ethanol inhibits one-carbon metabolism and suppresses methyltransferase enzymes. Thus, supplemental folate would be useful in minimizing ethanol toxicity during gestation. Curcumin is used to inhibit histone acetylation and could thus be used to combat the effect of ethanol in the appropriate context. In this review, we have gathered information about the known relationships between alcohol exposure and histone modifications. Future studies will serve to further explain the mechanistic details of numerous deleterious effects associated with ethanol exposure in utero.
Summary of alcohol-mediated histone modifications in utero. Gestational alcohol exposure causes epigenetic alteration in the developing fetus. Many histone marks are altered by alcohol during fetal development. Research suggests that alcohol can alter the expression level of several important epigenetic modulators that contribute to the alteration of histone marks. Current knowledge about alcohol toxicity indicates that it is able to affect acetylation/deacetylation, methylation/demethylation and phosphorylation of histone proteins. However, the correlation of these changes with the FASD phenotype remains unknown.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIP 2011-0030049 to Y.G.C.).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yao B, Christian KM, He C. et al. Epigenetic mechanisms in neurogenesis. Nat Rev Neurosci. 2016;17(9):537-49
2. Goyama S, Kitamura T. Epigenetics in Normal and Malignant Hematopoiesis: An Overview and Update 2017. Cancer Sci. 2017
3. Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. 2015;16(2):71-84
4. Moosavi A, Motevalizadeh Ardekani A. Role of Epigenetics in Biology and Human Diseases. Iran Biomed J. 2016;20(5):246-58
5. Kim S, Kaang BK. Epigenetic regulation and chromatin remodeling in learning and memory. Exp Mol Med. 2017;49(1):e281
6. Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693-705
7. Bhattacharjee D, Shenoy S, Bairy KL. DNA Methylation and Chromatin Remodeling: The Blueprint of Cancer Epigenetics. Scientifica (Cairo). 2016;2016:6072357
8. Bunkar N, Pathak N, Lohiya NK. et al. Epigenetics: A key paradigm in reproductive health. Clin Exp Reprod Med. 2016;43(2):59-81
9. Shukla SD, Velazquez J, French SW. et al. Emerging role of epigenetics in the actions of alcohol. Alcohol Clin Exp Res. 2008;32(9):1525-34
10. Dong X, Weng Z. The correlation between histone modifications and gene expression. Epigenomics. 2013;5(2):113-6
11. Ungerer M, Knezovich J, Ramsay M. In utero alcohol exposure, epigenetic changes, and their consequences. Alcohol Res. 2013;35(1):37-46
12. Mandal C, Kim SH, Chai JC. et al. RNA Sequencing Reveals the Alteration of the Expression of Novel Genes in Ethanol-Treated Embryoid Bodies. PLoS One. 2016;11(3):e0149976
13. Mandal C, Halder D, Chai JC. et al. Profiling ethanol-targeted transcription factors in human carcinoma cell-derived embryoid bodies. Gene. 2016;576(1 Pt 1):119-25
14. Halder D, Park JH, Choi MR. et al. Chronic ethanol exposure increases goosecoid (GSC) expression in human embryonic carcinoma cell differentiation. J Appl Toxicol. 2014;34(1):66-75
15. Mandal C, Park JH, Lee HT. et al. Reduction of Nfia gene expression and subsequent target genes by binge alcohol in the fetal brain. Neurosci Lett. 2015;598:73-8
16. Grewal J, Carmichael SL, Ma C. et al. Maternal periconceptional smoking and alcohol consumption and risk for select congenital anomalies. Birth Defects Res A Clin Mol Teratol. 2008;82(7):519-26
17. Mandal C, Park KS, Jung KH. et al. Ethanol-related alterations in gene expression patterns in the developing murine hippocampus. Acta Biochim Biophys Sin (Shanghai). 2015;47(8):581-7
18. Mandal C, Park JH, Choi MR. et al. Transcriptomic study of mouse embryonic neural stem cell differentiation under ethanol treatment. Mol Biol Rep. 2015;42(7):1233-9
19. Halder D, Mandal C, Lee BH. et al. PCDHB14- and GABRB1-like nervous system developmental genes are altered during early neuronal differentiation of NCCIT cells treated with ethanol. Hum Exp Toxicol. 2015;34(10):1017-27
20. Sanchez-Alvarez R, Gayen S, Vadigepalli R. et al. Ethanol diverts early neuronal differentiation trajectory of embryonic stem cells by disrupting the balance of lineage specifiers. PLoS One. 2013;8(5):e63794
21. Reynes B, Palou M, Palou A. Gene expression modulation of lipid and central energetic metabolism related genes by high-fat diet intake in the main homeostatic tissues. Food Funct. 2017
22. Boggan W. Alcohol and you. Kennesaw State University. 2003
23. Cederbaum AI. Alcohol metabolism. Clin Liver Dis. 2012;16(4):667-85
24. Henderson GI, Chen JJ, Schenker S. Ethanol, oxidative stress, reactive aldehydes, and the fetus. Front Biosci. 1999;4:D541-50
25. Zakhari S. Alcohol metabolism and epigenetics changes. Alcohol Res. 2013;35(1):6-16
26. Dong D, Zhong W, Sun Q. et al. Oxidative products from alcohol metabolism differentially modulate pro-inflammatory cytokine expression in Kupffer cells and hepatocytes. Cytokine. 2016;85:109-19
27. Dinis-Oliveira RJ. Oxidative and Non-Oxidative Metabolomics of Ethanol. Curr Drug Metab. 2016;17(4):327-35
28. Kinnaird A, Zhao S, Wellen KE. et al. Metabolic control of epigenetics in cancer. Nat Rev Cancer. 2016;16(11):694-707
29. Jaworski DM, Namboodiri AM, Moffett JR. Acetate as a Metabolic and Epigenetic Modifier of Cancer Therapy. J Cell Biochem. 2016;117(3):574-88
30. Hirschey MD, Shimazu T, Huang JY. et al. SIRT3 regulates mitochondrial protein acetylation and intermediary metabolism. Cold Spring Harb Symp Quant Biol. 2011;76:267-77
31. Choudhary C, Weinert BT, Nishida Y. et al. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 2014;15(8):536-50
32. Hannan A, Abraham NM, Goyal S. et al. Sumoylation of Sir2 differentially regulates transcriptional silencing in yeast. Nucleic Acids Res. 2015;43(21):10213-26
33. Mazzoccoli G, Pazienza V, Vinciguerra M. Clock genes and clock-controlled genes in the regulation of metabolic rhythms. Chronobiol Int. 2012;29(3):227-51
34. Matsushima S, Sadoshima J. The role of sirtuins in cardiac disease. Am J Physiol Heart Circ Physiol. 2015;309(9):H1375-89
35. French SW. Chronic alcohol binging injures the liver and other organs by reducing NAD(+) levels required for sirtuin's deacetylase activity. Exp Mol Pathol. 2016;100(2):303-6
36. Ruby CL, Palmer KN, Zhang J. et al. Differential Sensitivity to Ethanol-Induced Circadian Rhythm Disruption in Adolescent and Adult Mice. Alcohol Clin Exp Res. 2017;41(1):187-196
37. Wilson DA, Masiello K, Lewin MP. et al. Developmental ethanol exposure-induced sleep fragmentation predicts adult cognitive impairment. Neuroscience. 2016;322:18-27
38. De Nobrega AK, Lyons LC. Circadian Modulation of Alcohol-Induced Sedation and Recovery in Male and Female Drosophila. J Biol Rhythms. 2016;31(2):142-60
39. Varadinova MG, Valcheva-Traykova ML, Boyadjieva NI. Effect of Circadian Rhythm Disruption and Alcohol on the Oxidative Stress Level in Rat Brain. Am J Ther. 2016;23(6):e1801-e1805
40. Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer. 2016;16(10):650-62
41. Farias N, Ho N, Butler S. et al. The effects of folic acid on global DNA methylation and colonosphere formation in colon cancer cell lines. J Nutr Biochem. 2015;26(8):818-26
42. Crider KS, Yang TP, Berry RJ. et al. Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate's role. Adv Nutr. 2012;3(1):21-38
43. Grillo MA, Colombatto S. S-adenosylmethionine and its products. Amino Acids. 2008;34(2):187-93
44. Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13(8):572-83
45. Medici V, Halsted CH. Folate, alcohol, and liver disease. Mol Nutr Food Res. 2013;57(4):596-606
46. Young JK, Giesbrecht HE, Eskin MN. et al. Nutrition implications for fetal alcohol spectrum disorder. Adv Nutr. 2014;5(6):675-92
47. Tsukada Y, Fang J, Erdjument-Bromage H. et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439(7078):811-6
48. Xiao M, Yang H, Xu W. et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326-38
49. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251-62
50. Ziech D, Franco R, Pappa A. et al. Reactive oxygen species (ROS)-induced genetic and epigenetic alterations in human carcinogenesis. Mutat Res. 2011;711(1-2):167-73
51. Wu Q, Ni X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr Drug Targets. 2015;16(1):13-9
52. Afanas'ev I. New nucleophilic mechanisms of ros-dependent epigenetic modifications: comparison of aging and cancer. Aging Dis. 2013;5(1):52-62
53. Wang X, Gomutputra P, Wolgemuth DJ. et al. Acute alcohol exposure induces apoptosis and increases histone H3K9/18 acetylation in the mid-gestation mouse lung. Reprod Sci. 2010;17(4):384-90
54. Zhong L, Zhu J, Lv T. et al. Ethanol and its metabolites induce histone lysine 9 acetylation and an alteration of the expression of heart development-related genes in cardiac progenitor cells. Cardiovasc Toxicol. 2010;10(4):268-74
55. Pan B, Zhu J, Lv T. et al. Alcohol consumption during gestation causes histone3 lysine9 hyperacetylation and an alternation of expression of heart development-related genes in mice. Alcohol Clin Exp Res. 2014;38(9):2396-402
56. Wang L, Sun H, Pan B. et al. Inhibition of histone acetylation by curcumin reduces alcohol-induced expression of heart development-related transcription factors in cardiac progenitor cells. Biochem Biophys Res Commun. 2012;424(3):593-6
57. Peng C, Zhu J, Sun HC. et al. Inhibition of histone H3K9 acetylation by anacardic acid can correct the over-expression of Gata4 in the hearts of fetal mice exposed to alcohol during pregnancy. PLoS One. 2014;9(8):e104135
58. Gao W, Pan B, Liu L. et al. Alcohol exposure increases the expression of cardiac transcription factors through ERK1/2-mediated histone3 hyperacetylation in H9c2 cells. Biochem Biophys Res Commun. 2015;466(4):670-5
59. Peng C, Zhang W, Zhao W. et al. Alcohol-induced histone H3K9 hyperacetylation and cardiac hypertrophy are reversed by a histone acetylases inhibitor anacardic acid in developing murine hearts. Biochimie. 2015;113:1-9
60. Subbanna S, Nagre NN, Shivakumar M. et al. Ethanol induced acetylation of histone at G9a exon1 and G9a-mediated histone H3 dimethylation leads to neurodegeneration in neonatal mice. Neuroscience. 2014;258:422-32
61. Zhang W, Peng C, Zheng M. et al. Prenatal alcohol exposure causes the over-expression of DHAND and EHAND by increasing histone H3K14 acetylation in C57 BL/6 mice. Toxicol Lett. 2014;228(3):140-6
62. Subbanna S, Nagre NN, Umapathy NS. et al. Ethanol exposure induces neonatal neurodegeneration by enhancing CB1R Exon1 histone H4K8 acetylation and up-regulating CB1R function causing neurobehavioral abnormalities in adult mice. Int J Neuropsychopharmacol. 2014:18 (5)
63. Gangisetty O, Wynne O, Jabbar S. et al. Fetal Alcohol Exposure Reduces Dopamine Receptor D2 and Increases Pituitary Weight and Prolactin Production via Epigenetic Mechanisms. PLoS One. 2015;10(10):e0140699
64. Guo W, Crossey EL, Zhang L. et al. Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS One. 2011;6(5):e19351
65. Govorko D, Bekdash RA, Zhang C. et al. Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations. Biol Psychiatry. 2012;72(5):378-88
66. Bekdash RA, Zhang C, Sarkar DK. Gestational choline supplementation normalized fetal alcohol-induced alterations in histone modifications, DNA methylation, and proopiomelanocortin (POMC) gene expression in beta-endorphin-producing POMC neurons of the hypothalamus. Alcohol Clin Exp Res. 2013;37(7):1133-42
67. Veazey KJ, Carnahan MN, Muller D. et al. Alcohol-induced epigenetic alterations to developmentally crucial genes regulating neural stemness and differentiation. Alcohol Clin Exp Res. 2013;37(7):1111-22
68. Zhang CR, Ho MF, Vega MC. et al. Prenatal ethanol exposure alters adult hippocampal VGLUT2 expression with concomitant changes in promoter DNA methylation, H3K4 trimethylation and miR-467b-5p levels. Epigenetics Chromatin. 2015;8:40
69. Hoang M, Kim JJ, Kim Y. et al. Alcohol-induced suppression of KDM6B dysregulates the mineralization potential in dental pulp stem cells. Stem Cell Res. 2016;17(1):111-21
70. Veazey KJ, Parnell SE, Miranda RC. et al. Dose-dependent alcohol-induced alterations in chromatin structure persist beyond the window of exposure and correlate with fetal alcohol syndrome birth defects. Epigenetics Chromatin. 2015;8:39
71. Yuan F, Chen X, Liu J. et al. Up-regulation of Siah1 by ethanol triggers apoptosis in neural crest cells through p38 MAPK-mediated activation of p53 signaling pathway. Arch Toxicol. 2017;91(2):775-784
72. Rossetto D, Avvakumov N, Cote J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics. 2012;7(10):1098-108
73. Lee YJ, Shukla SD. Histone H3 phosphorylation at serine 10 and serine 28 is mediated by p38 MAPK in rat hepatocytes exposed to ethanol and acetaldehyde. Eur J Pharmacol. 2007;573(1-3):29-38
74. James TT, Aroor AR, Lim RW. et al. Histone H3 phosphorylation (Ser10, Ser28) and phosphoacetylation (K9S10) are differentially associated with gene expression in liver of rats treated in vivo with acute ethanol. J Pharmacol Exp Ther. 2012;340(2):237-47
75. McClain JA, Nixon K. Alcohol Induces Parallel Changes in Hippocampal Histone H3 Phosphorylation and c-Fos Protein Expression in Male Rats. Alcohol Clin Exp Res. 2016;40(1):102-12
76. Subbanna S, Shivakumar M, Umapathy NS. et al. G9a-mediated histone methylation regulates ethanol-induced neurodegeneration in the neonatal mouse brain. Neurobiol Dis. 2013;54:475-85
77. Subbanna S, Basavarajappa BS. Pre-administration of G9a/GLP inhibitor during synaptogenesis prevents postnatal ethanol-induced LTP deficits and neurobehavioral abnormalities in adult mice. Exp Neurol. 2014;261:34-43
Author contact
![]() Corresponding author: Young Gyu Chai, PhD, Department of Molecular & Life Science, Hanyang University, Ansan, 1271 Sa-dong, Republic of Korea. Tel: +82-31-400-5513, Email: ygchaiac.kr
Corresponding author: Young Gyu Chai, PhD, Department of Molecular & Life Science, Hanyang University, Ansan, 1271 Sa-dong, Republic of Korea. Tel: +82-31-400-5513, Email: ygchaiac.kr