Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(10):1329-1340. doi:10.7150/ijbs.19752 This issue Cite
Research Paper
Erythropoietin Reduces Insulin Resistance via Regulation of Its Receptor-Mediated Signaling Pathways in db/db Mice Skeletal Muscle
Yu Pan1, Xiu Hong Yang2, Li Li Guo3, Yan Hong Gu2, Qing Yan Qiao2, Hui Min Jin2 ![]()
1. Division of Nephrology, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China;
2. Division of Nephrology, Shanghai Pudong Hospital, Fudan University Pudong Medical Center, Shanghai, China;
3. Hemodialysis Center, Baoshan Branch of Shanghai No.1 People's Hospital, Shanghai, China.
Received 2017-2-21; Accepted 2017-8-8; Published 2017-10-17
Abstract

Erythropoietin (EPO) can reduce insulin resistance (IR) in adipocytes; however, it is unknown whether EPO can decrease IR in skeletal muscle. Here we investigated whether EPO could reduce IR in type 2 diabetic mouse skeletal muscle and its possible signaling mechanisms of action. Twelve-week-old db/db diabetic mice were employed in this study. Systemic use of EPO improved glucose profiles in type 2 diabetic mice after 4 and 8 weeks treatment. EPO up-regulated EPOR protein expression in skeletal muscle, and subsequently activated downstream signaling molecules such as JAK2, IRS-1, PI3K, AKT, and eNOS. We next constructed lentivirally-delivered shRNAs against EPOR and transfected skeletal muscle cells to knockdown EPOR. EPOR knockdown inhibited EPO induced JAK2, IRS-1, PI3K, AKT, eNOS signaling transduction, autophagy and Glut 4 translocation, as well as promoted apoptosis in skeletal muscle. Thus, EPO reduces skeletal muscle IR in type 2 diabetic mice via its specific receptor, EPOR. Possible mechanisms involved in its action may include increased autophagy and reduced apoptosis in type 2 diabetic skeletal muscles, which provides a new strategy for the treatment of IR.
Keywords: Erythropoietin (EPO), Erythropoietin receptor EPOR, Insulin resistance (IR), Skeletal muscle, type 2 diabetes, Insulin signaling.
Introduction
Type 2 diabetes mellitus is highly prevalent worldwide, and is characterized by insulin resistance (IR). IR occurs most commonly within peripheral adipocytes and skeletal muscle 1, and recently, these tissues have been of research interest due to the increasing importance of strategically targeting IR for the therapeutic prevention of type 2 diabetes.
Erythropoietin (EPO) is a glycoprotein produced during fetal development within kidney and liver tissue, and has been widely used in the clinic to treat anemia induced by chronic kidney disease, tumors and other diseases. EPO primarily activates and promotes erythroid cell proliferation and growth via its specific cell surface receptor, EPOR. However, many studies suggest EPO is also active locally in many other tissues, including cardiovascular, renal, endothelium, nerves, and muscle in response to physical and metabolic stress2-7. In non-hematopoietic cells, however, it remains controversial whether EPO exerts its effect by binding EPOR. In our previous research8, we confirmed that EPOR is expressed in 3T3-L1 adipocytes, and EPO improved IR in cultured adipocytes via EPOR. Blockade of EPOR with antagonists or siRNA completely reversed IR decrease induced by EPO, indicating EPO reduces IR via EPOR mediated signaling cascades in adipocytes.
The role and regulatory processes involving EPO and EPOR in skeletal muscle remain to be elucidated. Part of the difficulties in ascertaining this information is due to inconsistent results with respect to prior in vitro and in vivo studies. Evidence suggests C2C12 myoblasts show elevated JAK2, STAT5 and AKT phosphorylation when treated with EPO, similar to signaling responses observed in neural cells9, 10. Interestingly, STAT5 activation was not observed in rat skeletal muscle tissue in response to EPO stimulation11. In human skeletal muscle, acute EPO administration failed to induce phosphorylation and activation of STAT5, AKT and MAPK signaling pathways12. Discrepancies in the measurement of EPOR mRNA and protein levels may be related to potential species differences and inherent potential variability in in vitro and in vivo models. Using primary human muscle cells and examining mouse muscle in vivo will aid in clarifying these inconsistencies. Depending on this background information, we designed and executed experiments using db/db mice, a type 2 diabetic mouse model, to explore whether EPO therapy can reduce skeletal muscle IR, and elucidate potential signaling pathways that may be involved in the mechanism of EPO-EPOR signaling in type 2 diabetes skeletal muscle.
Materials and Methods
Animal model and in vivo experimental design
Twelve-week-old db/db mice and background control mice (db/m) were purchased from SLAC Laboratories (Shanghai, China). All mice were housed under controlled light (12 h light/12 h dark) and temperature conditions, and had ad libitum access to food (normal rodent chow) and water. All procedures were conducted in accordance with the guidelines of the Chinese Council on Animal Care and were approved by Shanghai Jiao Tong University Animal Care Committee.
All experiments involving db/db mice were performed at 12 weeks of age following determination that glucose concentrations in fasting blood samples indicated the mice were diabetic. Mice were divided into the following treatment groups (n=10/each, 5 male and 5 female): (1) non-treated db/db (treated with 0.9% saline), (2) EPO (0.4μg/kg/w) (recombinant human Epoetin alpha, 3SBIO, China), and (3) EPO (1.2μg/kg/w). Saline and EPO were administered to mice once a weekfor8 week via subcutaneous injection.
Fasting glycemia, glucose tolerance (IGTT) and insulin tolerance (ITT insulin) tests
Mice were subjected to glucose tolerance and insulin tolerance tests at days 0, 28 and 56. Briefly, mice were fasted for 15h, and a basal blood sample (20 µl) was collected from the tail vein (t=0). Serum glucose levels were determined using the One Touch Basic glucose meter (Lifescan Canada, Burnaby, British Columbia, Canada). Next, a glucose tolerance test was performed after mice were fasted overnight. Briefly, mice were injected intraperitoneally with 1g/kg glucose. Serum glucose was quantified in tail blood samples collected at 0 (prior to glucose administration), 30, 60, 90 and 120 min after glucose administration. An insulin tolerance tests was performed 3 days following tests for glucose tolerance. All mice with free access to water and food were injected intraperitoneally with insulin (1 IU/kg) and serum glucose was quantified in tail blood samples collected at 0 (prior to insulin administration), 30, 60, 90 and 120 min after insulin administration. Plasma insulin was assayed using an ELISA kit (Alpco, Salem, USA).
Muscle NO level measurement
Muscle NO levels were measured using a 280i Nitric Oxide Analyzer (GE Analytical Instruments), according to the manufacturer's instructions. In brief, ice-cold ethanol was added into muscle homogenate samples at a ratio of 2:1. The mixture was kept at 0°C for 30 min and then centrifuged at 14,000 RPM for 5 min. The supernatant was then used for NO analysis based on a gas-phase chemiluminescent reaction between NO and ozone.
Periodic Acid-Schiff staining and immunofluorescence
Mice were anesthetized and hind legs gastrocnemius muscles were immediately dissected. Tissues were then fixed in 4% buffered paraformaldehyde for 2 days, embedded in paraffin and processed for sectioning. Tissue sections (3µm) were assessed by Periodic Acid-Schiff staining. Immunofluorescence analyses of mouse muscle sections were accomplished with DAPI labeling of cell nuclei, and specific antibody according to standard protocols. Briefly, sections were incubated overnight at 4°C with rabbit anti-EPOR antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA).After extensive washes, sections were then incubated with anti-rabbit FITC-conjugated secondary antibody (DAKO, Carpinteria, CA, USA) for 30 min. Following labeling and coverslip mounting, images were acquired using a Nikon (ECLIPSE-E1000) microscope connected to a video camera equipped with a color monitor and ImagePlus software.
Preparation of primary muscle cells
Primary muscle cells were obtained from lower hind limb muscles of adult db/db animals (10 week old) using a protocol modified from previous description13. In brief, The muscles were excised from the lower limbs of mice, thoroughly minced and digested. And next the cells were collected via a 5-minute centrifugation at 1800g. The collected cells were exposed to trypsin-EDTA (solarbio) (0.25% with 1mM EDTA) at 37°C for 10 minutes. Fibroblasts adhere to uncoated culture dishes more readily than muscle cells, so fibroblast contamination was minimized by preplating the cells onto 100 mm tissue culture dishes for 3 to 4 hours at 37°C. The unattached cells were centrifuged at 1800g for 5 minutes, suspended in culture medium, plated onto dishes coated with 0.1% gelatin, and incubated at 37°C overnight. The attached cells were trypsinized, suspended in culture medium, and plated at 1.5 to 2×105 cells/mL in culture dishes (6-well or 24-well plates). Primary myotubes were used 4 to 5 days following differentiation induction when most cells were multinucleated, and then the cells were treated with LV-shRNA EPOR.
shRNA delivery via lentivirus in skeletal muscle cells
Paired oligonucleotides targeting human EPOR gene were synthesized and annealed into a linear pLKO lentiviral vector digested by EcoRI and AgeI. The recombined vector, identified by double digest with EcoRI and NdeI and DNA sequencing, was packaged in 293T cells together with psPAX2 and pMD2.G. Virus present in in culture supernatant was concentrated, three recombinant vectors (shRNA EPOR-1: 5'-CAGCCUACAACGUCUUGAA-3'; shRNA EPOR-2: 5'-CAGCCUACAACGUCUUGAA-3'; shRNA EPOR-3:5'-CACCGCAUCAUCCAUAUCA-3') and a control vectors (5'-TAGAAGGCACAGTCGAGG-3') were transfected into skeletal muscle cells, and the constitutive EPOR knockdown cells were selected by puromycin. The efficiency of RNA interference was detected by real- time PCR and Western blotting. Empty plasmid packing lentivirus as a control (LV-vehicle).
2-deoxy-d [3H] glucose (2-DG) uptake and GLUT4 translocation
Cell culture medium was replaced with Krebs-Ringer-phosphate buffer (25 mmol/l HEPES-NaOH [pH 7.4], 1.3mmol/l of CaCl2, 120 mmol/l of NaCl, 5 mmol/l of KCl, 1.3 mmol/l of KH2PO4, and 1.2 mmol/l of MgSO4) for 45 min at 37ºC. Cells were then either treated or not treated with 1 μmol/l of insulin for 20 min, and cellular 2-deoxy-d [3H] glucose (2-DG) uptake was measured as described previously8. GLUT4 translocation in the cells was analyzed with and without exposure to 2 μmol/l of insulin for 30 min. Next, membrane and cytosolic proteins were extracted using a Membrane Protein Extraction Reagent (Thermo Fisher, Waltham, MA, USA). Extracted proteins were used for Western blot analysis.
mRNA Isolation and real-time PCR
Gastrocnemius muscle tissues were isolated from db/db mice (4 and 8 week treatment). Total RNA from muscle tissue and cells was isolated using Trizol reagent (Invitrogen) according to manufacturer's instruction. Total RNA was quantified by absorbance at 260 nm. EPOR Primer F 5' CCGTGCGTTTCTGGTGTTC 3', Primer R 5' ATGCGGTGATAGCGAGGAG 3' was used to detect EPOR transcripts. mRNA was extracted using the RNAeasy mini kit (QIAGEN Science, Germantown, MD) and cDNA was synthesized from 1.5μg RNA with the first-strand cDNA synthesis kit (Amersham, Buckmgahamshire, UK). Each sample was run and analyzed in triplicate. Real-time PCR data were analyzed using the 2-ΔΔCT method with the SDS Software package (Applied Biosystems).
Western blot analysis
Western blot analysis was performed as previously described8. In brief, total protein samples extracted from untreated and treated tissues or cells were prepared using RIPA buffer containing a protease inhibitor cocktail (Roche, Branford, CT, USA) and a phosphatase inhibitor and were subjected to SDS PAGE (90μg protein per sample). The membranes were then incubated with the following primary antibodies: p-IRS-1 (Cell Signaling Inc. (CST), Danvers, MA, USA,1:800), eNOS (CST,1:1,000), p-eNOS (Thermo Fisher,1:800), IRS-1 (CST,1:1,000), p-AKT (CST,1:1000), AKT (CST,1:1000), p-PI3K (Abcam, Cambridge, MA, USA,1:500), PI3K(Abcam1:1000),GAPDH (CST, 1:1,500), anti-LC3 (1:200, Sigma-Aldrich, St. Louis, MO, USA), Beclin1 (1:200, Sigma-Aldrich), GLUT4 (1:3,000, Thermo Fisher), p-JAK2 and JAK2(CST,1:1000).
Statistical analysis
All data shown are presented as the mean ± SD of three or more independent experiments. Differences were considered statistically significant if p<0.05, as assessed using a Student's t test (for paired samples) or the one-way ANOVA (for more than two groups).
Results
rhEPOimproved glucose intolerance in db/db mice via unregulated EPOR/IRS-1/AKT/PI3K signaling
To test whether rhEPO could improve mouse glucose intolerance, db/dbmice were given subcutaneous injections of 0.4μg/kg/w or 1.2μg/kg/wrhEPO for 8 weeks. Results showed no significance in body weight of db/db mice treated with rhEPO compared to that of untreated db/db mice (Figure 1A, B). Hematocrit level was not elevated following 0.4μg/kg/w or1.2μg/kg/w rhEPO treatment.
Effect of EPO on body weight, plasma eNOS level and EPOR expression in db/db mice. Body weight of mice in db/m and db/dbgroupafter treatment with rhEPO for 4 and 8 weeks (A-B) (N=10in each group). Muscle No concentration in db/m and db/dbgroupafter treatment with rhEPO for 4 and 8 weeks measuring by a 280i Nitric Oxide Analyzer(C-D) (N=10in each group). Relative EPOR mRNA level in db/m and db/dbgroupafter treatment with rhEPO for 8 weeks by using real-time PCR. (E) Skeletal muscle Periodic Acid-Schiff staining and EPOR expression by immunofluorescence analysis treated with or without 14μgrhEPOfor 8weeks in db/dbmice (F). White arrows represent abundant EPOR expression in the db/m and rhEPO treated db/db skeletal muscle. Scale bar: 50μm.
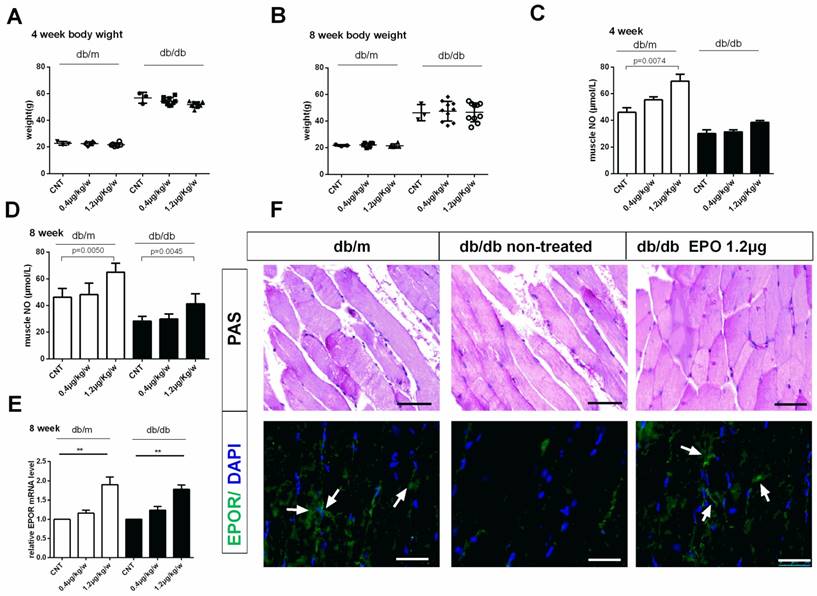
Insulin has the ability to stimulate the production of nitric oxide (NO) which is important for insulin-stimulated glucose disposal14. To clear whether EPO reduced insulin resistance through mediating the nitric oxide, we have examined the muscle NO level after rhEPO treatment for 4 and 8 weeks (Figure 1 C, D). Muscle NO increased after 8 weeks of 1.2μg/kg/w rhEPO treatment and rhEPO administration increased EPOR expression in skeletal muscle (Figure 1E, F).
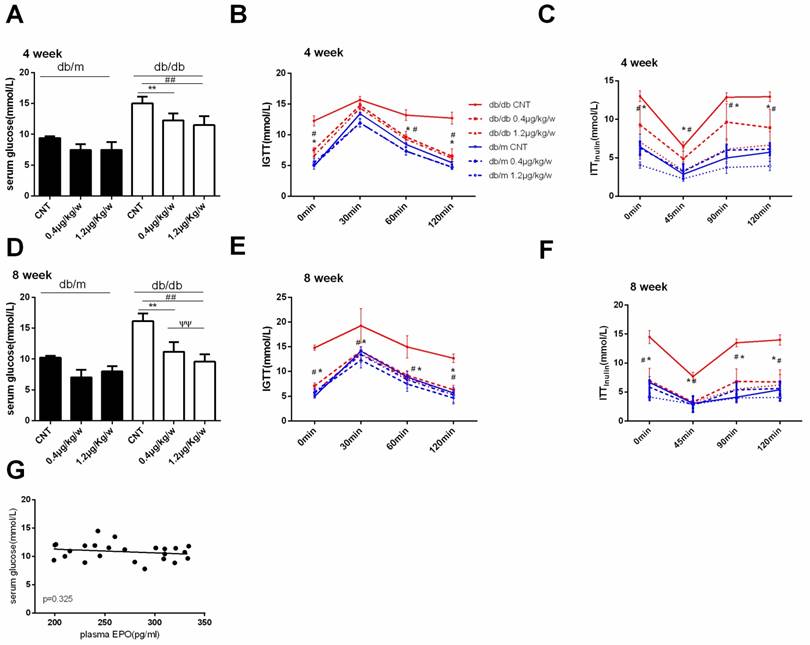
As shown in Figure 2, rhEPO treatment effectively decreased serum glucose and improved glucose intolerance (IGTT) in db/db mice (Figure 2A, B, D, and E). We also observed a significantly decreased insulin tolerance test (ITT insulin) in db/db mice that received 1.2μg/kg/w rhEPO treatment (Figure 2C, F). However, Pearson correlation analysis revealed a weak correlation between plasma EPO and serum glucose (Figure 2G).
rhEPO improved glucose intolerance in db/db mice. Serum glucose in db/m and db/db mice after treatment with rhEPO for 4 and 8 weeks (A, D). Glucose tolerance (IGTT) and insulin tolerance (ITTinsulin) in db/m and db/db mice after treatment with rhEPO for 4 and 8 weeks (B, C, E, F).The data were analyzed to determine the area under the curve (AUC) for rhEPO treatment in db/m mice (blue) and db/db mice (red). Data are presented as mean ± SD. N=10in each group.Thelinear regression results between plasma EPO and serum glucose (G). * db/db0.4μg/kg/w vs. db/db CNT P<0.05, ** p<0.01; # db/db 1.2μg/kg/w vs. db/db CNT P<0.05, ## p<0.01; ѱѱdb/db 1.2μg/kg/w vs. db/db0.4μg/kg/w, p<0.01.
We further evaluated phosphorylated Akt, PI3K, IRS-1 and eNOS levels in total protein fractions of skeletal muscle tissue from db/db mice. The ratio of phosphorylated IRS-1ser612/IRS1, AKTSer473/AKT and PI3K-p85/PI3Kas well as phosphorylated eNOSSer1177/eNOS was increased in db/db mice treated with rhEPO 1.2μg/kg/w for 8-week (Figure 3).
rhEPO unregulated IRS-1/AKT/PI3K activity and eNOS signaling in db/db skeletal muscle. Immunoblots showed rhEPO treatment up-regulated pIRS-1/IRS-1, pAKT/AKT, pPI3K/PI3K ratios and eNOS proteins (A); Quantified results expressed as the mean±SD, in at least three repeated experiments(B-E).
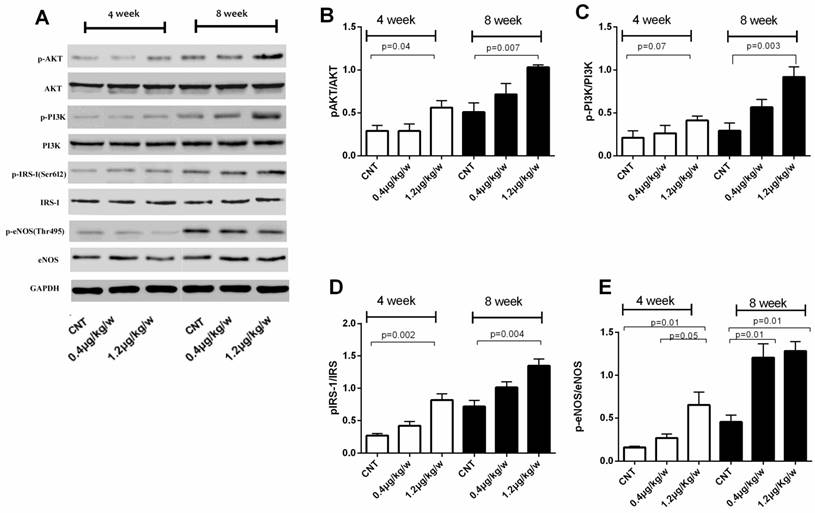
These results suggest that IRS-1/AKT/PI3K signal pathways were activated by rhEPO treatment and rhEPO could promote NO products and increased eNOS activity in skeletal muscle of type 2 diabetic mice.
EPOR knockdown impairs Glut4 translocation and glucose uptake (2-DG)
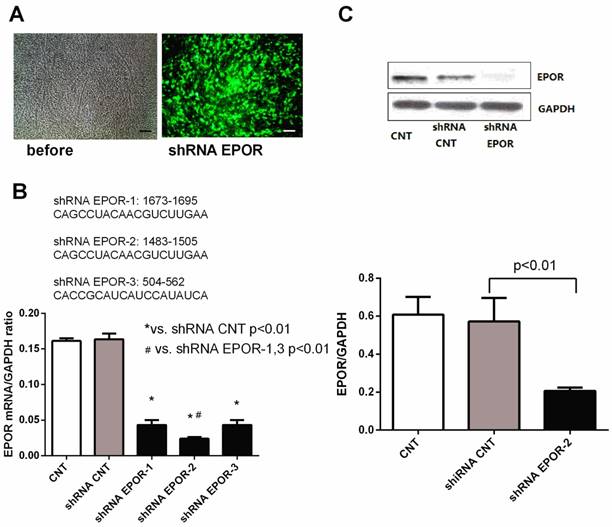
To further determine whether rhEPO protects skeletal muscle against hyperglycemia via its specific EPO receptor, we isolated and cultured primary skeletal muscle cells from 12-week-old db/db mice and knocked down EPOR in skeletal muscle cells with EPOR shRNA lentivirus (Figure 4). The results demonstrated a successful knockdown of EPOR has been obtained in our study.
The generation of lentivirus-delivered EPOR shRNA in skeletal muscle cells. After infection of lentivirally-delivered EPOR shRNA, skeletal muscle cells exhibited eGFP expression (A), Scalebar: 50μm. Sequence analysis of shRNA EPOR (B). qPCR analysis and Western blot analysis demonstrated successful induction of shRNA EPOR-2 in skeletal muscle cells, as demonstrated through EPOR knockdown (C). All data are expressed as the mean ± SD of at least three repeated experiments.
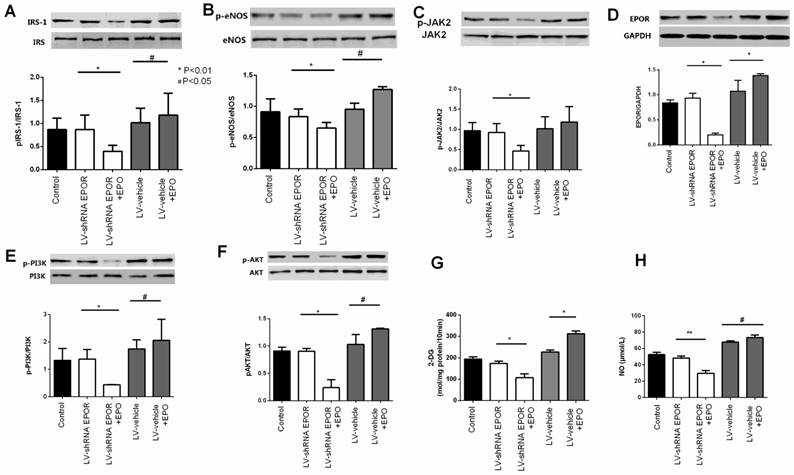
To clarify whether rhEPO activation of the PI3K/AKT pathway is involved in EPO/EPOR signal transduction in skeletal muscle cells, we examined phosphorylated IRS-1ser307/IRS-1, AKTSer473/AKT, PI3K-p85/PI3K, eNOSSer1177/eNOS and NO product in skeletal muscle cells transfected with EPOR shRNA. As shown in Figure 5, the ratios of IRS-1ser307/IRS-1, AKTSer473/AKT, and PI3K-p85/PI3K, JAK2 Tyr1007/1008/JAK2 were decreased in shRNA EPOR treated cells and phosphorylated eNOSSer1177/eNOS and NO products were also decreased. These results indicated EPOR may play a vital role in the activation of the PI3K/AKT signaling (Figure 5A-F).
Signaling molecule expression analyzed by Western blot. EPOR silencing using the shRNA induced down-regulation of IRS-1/AKT/PI3K and eNOS signaling, which were prevented by rhEPO treatment in skeletal muscle cells. Immunoblots and quantification of the results show shRNA EPOR-mediated down-regulation of IRS-1/AKT/PI3K and eNOS pathways, however, rhEPO treatment up-regulated these proteins (A-F). 2-DG uptake and supernatant NO levels were also decreased following EPOR silencing and recovered by rhEPO treatment (G-H). All results are expressed as the mean ± SD of at least three repeated experiments.
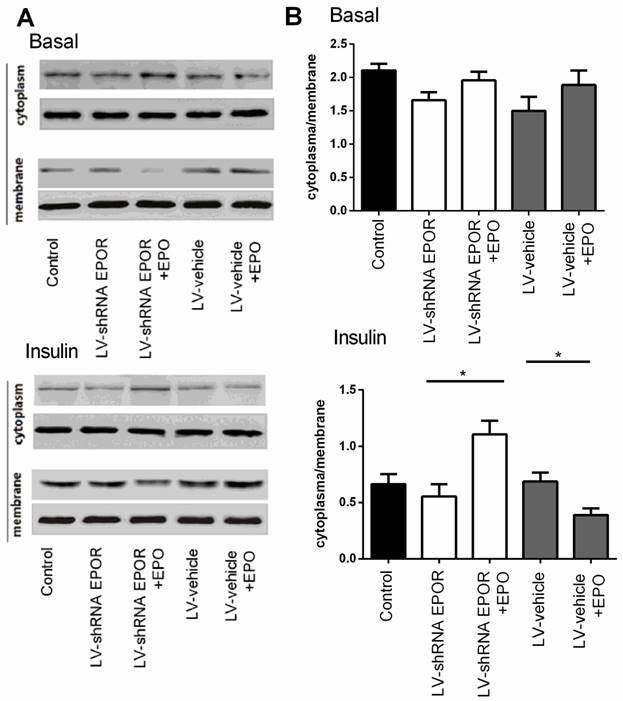
Suppression of EPOR resulted in a reduction in 2-DG uptake under insulin-stimulated conditions in skeletal muscle cell. Compared with the EPOR shRNA group, 2-DG uptake in rhEPO treated cells was significantly increased (P<0.01) (Figure 5G). Similar results were observed in NO production (Figure 5H). Furthermore, we observed that insulin-stimulated Glut4 translocation was also suppressed in cells transfected with EPOR shRNA, and on the contrary, rhEPO treatment increased Glut4 translocation to the cell membrane (measured as cytoplasm/membrane ratio). To further demonstrate this conclusion, we blocked JAK2 and STAT5 signaling with the JAK2 inhibitor, AG490 (10 μM, Sigma), and a STAT5 inhibitor (10 mg/ml, Santa Cruz) pre-incubated for 30 min. Similar results were obtained as EPOR silence (Figure 6 and Figure 7).
Knockdown of EPOR inhibited insulin-stimulated Glut4 translocation inskeletal muscle cells. Immunoblots showed GLUT4 expression with and without insulin stimulated on the membrane and in the cytosol of skeletal muscle cells (A). Quantification of the results from “A” (B).All data are expressed as the mean±SD of at least three repeated experiments.
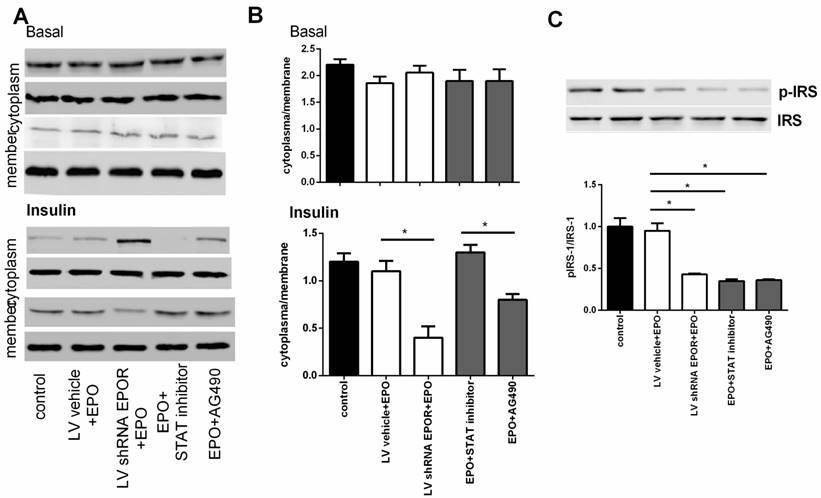
The effect of JAK2 inhibitor AG490 and STAT5 inhibitor on the phosphorylation of IRS1 and translocation of Glut4 Immunoblots showed GLUT4 expression with and without insulin stimulated on the membrane and in the cytosol of skeletal muscle cells (A). Quantification of the results from “A” (B). The phosphorylation of IRS1 measured by western bolt and quantification of the results(C). All data are expressed as the mean±SD of at least three repeated experiments.
EPOR is required for autophagy clearance
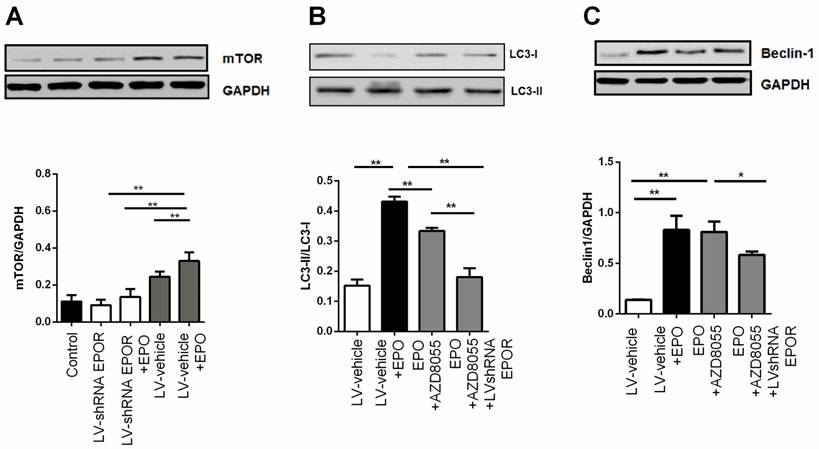
Skeletal muscle cells treated with rhEPO showed increased AKT phosphorylation at Ser473, and this mTOR complex 2 substrate is a key regulator of cell proliferation and autophagy in diabetes. As such, we assessed the induction of autophagy by examining the levels of the autophagosome marker, microtubule-associated protein-1 light chain 3 (LC3) and Beclin-1. As shown in Figure 8 A, B, and C, 100nM rhEPO significantly increased the expressions of mTOR, the ratio of LC3II/LC3I, and Beclin-1 in skeletal muscle cells. However, these changes were all decreased in cells transfected with EPOR shRNA lentivirus. Moreover, the autophagosome marker was further reduced following treatment with mTORC1/2 inhibitor, AZD8055 (500 nM). These results indicated that the protective effect of rhEPO might be mediated through increased mTOR/AKT ser473 signaling.
EPOR knockdown reduced autophagosomes in skeletal muscle cells. Immunoblots showed rhEPO significantly increased the expression of mTOR (A), the ratio of LC3II/LC3I (B), and Beclin-1 (C), all effects that were reversed in cells transfected with EPOR shRNAlentivirus or treatment with mTORC1/2 inhibitor, AZD8055. All results expressed as the mean±SD of at least three repeated experiments.*p<0.05, ** p<0.01.
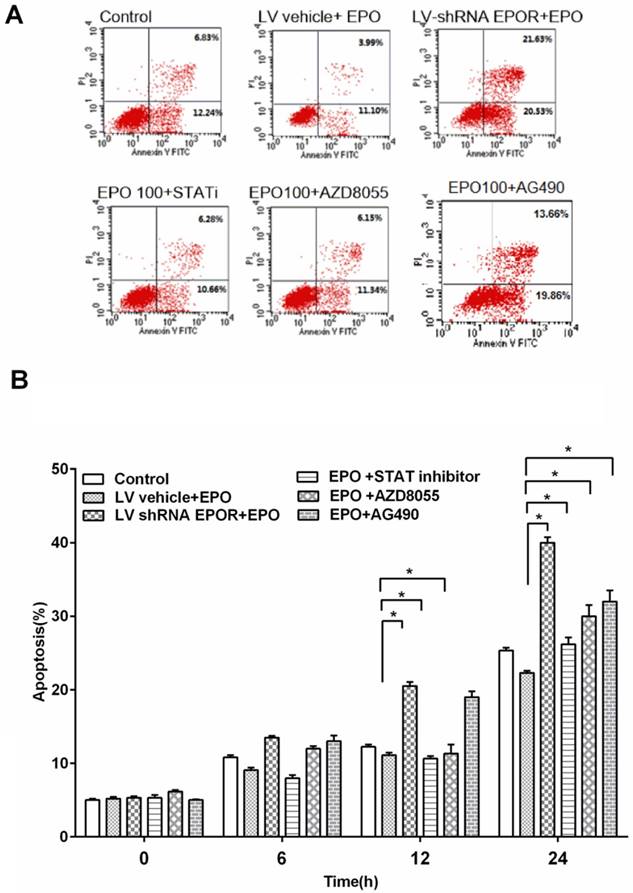
rhEPO inhibited cell apoptosis via AKT/PI3K, STAT5 and JAK2 pathways
In order to determine the effect of EPO on skeletal muscle cells apoptosis, we measured cell apoptosis treated with rhEPO. The results showed that the percentage of apoptotic cells was decreased after rhEPO treatment compared with scramble control.
To further study the mechanism on EPO inhibiting apoptosis, we measured cell apoptosis after EPOR silence and found that cell apoptosis increased in shRNA EPOR group. This indicated that EPO inhibited apoptosis through mediating the expression of EPOR.
In addition, in order to find the downstream molecular mediated by EPOR, we blocked mTOR/AKT ser473, JAK2 and STAT5 signaling with the mTOR inhibitor AZD8055 (500nM), JAK2 inhibitor, AG490(10 μM) and a STAT5 inhibitor (10 mg/ml) pre-incubated for 30 min. We found that when JAK2, AKT/PI3K and STAT5 were blocked, the apoptosis increased significantly at 12h and 24h.
Together, these results indicated that rhEPO protected skeletal muscle cells from apoptosis by mediating EPOR and its downstream JAK2, AKT/PI3K and STAT5pathways.
Discussion
In the present study, we demonstrate that EPO administration can reduce insulin resistance in type 2 db/db mice, and may activate its specific receptor, EPOR. It also appears to act via activation of subsequent signaling pathways, manifested as phosphorylation of AKT, IRS-1, and PI3K, Glut4 translocation, autophagy regulation and reduced apoptosis in db/db mouse primary skeletal muscle. Blockade of EPOR with shRNA inhibited all those EPO-mediated alterations, indicating EPO-EPOR is essential for glucose metabolism in mouse skeletal muscle, similar to results observed in cultured mouse 3T3-L1 adipocytes8.
EPO treatment improves glucose intolerance as shown in experimental rats and humans15-17. In a rat model of diet-induced glucose intolerance, chronic EPO treatment improved fasting glucose levels and glucose tolerance, and partially rescued insulin-stimulated AKT activation and reduced markers of oxidative stress15. When treated with EPO, patients with chronic kidney disease exhibited reduced IR16, 17. Reduced IR mostly likely relies on increase of glucose uptake in adipose tissue and skeletal muscle. In our prior in vivo research, our results showed that systemic EPO administration reduced IR, as manifested in decreased serum glucose, the area under curve from an IGTT, and increased glucose uptake in skeletal muscle.
rhEPO inhibited cell apoptosis by AKT/PI3K and STAT5 pathways. Flow cytometric analysis of the Annexin V-stained cells after treatment with rhEPO for 12 hours (A). Effects of different inhibitors on skeletal muscle cell apoptosis. Quantification of the results from “A” (B). All results expressed as the mean±SD of at least three repeated experiments.
It has long been disputed whether EPO activity effects in skeletal muscle occurs through its specific cell membrane, EPOR. The EPOR gene and protein has been shown to be expressed in primary satellite cells isolated from mouse skeletal muscle and in cultured C2C12 myoblasts, and was down-regulated during C2C12 myoblasts differentiation to myotubes9. EPOR mRNA and protein were also reported in rat L6 myoblasts and in human primary myoblast cultures18. In contrast, EPOR gene and protein expression were not detected in isolated normal ratmyoblasts19. Still, EPOR mRNA and protein localization in skeletal muscle biopsies has been documented while EPOR mRNA has been detected in isolated human muscle fibers and human satellite cells12, 20. Discrepancies in EPOR gene and protein measurement appear to be associated with possible species differences and investigation in in vivo versus in vitro experimental models. In our study, we detected EPOR protein expression by immunofluorescence and and western blot in skeletal muscle isolated from type 2 diabetic mice, and systemic EPO stimulation activated EPOR protein, as confirmed by immunofluorescence and Western blot. This evidence supports an important role for EPOR in skeletal muscle in type 2 diabetes.
It remains unclear, however, whether EPOR potentially activates and modulates similar signaling cascades in skeletal muscle as in hematopoietic cells. In vitro research indicates EPO treatment of C2C12 myoblast causes increased JAK2, STAT5, and AKT phosphorylation10. However, STAT5 activation was not detected in rat skeletal muscle tissue in response to EPO stimulation11. In addition, acute EPO stimulation does not appear to promote phosphorylation of AKT, STAT5, and MAPK signaling pathways in human skeletal muscle12. In our in vivo study involving type 2 diabetic mice, we observed that EPO stimulated increased EPOR expression, followed by subsequent phosphorylation of AKT, STAT5, PI3K, IRS-1, and e-NOS. The PI3K/AKT and JAK2/STAT5 signaling pathways mediated many cellular processes including proliferation, differentiation, and apoptosis and evidence that EPO induces these signal transductions were accumulating. Chan et al. reported EPOR knockdown abrogated human MDA-MB-231 and MDA-MB-435 cells growth by inactivation of PI3K/AKT pathway21. Here we confirmed blockade of EPOR with specific shRNA inhibited associated downstream signaling, indicating EPO-EPOR signaling is essential for improved IR in db/db mice skeletal muscle.
Autophagy has been suggested to protect against some diseases such as cancer, inflammatory diseases, aging, diabetes and insulin resistance in experimental animal models3, 22, 23. The ability of skeletal muscle to adapt to various loading and other environment conditional is substantial. Several factors such as aging, exercise, diet restriction, paraplegia, etc, could promote autophagy in skeletal muscle 24-26. The mammalian target of rapamycin, mTOR inhibitor, is an important regulator of programmed cell death involving autophagy and apoptosis, and in some cells, proliferation and survival. In the cardiovascular system, mTOR is one of several molecular modulators necessary for human embryonic stem cell-derived cardiomyocytesproliferation27. Growth factors such as EPO have previously been shown to be cytoprotective in cardiac muscle cells/tissue during ischemic stress28. It has been shown EPO's protective effect requires mTOR activation for the regulation of bone homeostasis involving osteoblastogenesis and osteoclastogenesis2. Differentiation of neural precursor cells with the potential for use in neurodegenerative disorders is also dependent upon EPO and mTORactivity29. EPO likely plays different roles in the induction or inhibition of autophagy in various cells and tissues. EPO has shown protective effects against rotenone-induced neurotoxicity in SH-SY5Y cells through the induction of autophagy30. However, in renal mesangial cells, EPO was observed to impart protective effects against lipopolysaccharide-induced autophagy6. It is unknown whether EPO increases or decreases skeletal muscle autophagy in physiologic or pathologic conditions.
In our study, we first observed a reduction in skeletal muscle autophagy in type 2 diabetic mice, and systemic EPO induced an elevation in skeletal muscle autophagy, as indicated by increased upstream autophagy regulatory proteinBeclin-1, and downstream regulators of autophagy, LC3. Modulation of LC3-II protein expression suggests that conversion of the non-lipidated form of LC3, LC3-I, to the autophagosome-membrane-associated lipidated form, LC3-II is similar to what is observed following exercise. Our findings indicate that EPO improvement of skeletal muscle insulin resistance may occur through induction of autophagy as a potential mechanism, as similar results were observed in dihydromyricetin31.Autophagy has been reported to improve insulin sensitivity during high fat diet administration in mice32.Autophagy and apoptotic pathways may complement each other to regulate cell survival, as observed in diabetes, since induction of autophagy may protect cardiomyocytes from apoptosis33.mTOR can influence both progression of apoptosis and autophagic pathways of programmed cell death4. mTOR can prevent apoptosis and reduce IR, and reduce vascular thrombosis in patients with metabolic syndrome5. EPO activates mTOR to increase cell survival during oxygen-glucose deprivation34, and reduces the detrimental processes in animal models of obesity7. In our study, EPO activated mTOR, and elevated Beclin-1, and LC3-II, autophagy markers, and further resulted in autophagy in type 2 diabetic skeletal muscle, accompanied by reduced IR and glucose transfer increase.
Together, our results from the present study provide supporting evidence that EPO-EPOR interaction is an essential process for the induction of glucose uptake, and the possible mechanisms of its beneficial actions involve activation of downstream signaling molecules including PI3K, AKT, IRS-1, and enhanced autophagy and reduced apoptosis in type 2 diabetic skeletal muscle. In summary, these findings shed light on possible new strategies for therapy of IR in diabetes and chronic kidney disease.
Abbreviations
EPO: Erythropoietin;
IR: insulin resistance;
IGTT: glucose tolerance;
ITT insulin: insulin tolerance test;
2-DG: 2-deoxy-d [3H] glucose;
NO: nitric oxide.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No: 81300652), Shanghai Science and Technology Commission (15ZR1437400), the Key Specialized Construction Project in Medicine of Shanghai (No: ZK2015A15), and Outstanding Leaders Training Program of the Pudong Health Bureau of Shanghai (No: PWRl 2012-05).
Author contributions
Pan Y, Yang XH, Guo LL, Qiao QY, Gu YH, and Gu HF performed research, experiments and data analysis. Pan Y and Jin HM wrote and reviewed/edited manuscript. All authors have approved the final version of the manuscript and agreed to submit for publication. Guo LL, Yang XH and Pan Y contributed equally to this paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lamon S, Russell AP. The role and regulation of erythropoietin (EPO) and its receptor in skeletal muscle: how much do we really know? Frontiers in physiology. 2013;4:176
2. Kim J, Jung Y, Sun H, Joseph J, Mishra A, Shiozawa Y. et al. Erythropoietin mediated bone formation is regulated by mTOR signaling. Journal of cellular biochemistry. 2012;113:220-8
3. Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K. et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell metabolism. 2008;8:325-32
4. Chong ZZ, Shang YC, Maiese K. Cardiovascular disease and mTOR signaling. Trends in cardiovascular medicine. 2011;21:151-5
5. Pasini E, Flati V, Paiardi S, Rizzoni D, Porteri E, Aquilani R. et al. Intracellular molecular effects of insulin resistance in patients with metabolic syndrome. Cardiovascular diabetology. 2010;9:46
6. Bi L, Hou R, Yang D, Li S, Zhao D. Erythropoietin protects lipopolysaccharide-induced renal mesangial cells from autophagy. Experimental and therapeutic medicine. 2015;9:559-62
7. Zhang Y, Wang L, Dey S, Alnaeeli M, Suresh S, Rogers H. et al. Erythropoietin action in stress response, tissue maintenance and metabolism. International journal of molecular sciences. 2014;15:10296-333
8. Pan Y, Shu JL, Gu HF, Zhou DC, Liu XL, Qiao QY. et al. Erythropoietin improves insulin resistance via the regulation of its receptor-mediated signaling pathways in 3T3L1 adipocytes. Molecular and cellular endocrinology. 2013;367:116-23
9. Ogilvie M, Yu X, Nicolas-Metral V, Pulido SM, Liu C, Ruegg UT. et al. Erythropoietin stimulates proliferation and interferes with differentiation of myoblasts. The Journal of biological chemistry. 2000;275:39754-61
10. Jia Y, Suzuki N, Yamamoto M, Gassmann M, Noguchi CT. Endogenous erythropoietin signaling facilitates skeletal muscle repair and recovery following pharmacologically induced damage. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012;26:2847-58
11. LeBaron MJ, Ahonen TJ, Nevalainen MT, Rui H. In vivo response-based identification of direct hormone target cell populations using high-density tissue arrays. Endocrinology. 2007;148:989-1008
12. Christensen B, Lundby C, Jessen N, Nielsen TS, Vestergaard PF, Moller N. et al. Evaluation of functional erythropoietin receptor status in skeletal muscle in vivo: acute and prolonged studies in healthy human subjects. PloS one. 2012;7:e31857
13. Metzinger L, Poindron P, Passaquin AC. A rapid preparation of primary cultures of mouse skeletal muscle cells. Cytotechnology. 1993;13:55-60
14. Reynolds LJ, Credeur DP, Manrique C, Padilla J, Fadel PJ, Thyfault JP. Obesity, type 2 diabetes, and impaired insulin-stimulated blood flow: role of skeletal muscle NO synthase and endothelin-1. Journal of applied physiology. 2017;122:38-47
15. Caillaud C, Mechta M, Ainge H, Madsen AN, Ruell P, Mas E. et al. Chronic erythropoietin treatment improves diet-induced glucose intolerance in rats. The Journal of endocrinology. 2015;225:77-88
16. Spaia S, Pangalos M, Askepidis N, Pazarloglou M, Mavropoulou E, Theodoridis S. et al. Effect of short-term rHuEPO treatment on insulin resistance in haemodialysis patients. Nephron. 2000;84:320-5
17. Mak RH. Correction of anemia by erythropoietin reverses insulin resistance and hyperinsulinemia in uremia. The American journal of physiology. 1996;270:F839-44
18. Launay T, Hagstrom L, Lottin-Divoux S, Marchant D, Quidu P, Favret F. et al. Blunting effect of hypoxia on the proliferation and differentiation of human primary and rat L6 myoblasts is not counteracted by Epo. Cell proliferation. 2010;43:1-8
19. Rotter R, Menshykova M, Winkler T, Matziolis G, Stratos I, Schoen M. et al. Erythropoietin improves functional and histological recovery of traumatized skeletal muscle tissue. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2008;26:1618-26
20. Rundqvist H, Rullman E, Sundberg CJ, Fischer H, Eisleitner K, Stahlberg M. et al. Activation of the erythropoietin receptor in human skeletal muscle. European journal of endocrinology. 2009;161:427-34
21. Chan KK, Matchett KB, Coulter JA, Yuen HF, McCrudden CM, Zhang SD. et al. Erythropoietin drives breast cancer progression by activation of its receptor EPOR. Oncotarget. 2017;8:38251-63
22. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42
23. Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell metabolism. 2010;11:467-78
24. Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, Leeuwenburgh C. Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Experimental gerontology. 2010;45:138-48
25. He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z. et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511-5
26. Fry CS, Drummond MJ, Lujan HL, DiCarlo SE, Rasmussen BB. Paraplegia increases skeletal muscle autophagy. Muscle & nerve. 2012;46:793-8
27. Foldes G, Mioulane M, Wright JS, Liu AQ, Novak P, Merkely B. et al. Modulation of human embryonic stem cell-derived cardiomyocyte growth: a testbed for studying human cardiac hypertrophy? Journal of molecular and cellular cardiology. 2011;50:367-76
28. Kiss K, Csonka C, Paloczi J, Pipis J, Gorbe A, Kocsis GF. et al. Novel, selective EPO receptor ligands lacking erythropoietic activity reduce infarct size in acute myocardial infarction in rats. Pharmacological research. 2016;113:62-70
29. Marfia G, Madaschi L, Marra F, Menarini M, Bottai D, Formenti A. et al. Adult neural precursors isolated from post mortem brain yield mostly neurons: an erythropoietin-dependent process. Neurobiology of disease. 2011;43:86-98
30. Jang W, Kim HJ, Li H, Jo KD, Lee MK, Yang HO. The Neuroprotective Effect of Erythropoietin on Rotenone-Induced Neurotoxicity in SH-SY5Y Cells Through the Induction of Autophagy. Molecular neurobiology. 2016;53:3812-21
31. Shi L, Zhang T, Liang X, Hu Q, Huang J, Zhou Y. et al. Dihydromyricetin improves skeletal muscle insulin resistance by inducing autophagy via the AMPK signaling pathway. Molecular and cellular endocrinology. 2015;409:92-102
32. Liu Y, Palanivel R, Rai E, Park M, Gabor TV, Scheid MP. et al. Adiponectin stimulates autophagy and reduces oxidative stress to enhance insulin sensitivity during high-fat diet feeding in mice. Diabetes. 2015;64:36-48
33. He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. 2013;62:1270-81
34. Shang YC, Chong ZZ, Wang S, Maiese K. Erythropoietin and Wnt1 govern pathways of mTOR, Apaf-1, and XIAP in inflammatory microglia. Current neurovascular research. 2011;8:270-85
Author contact
![]() Corresponding author: Hui Min Jin, MD., Address: Division of Nephrology, Shanghai Pudong Hospital, Fudan University, Pudong Medical Center, 2800 Gongwei Road, Huinan Town, Pudong, Shanghai 201399, China. Fax: +86 21 68036053. E-mail: hmjglicom
Corresponding author: Hui Min Jin, MD., Address: Division of Nephrology, Shanghai Pudong Hospital, Fudan University, Pudong Medical Center, 2800 Gongwei Road, Huinan Town, Pudong, Shanghai 201399, China. Fax: +86 21 68036053. E-mail: hmjglicom