International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Cyclic Peptidic Enzyme Inhibitors
uPA and cancer metastasis
Inhibitors of uPA
Catalytic domains of the...
Upain-1 derivatives, specific...
Mupain-1, a versatile peptidic...
Conclusion
Acknowledgements
References
Cyclic Peptidic Enzyme Inhibitors
uPA and cancer metastasis
Inhibitors of uPA
Catalytic domains of the...
Upain-1 derivatives, specific...
Mupain-1, a versatile peptidic...
Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(10):1222-1233. doi:10.7150/ijbs.21597 This issue Cite
Review
Structural Principles in the Development of Cyclic Peptidic Enzyme Inhibitors
Peng Xu1, Peter A. Andreasen2, Mingdong Huang1, 3 ![]()
1. State Key Laboratory of Structural Chemistry and Danish-Chinese Centre for Proteases and Cancer, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian, 350002, P.R. China;
2. Department of Molecular Biology and Genetics, Aarhus University, Aarhus, 8000, Denmark;
3. College of Chemistry, Fuzhou University, Fuzhou, Fujian, 350116, P.R. China.
Received 2017-6-23; Accepted 2017-8-28; Published 2017-9-21
Citation:
Xu P, Andreasen PA, Huang M. Structural Principles in the Development of Cyclic Peptidic Enzyme Inhibitors. Int J Biol Sci 2017; 13(10):1222-1233. doi:10.7150/ijbs.21597. https://www.ijbs.com/v13p1222.htm
Other stylesAbstract

This review summarizes our studies in the development of small cyclic peptides for specifically modulating enzyme activity. Serine proteases share highly similar active sites but perform diverse physiological and pathological functions. From a phage-display peptide library, we isolated two mono-cyclic peptides, upain-1 (CSWRGLENHRMC) and mupain-1 (CPAYSRYLDC), which inhibit the activity of human and murine urokinase-type plasminogen activators (huPA and muPA) with Ki values in the micromolar or sub-micromolar range, respectively. The following affinity maturations significantly enhanced the potencies of the two peptides, 10-fold and >250-fold for upain-1 and mupain-1, respectively. The most potent muPA inhibitor has a potency (Ki = 2 nM) and specificity comparable to mono-clonal antibodies. Furthermore, we also found an unusual feature of mupain-1 that its inhibitory potency can be enhanced by increasing the flexibility, which challenges the traditional viewpoint that higher rigidity leading to higher affinity. Moreover, by changing a few key residues, we converted mupain-1 from a uPA inhibitor to inhibitors of other serine proteases, including plasma kallikrein (PK) and coagulation factor XIa (fXIa). PK and fXIa inhibitors showed Ki values in the low nanomolar range and high specificity. Our studies demonstrate the versatility of small cyclic peptides to engineer inhibitory potency against serine proteases and to provide a new strategy for generating peptide inhibitors of serine proteases.
Keywords: serine protease, inhibitory mechanism, cyclic peptide, urokinase-type plasminogen activator.
Cyclic Peptidic Enzyme Inhibitors
In biology and pharmacy, the commonly accepted demarcations of drugs by molecular weights (MW) are “small molecules” (MW < 500 Da) and “macromolecules” (MW > 5000 Da) [1]. Drugs with molecular weights ranging from 200 to 600 Da predominate among drugs approved by the U.S. Food and Drug Administration (FDA) (>90%) [2]. Polypeptides with 10-20 amino acids are in the range between 500-5000 Da. Thus, peptide-based drugs are attracting increasing attention as they combine the advantages of small molecules and macromolecules, namely stability, potency, and specificity. Compared to linear peptides, cyclic peptides are more favorable candidates as pharmaceutics as they possess greater conformational constraint resulting in higher selectivity. Dysregulated enzyme activities cause physical disorders and are therapeutic targets of many pathological conditions. The only FDA-approved cyclic peptide-based enzyme inhibitor is cyclosporin A, an inhibitor of calcineurin [3]. Cyclosporin A interferes the activity and growth of T-cells and is therefore used as an immunosuppressant drug, which has an annual sale of >US $1.0 billion in the USA only [4]. Serine proteases are widely involved in many physiological and pathological pathways and are demonstrated as therapeutic targets of many important diseases [5-7]. In this article, we reviewed our journey in the development of cyclic peptidic inhibitors against serine proteases by combining de-novo screening and rational design. Our studies originate with two peptides, upain-1 (CSWRGLENHRMC) and mupain-1 (CPAYSRYLDC), which were screened from a phage-displayed peptide library as inhibitors of human and murine urokinase-type plasminogen activators (uPA), respectively.
uPA and cancer metastasis
Urokinase-type plasminogen activator (uPA) is a serine protease with a molecular weight of 55-kDa.[8, 9] uPA is originally secreted in the zymogen form, single chain uPA (sc-uPA), and is activated by plasmin or other proteases via the proteolysis of the peptide bond between Lys158 and Ile159 [10]. The disulfide-bridged two-chain uPA (tc-uPA) consists of the N-terminal amino terminal fragment (ATF), and the C-terminal catalytic domain. The N-terminal ATF, composed of a kringle domain and a growth factor-like domain [11, 12], binds to the uPA receptor (uPAR) tightly with a KD value of 0.3 nM [13, 14]. The catalytic domain performs the proteolytic function and activates the substrate, plasminogen [15]. uPA serves in the plasminogen activation (PA) system, which was originally known to perform fibrinolytic functions in blood by dissolving blood clots (fibrin) through the generation of plasmin [16-19]. A large number of studies have demonstrated that the PA system is also involved in many other physiological and pathological pathways, including wound healing [20], adhesion of cells [21], proliferation and metastasis of cancer [22], infectious diseases [23-25], and even neuropathology [26].
Plasmin is the central enzyme of PA system, which performs two main functions (Figure 1) [27]: 1) maintaining the fluidity of vessels by lysing the blood clots in blood [28, 29] and 2) turning over the extracellular matrix (ECM) components in tissue modeling [30], like wound healing. Plasmin is physiologically regulated by α2-antiplasmin [31] and generated through the activation of its zymogen, plasminogen, by tissue-type and the urokinase-type plasminogen activators (tPA and uPA) [32], which are regulated by plasminogen activator inhibitor 1 and 2 (PAI-1 and PAI-2) [33-35]. The uPA-triggered PA system is closely relevant to cancer proliferation and metastasis [30, 36, 37]. The role of uPA in cancer proliferation and metastasis has not been thoroughly validated but majorly accepted as illustrated in Figure 1: Sc-uPA is activated generating active tc-uPA after binding to the membrane-anchored receptor, uPAR [38, 39]. Tc-uPA on the cell surface then catalyzes the generation of plasmin, which consequently degrades the extracellular matrix (ECM). The overexpression of uPA and uPAR on the cell surface of tumor cells leads to aberrant plasminogen activation [40], resulting in the excessive degradation of ECM, which increases the mobility of cancer cells and facilitates cancer invasion.
Figure 1
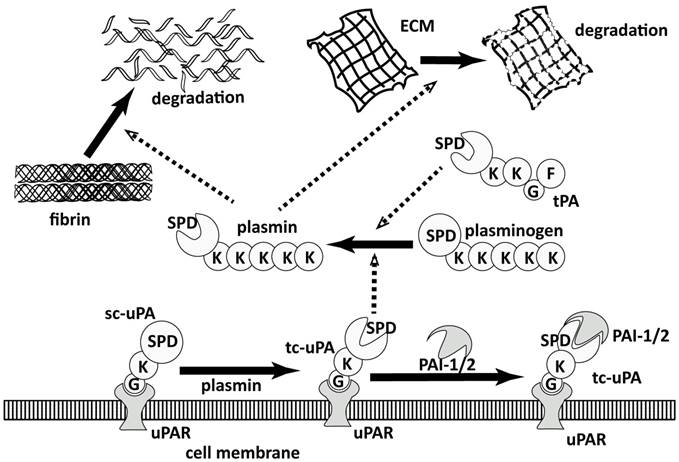
plasminogen activation system
Figure 2
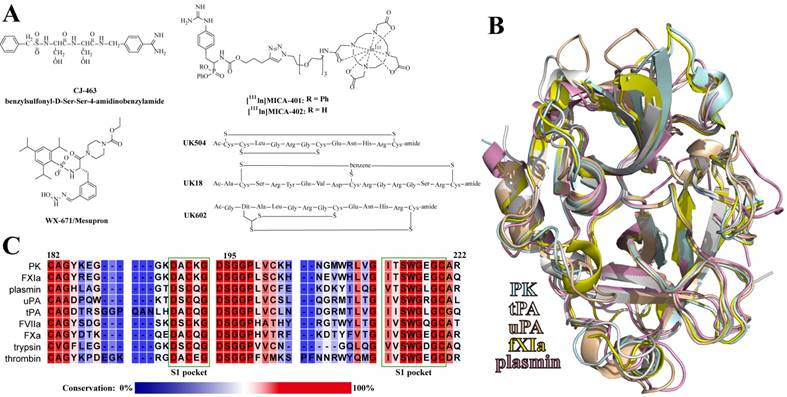
A: Chemical structures of the reported uPA peptide-based inhibitors. B: Alignment of the catalytic domains of plasma kallikrein (PK), tissue-type and urkinase-type plasminogen activators (tPA and uPA), coagulation factor XIa (fXIa), and plasmin. C: Sequence alignment of uPA-relevant serine proteases around the S1 pockets. The regions of the S1 pockets were marked with dark green frames. The residues are colored according to the conservation.
Inhibitors of uPA
Although the underlying biochemical processes for metastasis are still not fully understood, numerous studies have demonstrated that uPA and uPAR play critical roles in the processes of cancer proliferation and metastasis [36]. The attempts of inhibiting uPA activity can be dated from the mid-1960's [41], even before structural information of the enzyme was available [42]. After over 50-years study, Mesupron® (Figure 2A), an oral small-molecule uPA inhibitor, is now in Phase II clinical trial for the therapy of pancreatic and breast cancers [43]. Mesupron® was originally developed by WILEX AG, but was licensed to Link Health Group and RedHill Biopharma Ltd in 2014. Although performing obvious suppression of tumor metastasis in clinical trials, Mesupron® is not specific to uPA, as it inhibits uPA with a Ki value of 0.65 μM while showing comparable Ki values for the inhibition of many homogenous proteases, like plasmin (1.46 μM), thrombin (0.5 μM), coagulation factor Xa (0.98 μM), tPA (2.51 μM), and matriptase (0.37 μM) [44]. Similar to Mesupron®, other small-molecule uPA inhibitors have also shown insufficient selectivity [42, 45-47].
Peptides are also of considerable interest as modulators of uPA as peptides exhibit much higher specificity than small-molecule inhibitors do (Figure 2A). Sturzebecher and his colleagues reported a peptide-based uPA inhibitor CJ-463 (benzylsulfonyl-D-Ser-Ser-4- amidinobenzylamide) with a Ki value of 20 nM [48], which demonstrated good anti-tumor efficacy in a lung cancer animal model [49]. Haemers screened a peptide-based irreversible uPA inhibitor showing IC50 value of 57 nM and high selectivity over homogenic enzymes [50]. Later, the inhibitor was optimized and used for imaging breast cancer through the single photon emission computed tomography (SPECT) [51]. With phage-display screening, Heinis's group reported bicyclic peptide inhibitors of uPA with different cyclization styles, like via two disulfide bridges [52], via locking three cysteine residues by trifunctional tris(bromomethyl)benzene [53], and through dithiol amino acids [54].
Catalytic domains of the trypsin-like serine proteases
Serine proteases are widely involved in many important physiological and pathological pathways, and are considered as therapeutic targets of many diseases [5]. One critical issue for medicinal enzyme modulators is the specificity among homogenous enzymes, because the off-target inhibition of regulated enzyme might cause systemic damages. The failure of clinical trials of matrix metalloprotease inhibitors was just due to the lack of sufficient specificity [55]. Similarly, although performing diverse physiological functions, serine proteases are known to have an identical catalytic domain in sequences and structures (Figure 2B), leading to a main challenge to engineer sufficient specificity to one particular protease. The S1 specificity pocket accommodates the side chains of the positive P1 residues of substrates. Like Mesupron®, small-molecule inhibitors suffer from the low specificity because they mostly target the S1 pockets, which are highly conserved among the serine protease family (Figure 2C) [42, 56]. In contrast, peptide-based inhibitors showed higher selectivity for contacting multiple other regions outside the S1 pockets. These regions are much more variable among homologous enzymes than the S1 pockets, and are more specific for the recognition of substrates and pharmaceuticals [57].
Upain-1 derivatives, specific competitive inhibitors of human uPA
Screening of upain-1 from a phage-displayed library
One important source of peptide-based enzyme inhibitors is the de-novo peptide libraries [58, 59]. Phage display represents a biological method of rapidly generating large random peptide libraries [60]. To isolate uPA binding peptides, we used a phage-display peptide library with the formats of X7, CX7C, CX10C, and CX3CX3CX3C (C and X indicate cysteine and randomized amino acids), containing >109 unique inserts [61]. With the bait of human uPA (huPA) immobilized through monoclonal antibodies, mAb6, mAb12 or mAb390 [62], we identified a sequence of CSWRGLENHRMC [63] referred to as upain-1. The upain-1 sequence expressed in fusion with the D1 and D2 domains of M13 phage's pIII protein was referred to as upain-1-D1D2. The synthetic upain-1 peptide and upain-1-D1D2 fusion protein competitively inhibited the proteolytic activity of huPA and the plasminogen activation on the surface of histiocytic lymphoma cells. Upain-1-D1D2 (Ki = 2.2 μM) showed 13.6-fold higher inhibitory potency than upain-1 peptide (Ki = 29.9 μM) did. In addition, both upain-1 and upain-1-D1D2 showed high selectivity to huPA over many other human enzymes, including trypsin, fXa, tPA, plasmin, plasma kallikrein.
We further determined the crystal structure of upain-1 in complex with the catalytic domain of huPA with a resolution of 2.15 Å (PDB code: 2NWN, Figure 4A) [64]. upain-1 docks into the catalytic cleft of uPA blocking the substrate-binding sites. Crystal structure shows that Ser2, Leu6, Arg10, and Met11 are solvent exposed and have very few contacts (197 Å2) with uPA, consistent with the alanine-scanning analysis that inactivating these residues had very mild influence on upain-1's affinity to uPA. In contrast, residues 3-5 and 7-9 contact largely with uPA (939 Å2) and showed significant importance in the affinity to huPA according to the alanine-scanning analysis. Furthermore, the crystal structure explains why upain-1 is an inhibitor but not cleaved as a substrate: upain-1 docks in a non-canonical substrate-like conformation [65]. The bulky side chain of Trp3 hinders the scissile peptide bond (P1-P1') approaching the catalytic triad preventing the nucleophilic attack. Besides, the side-chain of Glu7 occupies the oxyanion hole, which typically accommodates the carbonyl oxygen of the P1 residues of substrates.
Improving inhibitory potency by increasing peptides' rigidity
Because of the insufficient inhibitory potency (Ki = 29.9 μM), we next performed affinity maturation to improve the affinity of upain-1 to huPA. The crystal structure shows that Arg10 and Met11 of upain-1 are solvent-exposed [64], and have minor contribution to the peptide-enzyme binding [63]. Hence, we mutated these two residues to alanine generating peptide upain-2 (CSWRGLENHAAC), which shows identical Ki values (35 μM) to that of upain-1 (Table 1) [66]. As mentioned, upain-1-D1D2 shows >10-fold higher inhibitory potency than upain-1. However, the only D1D2 recombinant protein shows non-measurable affinity [63]. Some specific residues in D1D2 domains might facilitate the upain-1-uPA binding. Thus, we extended the N and C termini of upain-1 according to the sequence of upain-1-D1D2 fusion protein resulting in peptide MGSADGA-CSWRGLENHAAC-GAAG, which shows affinity (Ki = 3.5 μM) comparable to that of upain-1-D1D2 (Ki = 2.2 μM).
Table 1
Sequences and Ki values of inhibiting huPA of upain-1 variants.
| Peptide code | Sequence* | Ki values of inhibiting huPA (μM) |
|---|---|---|
| upain-1 | CSWRGLENHRMC | 30 ± 1 (3) |
| D1D2-upain-1 | CSWRGLENHRMC-D1D2 | 2.2 ± 0.1 (3) |
| upain-2 | CSWRGLENHAAC | 35 ± 8 (3) |
| MGSADGA-upain-1-GAAG | MGSADGA-CSWRGLENHRMC-GAAG | 4.5 ± 0.8 (3) |
| MGSADGA-upain-2-GAAG | MGSADGA-CSWRGLENHAAC-GAAG | 3.5 ± 0.1 (3) |
| DGA-upain-2 | DGA-CSWRGLENHAAC | 4.6 ± 0.6 (3) |
| bicyclic peptide 24 | CSWRGCENHAAPA | 26 ± 13 (6) |
| bicyclic peptide 29 | CSWRGCENHAAAP | 32 ± 14 (6) |
| upain-1 W3A | CSARGLENHRMC | 313 ± 8 (3) |
*All synthetic peptides are of an acetylated N-terminus and an amidated C-terminus, and are cyclized through the underlined residues.
The crystal structure of MGSADGA-CSWRGLENHAAC-GAAG:huPA complex (PDB code: 3OY5) indicates the core region (CSWRGLENHAAC) has a conformation identical to that of upain-1 binding to huPA (Figure 4B). Interestingly, for the extended part, we only observed the electron density of the N-terminus extended tripeptide (Asp-3, Gly-2 and Ala-1, DGA), while all other extended residues were completely disordered. This DGA-tripeptide does not directly interact the enzyme surface but forms intramolecular hydrogen bonds stabilizing the conformation of the core peptide (Figure 4C), consistent with the SPR analysis that the N-terminus extension decreased the dissociation rate (koff) but rarely affected the association rate (kon). In addition, peptide DGA-CSWRGLENHAAC inhibits uPA with a Ki value of 4.6 μM, indicating that only the tripeptide DGA rather than other disordered extension residues improve the binding. One possible explanation is that DGA-extension provides a hydrogen-bond network enhancing the peptide's rigidity and causes declined entropic penalty. This is a nice example demonstrating the agreement between functional data and X-ray structures.
In an independent study, Heinis et al. reported a bicyclic peptide huPA inhibitor, UK-504, screened from another phage-displayed library [52]. UK-504 is cyclized by two disulfide bonds and has a sequence similar to upain-1 (UK-504: CLGRGCENHRCLC, upain-1: CSWRGLENHRMC, the same residues are underlined). Notably, the same residues are the residues forming large contacts with uPA. In addition, crystal structures demonstrate that upain-1 and UK-504 (PDB code: 4GLY) have highly similar conformations when binding to uPA (Figure 4D). UK-504 (Ki = 7.7 μM) binds to uPA with a stronger affinity than upain-1 (Ki = 29.9 μM) does, which is likely due to lower binding entropic penalty caused by the constraint of bicyclization. Similarly, UK-504 shows comparable affinity to that of DGA-upain-1 (Ki = 4.6 μM), which has additional hydrogen-bond network enhancing upain-1's rigidity. Thus, both examples demonstrate the importance the rigidity in the potencies of peptide inhibitors.
Rational design of bicyclic peptidic huPA inhibitors
In the same year when Heinis published UK-504, we also reported bicyclic peptide inhibitors of huPA based on upain-2 (CSWRGLENHAAC) [67]. With the aim of reducing entropic penalty during the binding event, we cyclized upain-2 through a disulfide bridge and a head-to-tail or side-chain-to-tail amide bond. Considering the head-to-tail or side-chain-to-tail cyclization might influence the overall ring size of the original mono-cyclic peptide, we synthesized these bicyclic peptides with the size ranged 11-13 residues. Accordingly, we synthesized 21 bicyclic peptides and measured the inhibitory potency. Only two peptides, CSWRGCENHAAPA (peptide 24, Ki = 26 μM) and CSWRGCENHAAAP (peptide 29, Ki = 32 μM), showed comparable inhibitory potency to that of the mono-cyclic upain-2 (Ki = 35.5 μM). ITC data showed that bicyclization indeed reduced peptides' binding entropic penalty, which, however, was partially offset by the increased enthalpy. We next solved the crystal structure of peptide 24 in complex with huPA and found and upain-1 bind to uPA in a mostly identical conformation, especially for the residues that directly interact with uPA (Figure 4E). Consistent with the higher enthalpy from ITC analysis, crystal structures also indicated that peptide 24 bound less tightly to uPA, as peptide 24 showed a higher relative B-factor than upain-1 did (1.86 vs. 1.58). Thus, even though our bicyclization decreased entropic penalty, the inhibitory potency was only moderately enhanced due to the less tight binding.
Switch of inhibitors to substrates
Based on our studies of the binding and inhibitory mechanisms of upain-1 derivatives, we reported the structural details of a pH-dependent proteolytic process of serine proteases (Figure 4F). Upain-1 is converted to a uPA substrate when Trp3 is mutated to alanine (CSARGLENHRMC).[63] We determined the high-resolution crystal structures (1.29-1.68 Å) of upain-1-W3A in complexes with active huPA and inactive huPA (huPA-S195A) at different pH values. At pH 4.6 and 5.5 (PDB code: 3M61 and 4ZKN), the electron density of all residues and peptide bonds were intact, showing the peptide was not cleaved. At pH 7.4 (PDB code: 4ZKO), only Ala3, Arg4, Gly5, and Leu6, (P2, P1, P1', and P2') were observed, and the rest part of the peptide was completely disordered. Notably, at pH 7.4, the P1-P1' peptide bond was hydrolyzed, because the distance between the carbonyl carbon of Arg4 and the amino group of Gly5 was 3.5 Å, much longer than the normal peptide amide bond (approximately 1.3 Å). This was the first report of the P' side residue conformation after proteolysis, suggesting that the P' side product release can be slow in some cases. In this particular case, the slow release was due to cyclic nature of the peptide. For comparison, we crystalized upain-1-W3A in complex with the inactive huPA-S195A at pH 7.4, in which, the peptide was intact without cleavage. At pH 9.0 (PDB code: 4ZKR), the proteolysis was even more thorough, because we only observed Arg4 (P1) in the catalytic cleft of uPA, the rest part of the peptide was disordered. This study reveals a new mechanism of proteolysis by serine proteases.
Mupain-1, a versatile peptidic scaffold for serine protease inhibitors
The phage-displayed screening of murine uPA inhibitors
Based on peptide-display screening, we also isolated a peptide with the sequence of CPAYSRYLDC (Figure 3B), termed mupain-1, by using the bait of murine uPA (muPA) [68]. Like upain-1, mupain-1 is also a mono-cyclic peptide disulfide-bridged by Cys1 and Cys10. Mupain-1 inhibits muPA with a Ki value of 0.55 μM. The only positive residue, Arg6, was speculated as the P1 residue, because substituting Arg6 with Ala resulted in a >2000-fold declined affinity. Like upain-1, mupain-1 also exhibited excellent specificity over uPAs in different species and other 17 human and murine serine proteases.
Chemical modification improving inhibitory potencies
The P1 residues of inhibitors and substrates, which insert into the S1 specificity pockets of proteases, are considered critical to the binding affinity. The P1 residues of upain-1 and mupain-1 are Arg4 and Arg6, respectively, which formed strong salt bridges to the carboxyl side chain of Asp189. Mutating P1 arginines to alanine led to the complete loss of measurable affinity to uPAs [63, 68]. In the following study, we synthesized 15 non-natural arginine analogues in order to improve peptides' inhibitory potencies [69]. Together with naturally occurring L-arginine and L-alanine, we studied the influence of different P1 residues on the binding of upain-1 and mupain-1 to huPA and muPA. The original upain-2 (CSWRGLENHAAC) inhibits huPA with a Ki value of 35 μM. However, none of the upain-2 derivatives with other 16 P1 residues showed higher or even comparable potency. Similarly, substituting Arg6 of mupain-1 (CPAYSRYLDC) with most of the synthetic P1 residues led to impaired affinities. However, mupain-1 variants with L-4-Guanidino-phenylalanine (12) and L-3-(N-Amidino-4-piperidyl)alanine (16), referred to as mupain-1-12 and mupain-1-16 (Figure 3C and 3D), showed 2-fold (Ki = 0.28 μM) and 10-fold (Ki = 0.045 μM) enhanced affinity to muPA, respectively (Table 2). Notably, substitution with 12 or 16 also enhanced the originally non-measurable affinity to huPA (Ki > 1000 μM) to the affinities with Ki values in μM range.
Figure 3
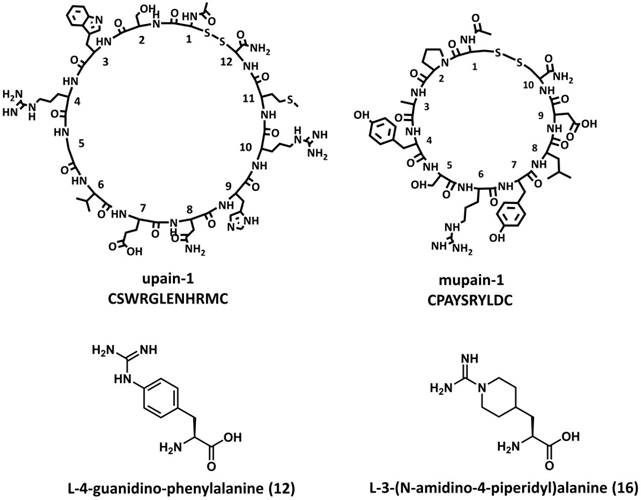
Chemical structures of upain-1, mupain-1, L-4-Guanidino-phenylalanine (12), and L-3-(N-Amidino-4-piperidyl)alanine (16).
Figure 4
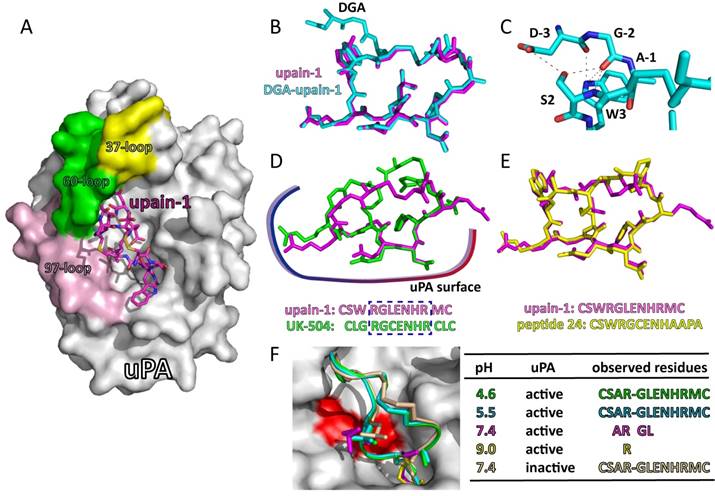
Structural features of upain-1 variants binding to huPA: A: the overview of the crystal structure of upain-1 (magenta sticks) in complex with the catalytic domain of human uPA (white surface) (PDB code: 2NWN), the solvent-exposed 37-, 60-, and 97-loops were colored yellow, green and light purple, respectively; B: the alignment of the N-terminus extended DGA-upain-1 (cyan sticks) and upain-1 (magenta sticks); C: the intramolecular hydrogen bond network between the extended DGA-tripeptide and the core upain-1 peptide; D: upain-1 (magenta sticks) and UK-504 (green sticks) form highly similar conformation on the surface of huPA, especially for the residues that form large contacts with huPA; E: upain-1 (magenta sticks) and bicyclic peptide 24 (yellow sticks) show mostly identical conformation when binding to huPA; F: upain-1-W3A was converted to a substrate of huPA. At pH 4.6 (green) and 5.5 (cyan), peptide was intact. At pH 7.4 (megenta), only electron density of Ala3, Arg4, Gly5, and Leu6 was observed, and the peptide bond between Arg4 and Gly5 was cleaved. At pH 9.0 (yellow), only electron density of Arg4 was observed, other residues of the peptide were disordered. At pH 7.4 (wheat), the inactive uPA did not cleave the peptide bond. Peptides at different pH were shown in cartoon, the side chains of Ala3, Arg4, Gly5, and Leu6 were shown in sticks, the residues that can be observed were listed in the table.
Table 2
Sequences and Ki values of inhibiting uPAs of mupain-1 variants.
| Peptide code | Sequence* | Ki values (μM) | ||
|---|---|---|---|---|
| muPA | huPA | huPA H99Y | ||
| mupain-1 | CPAYSRYLDC | 0.55 ± 0.08 (5) | > 1000 | 15.3 ± 0.2 (3) |
| mupain-1-12 | CPAYS[12]YLDC | 0.28 ± 0.02 (3) | 93 ± 19 (3) | 0.34 ± 0.06 (3) |
| mupain-1-16 | CPAYS[16]YLDC | 0.045 ± 0.01 (3) | 93 ± 18 (3) | 1.86 ± 0.74 (3) |
| mupain-1-IG | CPAYSRYIGC | 0.020 ± 0.004 (3) | 38 ± 23 (3) | 0.01 ± 0.001 (3) |
| mupain-1-12-IG | CPAYS[12]YIGC | 0.010 ± 0.002 (3) | 0.56 ± 0.12 (3) | 2.48 ± 0.07 (3) |
| mupain-1-16-IG | CPAYS[16]YIGC | 0.002 ± 0.0005 (3) | 0.53 ± 0.07 (3) | 0.01 ± 0.002 (3) |
| mupain-1-D9A | CPAYSRYLAC | 0.89 ± 0.02 (3) | > 1000 | 5.99 ± 0.43 (3) |
| mupain-1-12-D9A | CPAYS[12]YLAC | 0.19 ± 0.01 (3) | 11.7 ± 0.36 (3) | 0.19 ± 0.04 (3) |
| mupain-1-16-D9A | CPAYS[16]YLAC | 0.076 ± 0.003 (3) | 16.9 ± 1.8 (3) | 0.31 ± 0.01 (3) |
*All synthetic peptides are of an acetylated N-terminus and an amidated C-terminus, and are cyclized through the underlined residues.
Binding mode and inhibitory mechanism of mupain-1 derivatives
To illustrate the binding and inhibitory mechanisms of mupain-1 variants, we combined multiple biophysical and biochemical approaches, including crystallography, site-directed mutagenesis, NMR, ITC, SPR, etc [70]. For crystallization, we used a murinized huPA-H99Y mutant as a model and found that mupain-1 (PDB code: 4X1Q), mupain-1-12 (PDB code: 4X1R) and mupain-1-16 (PDB code: 4X1N) formed identical conformation on the catalytic cleft of uPA (Figure 5A), even though mupain-1-12 and mupain-1-16 showed obviously higher affinity (KD values: 1.21 and 0.84 vs. 14.1 μM according to SPR analysis). Crystal structures showed that peptides occupied the substrate-binding sites of uPA. Unlike the non-canonical substrate-like binding model of upain-1, mupain-1 forms a typical Laskowski conformation [71, 72]. The structure also explains why mupain-1 is not cleaved as a substrate (Figure 5B): the hydroxyl side-chain of Ser195 holds a distance of 3.91 Å to the carbonyl carbon of Arg6 (P1), which is unavailable for nucleophilic attack. Besides, the carbonyl oxygen of Arg6 does not correctly dock into the oxyanion hole. SPR analysis showed that the lower KD values of mupain-1-12 and mupain-1-16 were more dependent on the lower dissociation speed (koff) rather than the higher association speed (kon). The ITC data indicated that the higher affinities of mupain-1-12 and mupain-1-16 to muPA were due to the more favorable entropic changes induced by the substitution of non-natural P1 residues.
Optimizing mupain-1 derivatives by screening from a back-flip library
Alanine scanning analysis demonstrated that Leu8 and Asp9 had very minor contribution to the affinity to muPA. However, crystal structures showed that Leu8 and Asp9 are deeply embedded in the catalytic cleft of uPA. To screen the optimized peptide sequences, we established a “protease-peptide back-flip library” by expressing mupain-1 variants with randomized residues 8 and 9 (CPAYSRYXXC, X indicates the randomized natural residues except cysteine and proline) on the C-terminus of the catalytic domain of muPA (16-243) (Figure 5C) [73]. Between muPA and mupain-1, a linker of 14 amino acids containing two tobacco etch virus (TEV) protease cleaved sites were inserted, ensuring the sufficient flexibility for the peptide to flip back and dock into the active site of uPA. Thus, the muPA activity would decline resulting in a higher KM value of hydrolyzing chromogenic substrate. We measured the KM value of each muPA-peptide fusion protein and compared to that of muPA-mupain-1 fusion protein. Clones with higher KM values indicated the higher affinity of the fused peptides. To exclude the influence of the expression yield, clones with higher KM values were treated with the TEV protease afterwards. Clones with catalytic activity restored to that of muPA without peptide fusion were selected and sequenced. Among the screened sequences, a synthetic peptide with Ile8 and Gly9 (CPAYSRYIGC, termed mupain-1-IG) showed 0.02 μM Ki values of inhibiting muPA. To further improve potency, we substituted the P1 Arg6 of mupain-1-IG with non-natural amino acids, 12 and 16, leading to peptides mupain-1-12-IG and mupain-1-16-IG, which inhibited muPA with Ki values of 0.01 μM and 0.002 μM, respectively (Table 2).
Figure 5
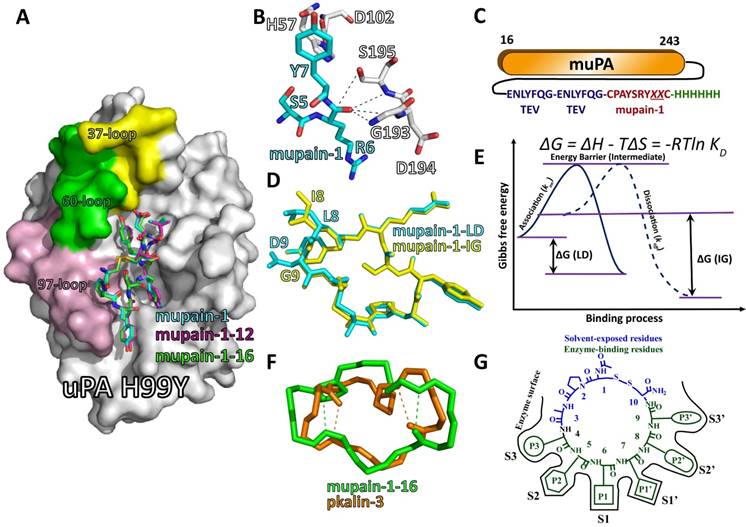
Structural features of mupain-1 variants binding to relevant proteases. A: the overview of the crystal structure of mupain-1 (cyan sticks), mupain-1-12 (magenta sticks), and mupain-1-16 (green sticks) in complexes with the catalytic domain of huPA-H99Y (white surface), the solvent-exposed 37-, 60-, and 97-loops were colored yellow, green and light purple, respectively; B: Arg6 (P1) showed a disordered non-proteolytic conformation in the catalytic region, key residues of mupain-1 and uPA were presented as cyan and white sticks, respectively. C: construction of the “protease-peptide back-flip library”. Mupain-1 variants with randomized reisudes 8 and 9 were fused in the C-terminus with a linker of two TEV protease cleaved sites. A 6xHis-tag was set for purification in the C-terminus of the fusion protein. D: the alignment of mupain-1-LD (cyan) and mupain-1-IG (yellow). E: Energetic changes of the LD-peptides and IG-peptides binding to uPA. F: the alignment of the backbones of mupain-1-16 (green) and pkalin-3 (brown), the tight β-turns were indicated as dash lines. G: The model for mupain-1 as the scaffold for developing serine protease inhibitors.
To our knowledge, mupain-1-16-IG is the most potent small-molecule inhibitor of uPAs among different species, only slightly weaker than two mono-clonal antibodies, mU1 and mU3.[74, 75] Unlike mupain-1-16, which has Ki values of 0.22 and 0.59 μM of inhibiting murine and human plasma kallikrein (PK), mupain-1-12-IG is much more specific for inhibiting many other serine proteases with Ki values in mM or sub-mM levels.
Improving peptides' potency and selectivity by increasing the flexibility
In contrast to upain-1 and many other peptide inhibitors, mupain-1-derived inhibitors are special because of the flexibility-dependent inhibitory potency, which challenges the traditional viewpoints of higher rigidity relevant to the higher affinity [73]. Inhibitors bind to serine proteases with entropic penalty. Thus, inhibitors with higher rigidity and reduced entropic penalty are favorable. The DGA-extended upain-1 and bicyclic peptides of Heinis are good examples. However, for mupain-1 variants, IG-peptides (peptides with Leu8 and Asp9) showed 20-25-fold enhanced affinity to muPA compared to LD-peptides (peptides with Ile8 and Gly9) with the same P1 residues. Similarly, another study showed that mutating Asp9 to Ala (CPAYS[P1]YLAC) also enhanced mupain-1's affinity to huPA, which conflicts the crystallographic data that mupain-1's carboxyl side-chain of Asp9 formed a salt bridge with the guanidino group of Arg35 in huPA. Both D9A-mutation and IG-substitution increased the flexibility but surprisingly enhanced the potency. ITC analysis demonstrated that both D9A-mutation and IG-substitution increased entropic penalty (TΔS)[70, 73], which, however, was compensated by the declined enthalpy changes (ΔH) finally leading to lower Gibbs free energy changes (ΔG) and lower KD values. Crystal structure of mupain-1-IG:huPA-H99Y (PDB code: 4ZHL) shows that mupain-1-IG bind to uPA (Figure 5D) with lower mobility (B-factor) than mupain-1 do, which is also consistent with the lower koff values determined via SPR analysis. To summarize, all the facts of the lower koff values (SPR), lower ΔH (ITC), and lower B-factor (crystallography) indicate that IG-peptides bind more tightly than LD-peptides do. Thus, we rationally conclude a unique feature of mupain-1-based peptides that the binding affinity can be improved by the increase of flexibility, opposite to the facts of upain-1 and Heinis's bicyclic peptides [52-54]. We hereby proposed a possible mechanism as described in Figure 5E: peptide and uPA needs to span an energy barrier to form the high-energy intermediate. The association energy (△Ga) is relevant to the association rate (kon) (△Ga=-RTlnKon). The peptide-uPA intermediate then transforms to final inhibitory complex with the release of energy (△Gd), which is also the energy needed for the complex dissociation (koff). The binding energy △G = △Ga - △Gd = -RT(lnKon/lnkoff) = RTln KD. Before binding, IG-peptides have higher entropy (S) and thus higher Gibbs free energy (G) in solution compared to LD-peptides. Thus, IG-peptides need lower △Ga to form intermediates indicating the higher kon values. On the other hand, the IG-peptide-uPA complexes are more stable and have lower energy because the higher flexibility results in higher adaptability. The IG-peptide-uPA complexes need higher energy for dissociation (△Gd) consistent with the lower koff, △H and B-factors. Thus, the lower association energy and higher dissociation energy results in lower △G and lower KD values. The flexibility-dependence also explains the fact that the affinity enhancement of IG-substitution is not specific to muPA but applicable to many other proteases, including plasmin, tPA, trypsin, etc.
Reengineering plasma kallikrein inhibitors with the scaffold of mupain-1[76]
One interesting feature of mupain-1 is the high versatility of engineering inhibitory potency to other homologous enzymes of uPA. Mupain-1 (CPAYSRYLDC) is a specific competitive inhibitor of muPA with a Ki value of 0.55 μM, while it shows no measurable inhibition of many other serine proteases, including plasma kallikrein (PK). However, substituting the P1 residue (Arg6) of mupain-1 with non-natural amino acid, L-3-(N-Amidino-4-piperidyl)alanine (16), resulted in peptide mupain-1-16 (CPAYS[16]YLDC), which not only showed a 10-fold enhanced affinity to muPA, but also inhibited human and murine PK (hPK, mPK) with measurable Ki values in μM range. We then wondered if it is possible to reengineer mupain-1-16 to a specific PK inhibitor. We superimposed the mupain-1-16 conformation from our crystal structure of mupain-1-16:huPA-H99Y complex (PDB code: 4X1N) to hPK (PDB code: 2AYN), and found that the peptide did not match well with the 37-, 97-, and 215-loops in terms of charge or van der Waals complementarity. To optimize the peptide-PK interactions at these sites, we modified residues 4, 5, 7, and 9 of mupain-1-16, resulting in peptide pkalin-3 (CPARF[16]ALWC), which inhibited hPK with a Ki value of 0.014 μM and showed high specificity over other serine proteases, including uPA. From mupain-1-16 to pkalin-3, we converted a specific uPA inhibitor to a specific PK inhibitor by just modifying 5 residues. Later, we confirmed our design strategy through crystallization, SPR analysis and site-directed mutagenesis.
The versatility of mupain-1
Small-molecule inhibitors often suffer from the lack of specificity because most of them target the S1 pockets, which are highly conserved among serine proteases [42, 56]. Peptide-based inhibitors contact the multiple specificity pockets, leading to higher selectivity [57]. The catalytic domains of serine proteases have common surface-exposed loops around residues 37, 60, 97, 110, 170, and 185, which are highly variable in length and sequence among different proteases [77]. These high variations can be identifiable features for the recognition of substrates, and can be the targets of engineering specificity of inhibitors. Mupain-1-derived inhibitors directly interact with 37-, 60-, and 97-loops (Figure 5A): the side-chains of residue 4 and 5 point to 97-loop, residues 7 and 9 stretch to 37-loop and 60-loop. Thus, residues 4, 5, 7, and 9 are likely to be critical for the selectivity of the peptide. These residues are just the changed non-P1 residues from mupain-1-16 to pkalin-3. However, even though upain-1 also forms large contacts with 37-, 60-, and 97-loops, our efforts on engineering affinity of upain-1 to other proteases failed. The reason of the versatility of mupain-1 is still unclear. One possible explanation could be that upain-1 binds uPA with a highly constrained conformation, mutating residues might lead to unfavorable conformational changes. In contrast, the potencies of mupain-1 derivatives are highly dependent on the high flexibility, which facilitate the backbone to adapt to the new situation when the residues are mutated. For mupain-1 variants, we enhanced the inhibitory potencies to muPA to a Ki value in the nano-molar range (Table 2) [73]. Besides, we also converted mupain-1 to inhibitors of PK and activated coagulation factor XI (fXIa) with Ki values in the low nM range (unpublished work). In addition, the affinity enhancement by modification of non-natural P1 residues only works in the case of mupain-1 mutants rather than that of upain-1 derivatives. Therefore, mupain-1, in contrast to upain-1, is a more interesting peptide scaffold with high versatility of engineering inhibitory potency to serine proteases.
Reengineering potencies to serine proteases with the scaffold of mupain-1
Here, we propose a principle for developing peptide inhibitors of serine proteases based on the scaffold of mupain-1. In crystal structures, all the peptides (mupain-1, mupain-1-12, mupain-1-16, mupain-1-IG, mupain-1-16-IG, mupain-1-D9A, and mupain-1-16-D9A) form mostly identical conformations in complexes with uPA although with highly variable Ki values. In addition, because of the constraint by two tight β-turns, the backbones of mupain-1 and pkalin-3 form largely similar conformations (Figure 5F) making the side chains of the two peptides stretch to the same sites of PK and uPA. Here, we nominate residues 1-3 and 10 as “solvent-exposed residues” because they have minor contacts with enzyme surface. In contrast, residues 4-9, which are deeply embedded into the catalytic cleft, are termed “enzyme-binding residues”. From mupain-1 (CPAYSRYLDC) to pkalin-3 (CPARF[16]ALWC), all the changed residues (residues 4-7 and 9) are “enzyme-binding residues”, suggesting that engineering the peptide-enzyme interactions should be focused on these “enzyme-binding residues”. Mupain-1 derivatives bind to proteases in a substrate-like conformation, in which residues 4-9 act as P3, P2, P1, P1', P2', and P3', respectively, and interact with the corresponding S and S' pockets of proteases (Figure 5G). Thus, to engineer the affinity, the “enzyme-binding residues” should be selected to match the properties of targeted protease surface, including optimizing the formation of salt bridges, hydrophobic interactions, etc. However, structure-based rational design cannot handle the entropy term very well and thus should be complemented with stochastic approaches, e.g. screening inhibitors from libraries with mupain-1 mutants with randomized residues 4-9. The back-flip peptide-protease fusion library mentioned above is one such successful example.
Conclusion
In this review, we summarized our studies on the mono-cyclic peptides targeting serine proteases. upain-1 (CSWRGLENHRMC) and mupain-1 (CPAYSRYLDC) are two peptidic inhibitors of huPA and muPA screened from a phage-displayed peptide library. Based on these two peptides, we generated uPA inhibitors with potency and specificity comparable to mono-clonal antibodies. Furthermore, we revealed the high versatility of mupain-1 as a scaffold for developing inhibitors of other serine proteases. We then generated PK and fXIa inhibitors with Ki values in low nM levels. Since serine proteases are potent therapeutic targets of many important diseases, in future, in addition to continuously generating inhibitors against more proteases, we will also evaluate the therapeutic efficacies of these inhibitors in corresponding disease animal models.
Acknowledgements
This work is in memory of Professor Peter A. Andreasen of Aarhus University, who passed away on Nov 15, 2016. He conceived and led this project, and made major contributions to this series of works. This work was carried out in the consortium of Danish-Chinese Center for Proteases and Cancer, supported by grants from Natural Science Foundation of China (21708043, 30811130467, 31161130356, 31370737, 31400637, 31570745, 31670739, 81572944, U1405229), and Danish National Research Foundation Grant 26-331-67. PAA was awarded Chinese Academy of Sciences visiting professorship for senior international scientist (Grant No 2012T1G0023). MH was awarded an Aarhus University Research Foundation visiting professorship. This work is also supported by the Strategic Priority Research Program of the CAS (XDA09030307), and the CAS/SAFEA International Partnership Program for Creative Research Teams.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lipinski CA. Lead- and drug-like compounds: the rule-of-five revolution. Drug discovery today Technologies. 2004;1:337-41
2. Duffy FJ, Devocelle M, Shields DC. Computational Approaches to Developing Short Cyclic Peptide Modulators of Protein-Protein Interactions. In: (ed.) Zhou P, Jiang H. Computational Peptidology. New York: Springer New York. 2015:241-71
3. Conti-Fine BM, Diethelm-Okita B, Ostlie N, Wang W, Milani M. Immunopathogenesis of Myasthenia Gravis. New Jersey, USA: Humana Press. 2003
4. Giroux R. Cyclosporine. Chem Eng News. 2005;83:56 -
5. Turk B. Targeting proteases: successes, failures and future prospects. Nat Rev Drug Discov. 2006;5:785-99
6. Puente XS, Ordóñez GR, López-Otín C. Protease Genomics and the Cancer Degradome. New York, USA: Springer New York. 2008
7. Cudic M, Fields GB. Extracellular Proteases as Targets for Drug Development. Curr Protein Pept Sc. 2009;10:297-307
8. Crippa MP. Urokinase-type plasminogen activator. The international journal of biochemistry & cell biology. 2007;39:690-4
9. Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Bio. 2010;11:23-36
10. Kasai S, Arimura H, Nishida M, Suyama T. Proteolytic Cleavage of Single-Chain Pro-Urokinase Induces Conformational Change Which Follows Activation of the Zymogen and Reduction of Its High-Affinity for Fibrin. J Biol Chem. 1985;260:2377-81
11. Vassalli JD, Baccino D, Belin D. A Cellular-Binding Site for the Mr 55,000 Form of the Human-Plasminogen Activator, Urokinase. J Cell Biol. 1985;100:86-92
12. Appella E, Robinson EA, Ullrich SJ, Stoppelli MP, Corti A, Cassani G. et al. The Receptor-Binding Sequence of Urokinase - a Biological Function for the Growth-Factor Module of Proteases. J Biol Chem. 1987;262:4437-40
13. Ploug M, Ellis V. Structure-function relationships in the receptor for urokinase-type plasminogen activator. Comparison to other members of the Ly-6 family and snake venom alpha-neurotoxins. FEBS letters. 1994;349:163-8
14. Huai Q, Mazar AP, Kuo A, Parry GC, Shaw DE, Callahan J. et al. Structure of human urokinase plasminogen activator in complex with its receptor. Science. 2006;311:656-9
15. Steffens GJ, Gunzler WA, Otting F, Frankus E, Flohe L. The complete amino acid sequence of low molecular mass urokinase from human urine. Hoppe-Seyler's Zeitschrift fur physiologische Chemie. 1982;363:1043-58
16. Macfarlane RG, Pilling J. Observations on Fibrinolysis - Plasminogen, Plasmin, and Antiplasmin Content of Human Blood. Lancet. 1946;251:562-5
17. Camiolo SM, Thorsen S, Astrup T. Fibrinogenolysis and Fibrinolysis with Tissue Plasminogen Activator, Urokinase, Streptokinase-Activated Human Globulin, and Plasmin. P Soc Exp Biol Med. 1971;138:277
18. Lijnen HR. Elements of the fibrinolytic system. Ann Ny Acad Sci. 2001;936:226-36
19. Foley JH. Plasmin(ogen) at the Nexus of Fibrinolysis, Inflammation, and Complement. Semin Thromb Hemost. 2017;43:135-42
20. Schafer BM, Maier K, Eickhoff U, Todd RF, Kramer MD. Plasminogen Activation in Healing Human Wounds. Am J Pathol. 1994;144:1269-80
21. Andreasen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci. 2000;57:25-40
22. Law RHP, Abu-Ssaydeh D, Whisstock JC. New insights into the structure and function of the plasminogen/plasmin system. Curr Opin Struc Biol. 2013;23:836-41
23. Boyle MDP, Lottenberg R. Plasminogen activation by invasive human pathogens. Thromb Haemostasis. 1997;77:1-10
24. van Gorp EC, Suharti C, ten Cate H, Dolmans WM, van der Meer JW, ten Cate JW. et al. Review: infectious diseases and coagulation disorders. The Journal of infectious diseases. 1999;180:176-86
25. Lahteenmaki K, Edelman S, Korhonen TK. Bacterial metastasis: the host plasminogen system in bacterial invasion. Trends Microbiol. 2005;13:79-85
26. Teesalu T, Kulla A, Asser T, Koskiniemi M, Vaheri A. Tissue plasminogen activator as a key effector in neurobiology and neuropathology. Biochem Soc T. 2002;30:183-9
27. Andreasen PA, Kjoller L, Christensen L, Duffy MJ. The urokinase-type plasminogen activator system in cancer metastasis: A review. Int J Cancer. 1997;72:1-22
28. Thorsen S, Bangert K. Plasminogen and plasmin inhibitor: a review of their biosynthesis, biochemistry and role in fibrinolysis. Int Congr Ser. 1997;1129:47-56
29. Kwaan HC. From Fibrinolysis to the Plasminogen-Plasmin System and Beyond: A Remarkable Growth of Knowledge, with Personal Observations on the History of Fibrinolysis. Semin Thromb Hemost. 2014;40:585-91
30. Duffy MJ, McGowan PM, Gallagher WM. Cancer invasion and metastasis: changing views. The Journal of pathology. 2008;214:283-93
31. Wiman B, Collen D. On the mechanism of the reaction between human alpha 2-antiplasmin and plasmin. J Biol Chem. 1979;254:9291-7
32. Wyganowska-Swiatkowska M, Surdacka A, Skrzypczak-Jankun E, Jankun J. The plasminogen activation system in periodontal tissue (Review). International journal of molecular medicine. 2014;33:763-8
33. Sprengers ED, Kluft C. Plasminogen-Activator Inhibitors. Blood. 1987;69:381-7
34. Kruithof EK. Plasminogen activator inhibitors-a review. Enzyme. 1988;40:113-21
35. Chorostowska-Wynimko J, Skrzypczak-Jankun E, Jankun J. Plasminogen activator inhibitor type-1: its structure, biological activity and role in tumorigenesis (Review). International journal of molecular medicine. 2004;13:759-66
36. Foekens JA, Peters HA, Look MP, Portengen H, Schmitt M, Kramer MD. et al. The urokinase system of plasminogen activation and prognosis in 2780 breast cancer patients. Cancer Res. 2000;60:636-43
37. Duffy MJ. Urokinase-type plasminogen activator: a potent marker of metastatic potential in human cancers. Biochem Soc T. 2002;30:207-10
38. Cubellis MV, Nolli ML, Cassani G, Blasi F. Binding of single-chain prourokinase to the urokinase receptor of human U937 cells. J Biol Chem. 1986;261:15819-22
39. Ellis V, Pyke C, Eriksen J, Solberg H, Dano K. The urokinase receptor: involvement in cell surface proteolysis and cancer invasion. Annals of the New York Academy of Sciences. 1992;667:13-31
40. Tang LL, Han XZ. The urokinase plasminogen activator system in breast cancer invasion and metastasis. Biomed Pharmacother. 2013;67:179-82
41. Misheneva VS. [Effect of Some Inhibitors on the Urokinase Activity of the Liver of Normal and Tumor-Bearing Animals]. Ukrains'kyi biokhimichnyi zhurnal. 1963;35:566-72
42. Rockway TW, Nienaber V, Giranda VL. Inhibitors of the protease domain of urokinase-type plasminogen activator. Curr Pharm Design. 2002;8:2541-58
43. Mack GS, Marshall A. Lost in migration. Nat Biotechnol. 2010;28:214-29
44. Setyono-Han B, Sturzebecher J, Schmalix WA, Muehlenweg B, Sieuwerts AM, Timmermans M. et al. Suppression of rat breast cancer metastasis and reduction of primary tumour growth by the small synthetic urokinase inhibitor WX-UKI. Thromb Haemostasis. 2005;93:779-86
45. Nienaber VL, Davidson D, Edalji R, Giranda VL, Klinghofer V, Henkin J. et al. Structure-directed discovery of potent non-peptidic inhibitors of human urokinase that access a novel binding subsite. Struct Fold Des. 2000;8:553-63
46. Mackman RL, Katz BA, Breitenbucher JG, Hui HC, Verner E, Luong C. et al. Exploiting subsite S1 of trypsin-like serine proteases for selectivity: Potent and selective inhibitors of urokinase-type plasminogen activator. J Med Chem. 2001;44:3856-71
47. Zhu M, Gokhale VM, Szabo L, Munoz RM, Baek H, Bashyam S. et al. Identification of a novel inhibitor of urokinase-type plasminogen activator. Mol Cancer Ther. 2007;6:1348-56
48. Schweinitz A, Steinmetzer T, Banke IJ, Arlt MJE, Sturzebecher A, Schuster O. et al. Design of novel and selective inhibitors of urokinase-type plasminogen activator with improved pharmacokinetic properties for use as antimetastatic agents. J Biol Chem. 2004;279:33613-22
49. Henneke I, Greschus S, Savai R, Korfei M, Markart P, Mahavadi P. et al. Inhibition of Urokinase Activity Reduces Primary Tumor Growth and Metastasis Formation in a Murine Lung Carcinoma Model. Am J Resp Crit Care. 2010;181:611-9
50. Joossens J, Van der Veken P, Lambeir AM, Augustyns K, Haemers A. Development of irreversible diphenyl phosphonate inhibitors for urokinase plasminogen activator. J Med Chem. 2004;47:2411-3
51. Vangestel C, Thomae D, Van Soom J, Ides J, Wyffels L, Pauwels P. et al. Preclinical evaluation of [111 In]MICA-401, an activity-based probe for SPECT imaging of in vivo uPA activity. Contrast media & molecular imaging. 2016
52. Chen SY, Rebollo IR, Buth SA, Morales-Sanfrutos J, Touati J, Leiman PG. et al. Bicyclic Peptide Ligands Pulled out of Cysteine-Rich Peptide Libraries. J Am Chem Soc. 2013;135:6562-9
53. Angelini A, Cendron L, Chen SY, Touati J, Winter G, Zanotti G. et al. Bicyclic Peptide Inhibitor Reveals Large Contact Interface with a Protease Target. Acs Chem Biol. 2012;7:817-21
54. Chen SY, Gopalakrishnan R, Schaer T, Marger F, Hovius R, Bertrand D. et al. Dithiol amino acids can structurally shape and enhance the ligand-binding properties of polypeptides. Nat Chem. 2014;6:1009-16
55. Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295:2387-92
56. Drag M, Salvesen GS. Emerging principles in protease-based drug discovery. Nat Rev Drug Discov. 2010;9:690-701
57. Fergal J. Duffy MD, Denis C. Shields. Computional Approaches to Developing Short Cyclic Peptide Modulators of Protein-Protein Interactions. In: (ed.) Peng Zhou JH. Computational Peptidology. New York: Springer New York. 2015:241-71
58. Cwirla SE, Peters EA, Barrett RW, Dower WJ. Peptides on Phage - a Vast Library of Peptides for Identifying Ligands. P Natl Acad Sci USA. 1990;87:6378-82
59. Lowman HB. Bacteriophage display and discovery of peptide leads for drug development. Annu Rev Bioph Biom. 1997;26:401-24
60. Smith GP. Filamentous Fusion Phage - Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science. 1985;228:1315-7
61. Koivunen E, Wang BC, Dickinson CD, Ruoslahti E. Peptides in Cell-Adhesion Research. Method Enzymol. 1994;245:346-69
62. Petersen HH, Hansen M, Schousboe SL, Andreasen PA. Localization of epitopes for monoclonal antibodies to urokinase-type plasminogen activator - Relationship between epitope localization and effects of antibodies on molecular interactions of the enzyme. Eur J Biochem. 2001;268:4430-9
63. Hansen M, Wind T, Blouse GE, Christensen A, Petersen HH, Kjelgaard S. et al. A urokinase-type plasminogen activator-inhibiting cyclic peptide with an unusual P-2 residue and an extended protease binding surface demonstrates new modalities for enzyme inhibition. J Biol Chem. 2005;280:38424-37
64. Zhao GX, Yuan C, Wind T, Huang ZX, Andreasen PA, Huang MD. Structural basis of specificity of a peptidyl urokinase inhibitor, upain-1. J Struct Biol. 2007;160:1-10
65. Lin Z, Jiang L, Yuan C, Jensen JK, Zhang X, Luo Z. et al. Structural basis for recognition of urokinase-type plasminogen activator by plasminogen activator inhibitor-1. J Biol Chem. 2011;286:7027-32
66. Jiang LG, Svane ASP, Sorensen HP, Jensen JK, Hosseini M, Chen Z. et al. The Binding Mechanism of a Peptidic Cyclic Serine Protease Inhibitor. J Mol Biol. 2011;412:235-50
67. Roodbeen R, Paaske B, Jiang LG, Jensen JK, Christensen A, Nielsen JT. et al. Bicyclic Peptide Inhibitor of Urokinase-Type Plasminogen Activator: Mode of Action. Chembiochem. 2013;14:2179-88
68. Andersen LM, Wind T, Hansen HD, Andreasen PA. A cyclic peptidylic inhibitor of murine urokinase-type plasminogen activator: changing species specificity by substitution of a single residue. Biochem J. 2008;412:447-57
69. Hosseini M, Jiang LG, Sorensen HP, Jensen JK, Christensen A, Fogh S. et al. Elucidation of the Contribution of Active Site and Exosite Interactions to Affinity and Specificity of Peptidylic Serine Protease Inhibitors Using Non-Natural Arginine Analogs. Mol Pharmacol. 2011;80:585-97
70. Zhao BY, Xu P, Jiang LG, Paaske B, Kromann-Hansen T, Jensen JK. et al. A Cyclic Peptidic Serine Protease Inhibitor: Increasing Affinity by Increasing Peptide Flexibility. Plos One. 2014:9
71. Laskowski M Jr, Kato I. Protein inhibitors of proteinases. Annual review of biochemistry. 1980;49:593-626
72. Laskowski M, Qasim MA. What can the structures of enzyme-inhibitor complexes tell us about the structures of enzyme substrate complexes? Bba-Protein Struct M. 2000;1477:324-37
73. Sorensen HP, Xu P, Jiang LG, Kromann-Hansen T, Jensen KJ, Huang MD. et al. Selection of High-Affinity Peptidic Serine Protease Inhibitors with Increased Binding Entropy from a Back-Flip Library of Peptide-Protease Fusions. J Mol Biol. 2015;427:3110-22
74. Liu Z, Kromann-Hansen T, Lund IK, Hosseini M, Jensen KJ, Hoyer-Hansen G. et al. Interconversion of Active and Inactive Conformations of Urokinase-Type Plasminogen Activator. Biochemistry-Us. 2012;51:7804-11
75. Kromann-Hansen T, Lund IK, Liu Z, Andreasen PA, Hoyer-Hansen G, Sorensen HP. Allosteric Inactivation of a Trypsin-Like Serine Protease by An Antibody Binding to the 37-and 70-Loops. Biochemistry-Us. 2013;52:7114-26
76. Xu P, Xu M, Jiang L, Yang Q, Luo Z, Dauter Z. et al. Design of Specific Serine Protease Inhibitors Based on a Versatile Peptide Scaffold: Conversion of a Urokinase Inhibitor to a Plasma Kallikrein Inhibitor. J Med Chem. 2015;58:8868-76
77. Perona JJ, Craik CS. Structural Basis of Substrate-Specificity in the Serine Proteases. Protein Sci. 1995;4:337-60
Author contact
![]() Corresponding author: Mingdong Huang, 155 Yangqiao Road West, Fuzhou, Fujian 350002, China, Tel: +86 591 8370 5814. Email: mhuangac.cn
Corresponding author: Mingdong Huang, 155 Yangqiao Road West, Fuzhou, Fujian 350002, China, Tel: +86 591 8370 5814. Email: mhuangac.cn
Citation styles
APA
Xu, P., Andreasen, P.A., Huang, M. (2017). Structural Principles in the Development of Cyclic Peptidic Enzyme Inhibitors. International Journal of Biological Sciences, 13(10), 1222-1233. https://doi.org/10.7150/ijbs.21597.
ACS
Xu, P.; Andreasen, P.A.; Huang, M. Structural Principles in the Development of Cyclic Peptidic Enzyme Inhibitors. Int. J. Biol. Sci. 2017, 13 (10), 1222-1233. DOI: 10.7150/ijbs.21597.
NLM
Xu P, Andreasen PA, Huang M. Structural Principles in the Development of Cyclic Peptidic Enzyme Inhibitors. Int J Biol Sci 2017; 13(10):1222-1233. doi:10.7150/ijbs.21597. https://www.ijbs.com/v13p1222.htm
CSE
Xu P, Andreasen PA, Huang M. 2017. Structural Principles in the Development of Cyclic Peptidic Enzyme Inhibitors. Int J Biol Sci. 13(10):1222-1233.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.