International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(11):1458-1469. doi:10.7150/ijbs.21300 This issue Cite
Research Paper
Overexpression of selenoprotein H prevents mitochondrial dynamic imbalance induced by glutamate exposure
Yan-Mei Ma1*, Yong-Zhen Guo1*, Gordon Ibeanu2, Li-Yao Wang3, Jian-Da Dong1, Juan Wang1, Li Jing1, Jian-Zhong Zhang1 ![]() , P. Andy Li2
, P. Andy Li2 ![]()
1. School of Basic Medical Sciences, Department of Pathology, Ningxia Medical University; Ningxia Key Laboratory of Cerebrocranial Diseases, Yinchuan, Ningxia, P. R. China.
2. Department of Pharmaceutical Sciences, Biomanufacturing Research Institute and Technological Enterprise (BRITE), North Carolina Central University, Durham, North Carolina, USA.
3. Department of Pathology, Shanxi Traditional Chinese Medicine Hospital, Xi'an, Shanxi, P. R. China.
* These authors contributed equally to this work.
Received 2017-6-2; Accepted 2017-9-21; Published 2017-11-1
Citation:
Ma YM, Guo YZ, Ibeanu G, Wang LY, Dong JD, Wang J, Jing L, Zhang JZ, Li PA. Overexpression of selenoprotein H prevents mitochondrial dynamic imbalance induced by glutamate exposure. Int J Biol Sci 2017; 13(11):1458-1469. doi:10.7150/ijbs.21300. https://www.ijbs.com/v13p1458.htm
Other stylesAbstract

Selenium and selenoproteins play important roles in neuroprotection against glutamate‑induced cell damage, in which mitochondrial dysfunction is considered a major pathogenic feature. Recent studies have revealed that mitochondrial fission could activates mitochondrial initiated cell death pathway. The objectives of the study are to determine whether glutamate induced cell death is mediated through mitochondrial initiated cell death pathway and activation of autophagy, and whether overexpression of selenoprotein H can protect cells from glutamate toxicity by preserving mitochondrial morphology and suppressing autophagy. Vector- or human selenoprotein H (SelH)-transfected HT22 cells (V-HT22 and SelH-HT22, respectively) were exposed to glutamate. The results showed that glutamate-induced cytotoxicity was associated with increased ROS production and imbalance in mitochondrial dynamics and autophagy. These alterations were reversed and cellular integrity restored by overexpression of SelH in HT22 cells.
Keywords: Autophagy, Glutamate, Mitochondrial dynamics, Mitochondrial fission, Selenoprotein H.
1. Introduction
Approximately 25 selenoproteins have been identified in humans [1,2]. Selenium is incorporated into selenoproteins in the form of the amino acid selenocysteine (Sec) [1]. Sec is a central component at the catalytic sites of various selenium-dependent enzymes. The redox-sensitive Sec residues function as scavengers that limit the adverse effects of oxidative stress within the cell and protect against damage to cellular components. Although the functions of a majority of selenoproteins remains to be fully defined, emerging evidence suggests that impaired metabolism of selenoproteins contributes to a variety of diseases including defect in the regulation of cardiac function [3], ischemic stroke [4], cancer [5,6], and neurological disorders [7,8].
Selenoprotein H (SelH) is a 14 kDa thioredoxin-like responsive nuclear DNA binding protein that plays a role in gene regulation [9]. While the precise physiological role remains obscure, recent reports highlight the importance of SelH in cell survival and proliferation. This is based on the fact that SelH has been shown to promote transactivation of glutathione and expression of other genes that enhance antioxidant capacity of the cell [9], protect neurons against UVB-induced damage [10], prevent activation of the apoptotic cell death pathway by stabilizing membrane potential [11], and elevate mitochondrial biogenesis and mitochondrial functions [12]. In addition, a role for SelH to suppress oxidative stress-induced cellular senescence through genome maintenance and redox regulation was shown in tumorigenic cells [13].
The genetic inactivation of selenoproteins in neurons results in progressive neurodegeneration and impaired activities of selenoenzymes enhanced neuronal loss and dysfunction [14]. Conversely, a number of studies have suggested that increased selenoenzyme associated activities alleviates stress, hypoxia, trauma and neurodegenerative disorders including ischemic stroke [15,16,17]. Cerebral ischemia causes damage to neurons by activating a cascade of events including excitotoxicity, oxidative stress, apoptosis, and autophagy [18]. These events lead to increased reactive oxygen species (ROS) production, which disrupts antioxidant defense mechanisms and negatively impacts mitochondrial homeostasis and energy production, eventually leading to cell death [19,20]. Therefore, cell survival is critically dependent on the cells capacity to maintain a balance between ROS production and elimination.
Neuronal injury caused by ischemia is believed to be mediated by excessive activation of glutamate receptors. Glutamate is the most abundant primary excitatory neurotransmitter in the nervous system. However, at high concentrations it triggers hyperactive signaling events that frequently culminate in cell death both in vitro and in vivo. Neurotoxic level of glutamate contributes to the etiology of many neurological disorders such as Alzheimer's disease (AD), Parkinson's disease (PD), and ischemic stroke [21]. Glutamate induces cell death by mechanisms that involve well defined, receptor-initiated excitotoxicity and non-receptor mediated oxidative toxicity pathways [22,23]. Chronic exposure to extracellular glutamate initiates glutamate-evoked oxidative toxicity due to depletion of cysteine and glutathione. Depletion of glutathione results in ROS production, promotes oxidative stress mitochondrial dysfunction, and alterations in mitochondrial dynamics [24,25,26].
Balancing fission and fusion events is essential for proper maintenance of mitochondrial dynamics and function. In general, mitochondrial fusion refers to fusion of two mitochondria for exchanging their contents and signals and fission is division of a mitochondrion to daughter mitochondria. Fission is used by the mitochondrion as a way to package degenerated proteins and metabolic wastes and then to eliminate them through mitochondrial autophagy process [25]. We have demonstrated that glutamate induces mitochondrial fission and formation of mitochondrial permeability transition pore [26]. In the present study we sort to determine the mechanisms by which SelH protects neurons against excitotoxic damage induced by chronic glutamate exposure. Glutamate toxicity was simulated by exposing HT22 neuronal cells stably expressing the SelH gene and empty control vector to 6 mM glutamate. Cell viability, ROS production, and biomarkers for autophagy and mitochondrial fission were quantified.
2. Materials and Methods
2.1 Cell culture and treatment
Vector control and human SelH-transfected murine hippocampal neuronal HT22 cells (V-HT22 and SelH-HT22, respectively) were cultured in Dulbecco's Modified Eagle Medium (DMEM)/F12, containing 10% fetal bovine serum (FBS, HyClone Cell Culture and Bioprocessing), 100 nM streptomycin /penicillin (HyClone Cell Culture and Bioprocessing), and cultivated at 90% relative humidity in 5% CO2 at 37 ○C. The culture medium was renewed every 2 days. Glutamate toxicity was induced by incubating the V-HT22 and SelH-HT22 cells with 6 mM glutamate (Sigma-Aldrich Co.) for 24 h. Cell viability was assessed in 96-well cell culture plates. The transfection procedures and efficacy of transfection have been previously reported [9,10].
2.2 Cell viability assay
Cell viability was assessed with Cell Counting Kit-8 (CCK-8) (Dojindo Molecular Tech. Inc., Rockville, MD) according to manufacturer's instructions. In brief, V-HT22 and SelH-HT22 cells were plated into 96-well plates (5000/well, Corning Inc., New York, NY). After seeding 24h the cultures exposed to 6 mM glutamate and the viabilities were measured in the SpectraMax microplate reader (Molecular Devices, Sunnyvale, CA). The results were pooled from 2 separate experiments with triplicate sets for each condition in each experiment.
2.3 Electron Microscopy
Cells were fixed with glutaraldehyde solution, dimethyl sodium arsenate buffer and osmic acid for 24 h and dehydrated in ascending ethanol concentrations, rinsed in propylene oxide, and incubated in propylene oxide and resin before embedding resin. Samples were sliced and imaged on a transmission electron microscope.
2.4 Determination of ROS
Superoxide anion production was measured with dihydroethidium (DHE) in V-HT22 and SelH-HT22 cells exposed to glutamate (6 mM). Briefly, cells (1x105/ml) were seeded on glass slides. After glutamate treatment for 24 h, the slides were incubated with DHE (50 μM) for 45 min at 37 °C, washed with PBS, and fixed with 4% paraformaldehyde. Images were captured under magnification (400x) using a fluorescent microscope (Olympus CHC-212) and fluorescent intensity were measured by IPP6.0 software.
2.5 Western blotting
Cells were pooled from 3 separate experiments. The cells were cultured in 6-well plates and treated with control or glutamate. The cells were collected after 24h of treatment, rinsed, and lysed in RIPA buffer containing protease (Roche Diagnostics Corp., Indianapolis, IN) and phosphatase (Thermo Fisher Scientific, Waltham, MA) inhibitor cocktails. After various peed of centrifugations subcellular fractions of the cytosol and nucleus were isolated. The cytosol fraction was lysed in mitochondria protein extraction buffer (Enogene, China). After various peed of centrifugations mitochondria protein was isolated. The cell lysates containing 20 µG proteins were separated on 4%-12% NuPAGE SDS-PAGE gels (Invitrogen), transferred to nitrocellulose membrane, and probed overnight at 4 ○C with antibodies against Apaf-1 (1:500), total caspase-3 (1:1000), cleaved caspase-3 (1:500), total caspase-9 (1:1000), cleaved caspase-9 (1:500), β-tubulin (1:1000) from Cell Signaling Technologies, and LC3 (1:1000), Fis1 (1:2000), Drp1 (1:2000), p-Drp1 (1:1000) from Abcam Inc. After washing, the membranes were incubated with peroxidase-conjugated secondary antibody (1:5000, Jackson Lab) for 3h at room temperature. Antibody binding was detected with enhanced chemiluminescence kit (Thermo Fisher Scientific) and images obtained with gel imaging analyzer (BIO-RAD Inc). The intensities of bands were quantified using Image J software.
2.6 Immunofluorescence
Cells (1x105/ml) seeded on glass slides for 24 h were treated with glutamate for additional 24 h at 37 ○C in a humidified incubator in 5% CO2. The slides were washed with PBS, fixed with 4% paraformaldehyde for 15 min and 1% TritonX-100 for 30 min. The nonspecific binding sites were blocked with 10% goat serum and slides were incubated overnight at 4 ○C with antibodies against Apaf-1 (1:100), cleaved caspase-9 (1:100), or cleaved caspase-3 (1:100). The slides were washed 3 times, transferred to secondary antibody (1:200) for 1 h at 4 ○C in dark and stained with DAPI for 5 min. Images were captured in each well at the magnification of 400x. The experiments were repeated twice.
2.7 Detection of autophagy
Mitophagy was measured by immunofluorescence double labeling with LysoTracker Red and Mito Tracker Green in control and experimental cells exposed to glutamate (6 mM). The cells were seeded in 12-wells plate for 24 h before glutamate exposure. At the end of the treatment duration the slides were incubated in preheated culture medium containing LysoTracker red (75 nM) and MitoTracker Green (100 nM) for 45 min in a 37 ○C humidified cell culture incubator. The slides were washed with PBS and fixed with 4% paraformaldehyde. Images were captured under magnification (400x) using a fluorescent microscope (Olympus CHC-212) and fluorescent intensity were measured by IPP6.0 software.
2.8 Statistical Analysis
All values were expressed as mean ± SEM. IBM SPSS Statistics 18.0 software was used for statistical analysis. Data in Figure 1 and Figures 3 to 8 were analyzed using one-way ANOVA followed by post hoc Scheffe's test. A p value of <0.05 was considered as statistical significant.
3. Results
3.1 SelH enhanced cell viability in the presence of glutamate
In order to measure the neuroprotective effect of SelH, HT22 cells were exposed to various concentrations of glutamate. The data shows that 6 mM of glutamate reduced the cell viability to about 60% of control values. This concentration of glutamate was used for subsequent experiments. The viabilities of cells following glutamate treatment were assessed after 6, 10 and 24 h of incubation. The cell growth rate was slightly higher in SelH-HT22 cells compared to V-HT22 under normal culture conditions, whereas glutamate exposure resulted in a significant decrease in HT22 viability after 10 h of glutamate exposure (Figure 1). At both time points the viability was reduced to about 80% of and further decreased about 60% of the control values at 24 h following glutamate treatment in V-HT22 cells. In contrast, the growth of SelH-HT22 cells was not impeded by glutamate incubation, and its viability was similar to non-glutamate treated V-HT22 control cells at all three end points.
3.2 SelH protected HT22 cells against glutamate induced structural damage
Consistent with the results obtained in the viability assay, cell images captured from inverted microscope demonstrated normal cell morphology in non-glutamate treated control V-HT22 and SelH-HT22 cells. Glutamate exposure caused cell shrinkage and nuclear condensation in V-HT22 cells, indicative of cell death (Figure 2A). Overexpression of SelH preserved cell density and restored normal morphology features. Ultrastructural study revealed comparable morphology in V-HT22 and SelH-HT22 cells without glutamate incubation (Figure 2B). After glutamate treatment, there is an apparent decrease in the number of organelles observed in the cells. In addition, mitochondrial swelling (red arrows), rough endoplasmic reticulum distension and degranulation (Blue arrow), and increased appearance of lysosomes (green arrows) were observed. In contrast, glutamate-treated SelH-HT22 showed normal mitochondrial and normal rough endoplasmic reticulum morphology.
Figure 1
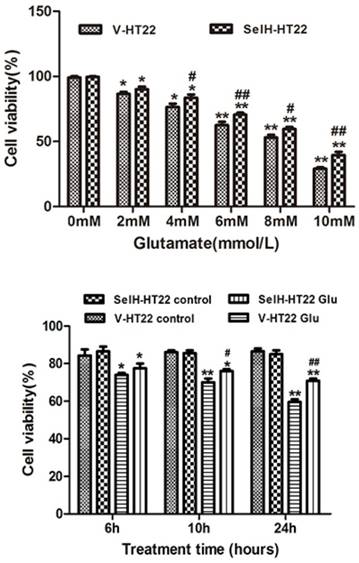
SelH protects HT22 cells against glutamate-induced toxicity. Upper panel, Vector transfected (V-HT22) and SelH expressing cells (SelH-HT22) were exposed to various concentrations of glutamate. Viability at 24 hrs of glutamate exposure showed a dose dependent decreased. Lower panel, V-HT22 and SelH-HT22 were exposed to 6 mM glutamate and cell viabilities were assessed after 6, 10 and 24 hrs. Data were expressed as mean ± SEM. One Way ANOVA followed by Scheffe's text. *p<0.05 and **p<0.01 vs. control (0 mM glutamate) group, #p<0.05 and ##p<0.01 vs. V-HT22 glutamate treatment.
3.3 SelH reduced glutamate-induced ROS production
ROS production was measured by DHE following glutamate incubation. As shown in Figure 3, faint DHE immunofluorescence was detected in normal V-HT22 and SelH-HT22 cells. Glutamate exposure led to increased DHE signal in V-HT22 cells, suggesting increased superoxide production. Overexpression of SelH reduced the DHE signal, suggesting a reduction in superoxide production by SelH (Figure 3).
Figure 2
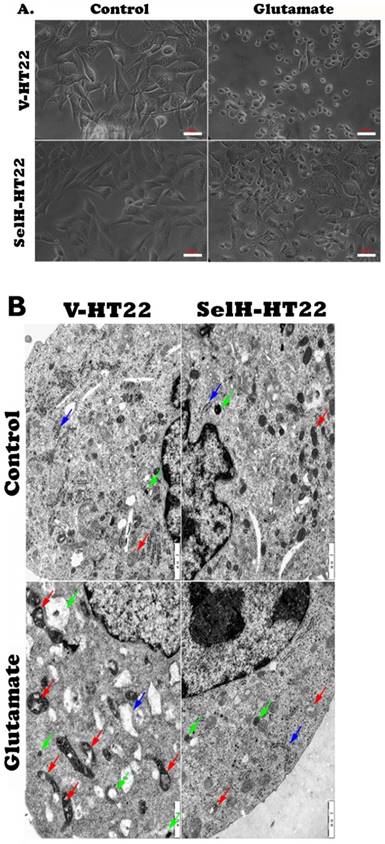
Effect of glutamate treatment on cell morphology. Cells treated with 6 mM glutamate for 24 hrs were processed and visualized under light and electron microscope. (A) Light microscopic images showing normal morphology in V-HT22 and SelH-HT22 cells and cell condensation after glutamate treatment. Overexpression of SelH partially ameliorated glutamate induced cell death. Scale bar = 50 µm. (B) electron microscopic view of V-HT22 and SelH-HT22 neurons under normal and glutamate stress conditions. Glutamate treatment reduced number of subcellular organelles, caused mitochondrial swelling (red arrows) in glutamate treated V-HT22, rough endoplasmic reticulum distension and degranulation (blue arrow) and increased the presence of lysosomes (green arrows). Scale bar = 1 µm.
Figure 3
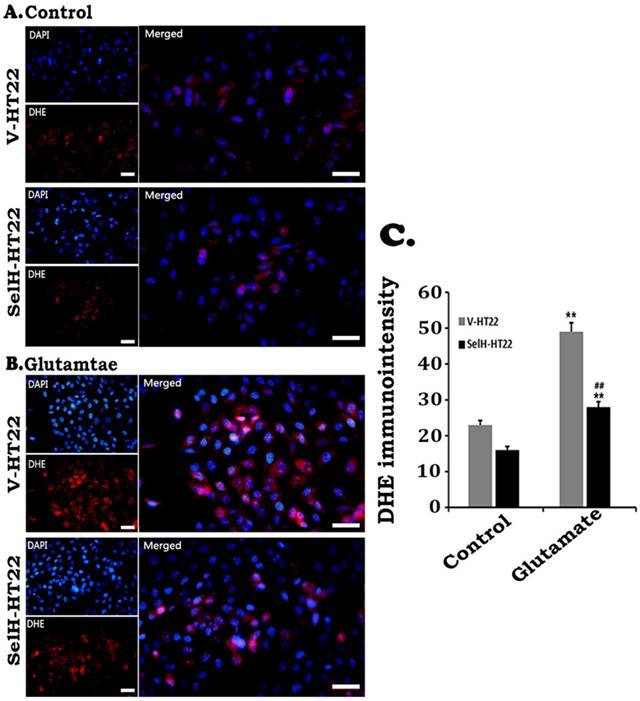
Effect of glutamate on the production of superoxide. Cells were treated with glutamate for 24 h and level of superoxide ions detected with DHE. Glutamate increased the production of superoxide (red) while SelH reduced level of superoxide. Nuclei were stained with DAPI (blue). Immunointensity of the superoxide ions depicted by red fluorescence signals were measured in 4 experimental groups and presented in bar graph. **p<0.01 vs. non-glutamate control and ##p<0.01 vs. V-HT22 cell with the same treatment. Scale bar = 100 µm.
3.4 SelH alleviated the mitochondria-initiated cell death pathway in HT22 cells activated by glutamate exposure
To study the involvement of intrinsic cell death pathway we measured Apaf-1, cleaved caspase-9, and caspase-3 using immunocytochemistry and Western blotting. The results showed that there were very few faintly stained Apaf-1 positive neurons in both V-HT22 and SelH-HT22 cells without glutamate incubation (Figure 4). The positive stains were localized in the cytoplasm. After glutamate exposure, the immuno-intensity of Apaf-1 was enhanced in V-HT22 cells. Compared to V-HT22, Apaf-1 immuno-reactivity was reduced in SelH-HT22 cells treated with glutamate. Western blot using cytosolic fraction further support the findings showing that Apaf-1 protein levels were slightly decreased in SelH-HT22 compared to V-HT22 under control conditions. After glutamate exposure, Apaf-1 protein content significantly increased in the cytosolic fraction and the increase was blunted in SelH-HT22 cells.
Similarly, there were very few stained cleaved caspase-9 positive cells in V-HT22 and SelH-HT22 cells without glutamate treatment. After glutamate exposure for 24h, the immuno-reactivity of cleaved caspase-9 moderately increased in V-HT22 cells (Figure 5). Glutamate enhanced cleaved caspase-9 immuno-reactivity was reduced in SelH-HT22 cells. Western blotting of total and cleaved capase-9 using cytosolic fraction support the findings. Thus, glutamate caused significant elevation of both total and cleaved caspase-9 in V-HT22 cells and overexpression of SelH blocked the increases. Cleaved caspase-3 immunoreactivity increased after glutamate treatment in V-HT22 cells and lowered by SelH overexpression (Figure 6). Immunoblotting showed that both total and cleaved caspase-3 increased significantly after incubation of V-HT22 cells in glutamate, while SelH overexpression blocked the increase (Figure 6).
3.5 SelH reduces glutamate-induced mitochondrial fission
Mitochondrial fission related proteins, including Fis1, Drp1 and p-Drp1 were quantified using Western blotting of mitochondrial fractions. As shown Figure 7, there was no difference among the three proteins between V-HT22 and SelH-HT22 cells under control status. However, after glutamate incubation for 24h, all three proteins markedly increased in V-HT22 and SelH-HT22 cells. However, the glutamate-dependent increases in the protein levels were significantly decreases in SelH-HT22 cells, suggesting that glutamate induced mitochondrial fission was blocked by SelH expressing cells.
Figure 4
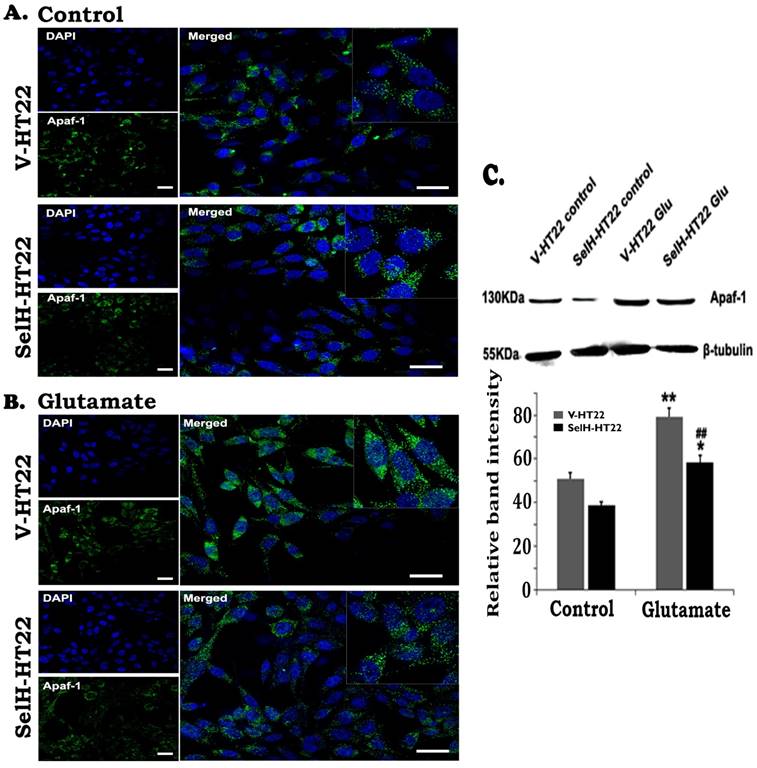
Expression of SelH in HT22 cells attenuated glutamate-induced upregulation of Apaf-1 protein expression. (A) Representative immunostaining images from control V-HT22 and SelH-HT22. (B) Representative immunostaining images from glutamate treated V-HT22 and SelH-HT22. (C) Western blot analysis of Apaf-1. HT22 neurons were exposed to 6 mM glutamate for 24 hrs as previously described in material and methods. Apaf-1 immunoreactivity (green color) were detected at higher levels in vector transfected HT22 cells (V-HT22) compared to cells overexpressing SelH (SelH-HT22) under glutamate tension. Glutamate-enhanced expression of the Apaf-1 was significantly attenuated in SelH-HT22 cell in immunoblots. There was no significant difference in Apaf-1 protein expression in cells not treated with glutamate. **p<0.01 vs. non-glutamate control and ##p<0.01 vs. V-HT22 cell with the same treatment. DAPI (blue color) was used to stain cell the nuclei. Scale bar = 125 µm.
Figure 5
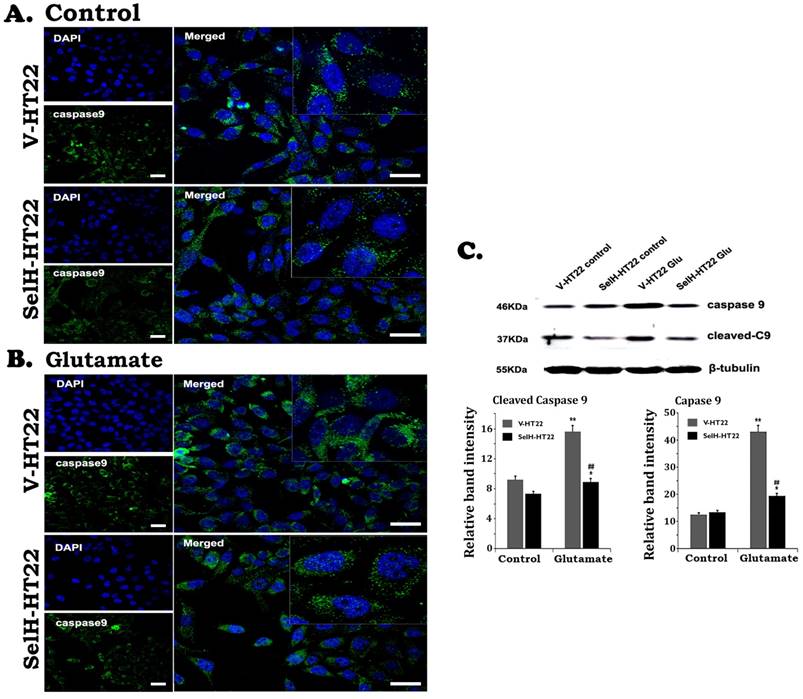
Expression of SelH reduced caspase-9 levels in glutamate exposed HT22 cells. (A) Representative immunostaining images from control V-HT22 and SelH-HT22. (B) Representative immunostaining images from glutamate treated V-HT22 and SelH-HT22. (C) Western blot analysis of cleaved and total capsase-9. HT22 neurons were exposed to glutamate for 24 h. The cleaved caspase-9 immunoreactivity (green color) reduced in SelH overexpressed cells under control and glutamate exposure conditions compared with V-HT22. Glutamate significantly increased the protein levels of total capase-9 and cleaved caspase-9 in the cytosolic fraction of the V-HT22 cell. Overexpression of SelH completely blocked the glutamate-induced elevations of total and cleaved caspase-9. **p<0.01 vs. non-glutamate control and ##p<0.01 vs. V-HT22 cell with the same treatment. DAPI (blue color) was used to stain cell the nuclei. Scale bar=125 µm.
3.6 SelH decreased lysosome staining
The last step of mitochondrial autophagy processes is fusion of lysosome with autophagosome which contains the mitochondria. We performed lysosome and mitochondrial labeling using commercially available LysoTracker Red and MitoTracker Green molecular probes. As shown in Figure 8, there were few punctate MitoTracker Green labeling, but almost no LysoTracker Red positive staining, in the cytosol of both V-HT22 and SelH-HT22 cells. After glutamate incubation, the appearance of LysoTracker staining was significantly increased in V-HT22 cells. Most of the lysosomes were co-localized with the mitochondrial marker as reflected by the orange color in merged image. Overexpression of SelH reduced the labeling of LysoTracker. Immunoblotting showed that LC3-II to LC3-I ratio increased significantly after incubation of V-HT22 cells in glutamate, while SelH overexpression blocked the increase (Figure 8).
4. Discussion
Although the functions of many selenoproteins remain largely unknown, it is widely accepted that they are key players in diverse of cell processes and diseases. The role of selenoproteins in initiation and progression of diseases has been supported, albeit controversial in some events, by findings showing that selenium deficiency or perturbation of cellular selenoprotein dynamics result in immune dysfunction [27,28], cardiovascular disorders [29,30,31], cancer [32,33], impairments in endocrine function [34], and neurological disorders [35,36,37]. SelH is a selenocystein protein that plays a role in the regulation of genes engaged in cellular detoxification and protection against oxidative cell damage [38]. The present study explores the neuroprotective potential of SelH in a glutamate excitotoxicity model to evaluate its prospects as a disease-modifying agent for ischemic stroke.
Figure 6
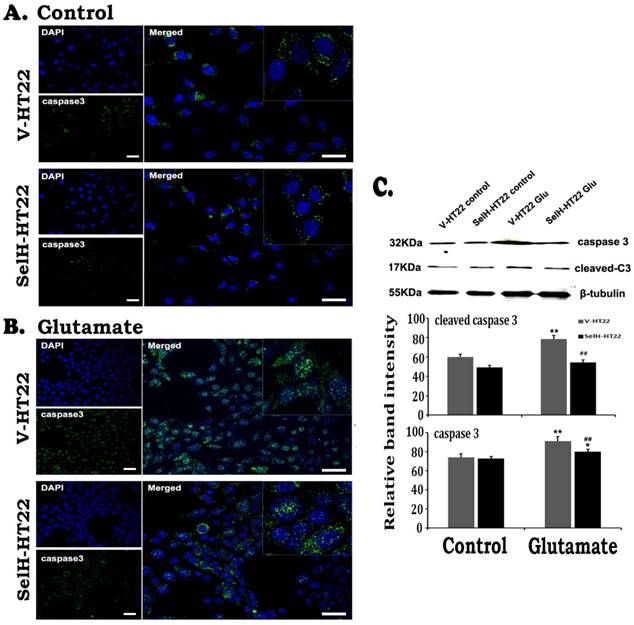
Expression of SelH reduced caspase-3 levels in glutamate exposed HT22 cells. (A) Representative immunostaining images from control V-HT22 and SelH-HT22. (B) Representative immunostaining images from glutamate treated V-HT22 and SelH-HT22. (C) Western blot analysis of cleaved and total capsase-3. HT22 neurons were exposed to glutamate for 24 h. A few caspase-3 positive neurons presented in both vector and SelH transfected cells under control conditions, with no significant difference. The number of caspase-3 positive cells (green color) significantly increased in glutamate treated V-HT22 cells, which is reduced in SelH-HT22 cells exposed to glutamate. Western blotting showed that both cleave and total caspase-3 were increased in glutamate-treated cells and these increases were blocked by overexpression of SelH. **p<0.01 vs. non-glutamate control and ##p<0.01 vs. V-HT22 cell with the same treatment. DAPI (blue color) was used to stain cell the nuclei. Scale bar=125 µm.
Neurodegenerative diseases lead to abrupt or progressive neuronal cell injury and death. Stroke in particular, has an immediate adverse effect on neuronal cells and surrounding tissues as a result of dysfunction in the homeostasis of glutamate that often accompany stroke. Numerous studies including ours have demonstrated that glutamate toxicity in neurons is mediated by oxidative stress as a consequence glutathione (GSH) depletion initiated by inhibition of the uptake of cystine into the cells via the cystine/glutamate antiporter system [23,39,40]. Depletion of GSH activates the mitochondria-initiated apoptotic death pathway leading to ultrastructural changes within the mitochondria, loss of mitochondrial transmembrane potential, increase in ROS, activation of caspases, and aberrant fission of the mitochondria. Our current findings reveal that overexpression of SelH in HT22 cells alleviated glutamate activated mitochondria-dependent cell death by inhibiting the mitochondrial initiated death pathway, and restoration of balance in mitochondrial dynamics.
The neuroprotective actions of selenoproteins are mediated in part through complex cellular antioxidant defense mechanisms that is critical for protection against stress-inducing reactive oxygen species (ROS). ROS including superoxide anions, hydroxyl radicals and hydrogen peroxide are produced as natural byproducts of aerobic respiration by oxidoreductase enzymes via the mitochondrial electron transport system and are important mediators of cell signaling events [41]. However, a disparity in ROS homeostasis favoring the pro-oxidative state has the potential to trigger deleterious events that damage the cell components leading to apoptosis. Glutamate, the most abundant neurotransmitter in the CNS plays a major role in the initiation and progression of neurological disorders. Acute neuronal excitotoxicity induced by glutamate is thought to occur during cerebral ischemia, traumatic brain injury (TBI), and epilepsy [42,43]. In the present experiments we utilized high concentrations of glutamate to model the effect of ischemia in a cultured cell system. Hyper-physiological concentrations of extracellular glutamate triggers two pathways of cell death, a non-receptor mediated oxidative pathway originating from mitochondrial release of cytochrome c and subsequent activation of caspase-3 and the receptor initiated excitotoxic pathway that is caspase independent. ROS production, a major contributor of cell death, is influenced by the glutamate-dependent blockade of the glutamate/cystine antiporter, and cysteine exhaustion and depletion of glutathione [44,45,46]. Under normal conditions, toxic radicals and hydrogen peroxide are detoxified to water and carbon dioxide by antioxidant components of the cell including selenium containing glutathione peroxidases. However, during stress conditions such as present in glutamate toxicity, the cell is overwhelmed by accumulation of ROS provoked by increased production or reduced clearance by the antioxidant system. It is widely accepted that the primary source of ROS is the mitochondria. Mitochondria ROS is produced by glutamate excitotoxicity, hypoxia, and calcium overload after ischemic stroke [47], and several studies have demonstrated the role of ROS in glutamate induced toxicity in HT22 cells [26,40,46,47,48,49]. Alterations in mitochondrial morphology have also been associated with glutamate toxicity. We established in this research that overexpression of SelH was sufficient to ameliorate the production of ROS in HT22 and rescue the cells from glutamate-induced death. SelH considerably moderated mitochondrial cytoskeletal collapse and restored structural integrity to damaged membrane bound components of mitochondria.
Apoptosis, the orderly process of programmed cell death, plays and important role in cellular remodeling. It is generally characterized by prominent morphological features that include cell condensation, fragmentation, cytoskeletal collapse, disassembly of the nuclear envelope, and formation of apoptotic bodies [50,51]. Glutamate-dependent oxidative toxicity leads to failure in the bioenergetics of the cell. Failure of the mitochondrial potential trigger morphological changes in the membrane that result in formation of membrane permeability transition pores (MPTP) and the release of cytochrome c and other pro-apoptotic factors to the cytosol [52]. The cytochrome c discharged in the cytosol binds and activates apoptotic protease activating factor-1 (Apaf-1) and procaspase-9 to form an apoptosome leading to downstream activation of effector caspase-3. Glutamate treatment of vector bearing HT22 cells significantly increased the expression of Apaf-1, cleaved caspase-9, and cleaved caspase-3.
Figure 7
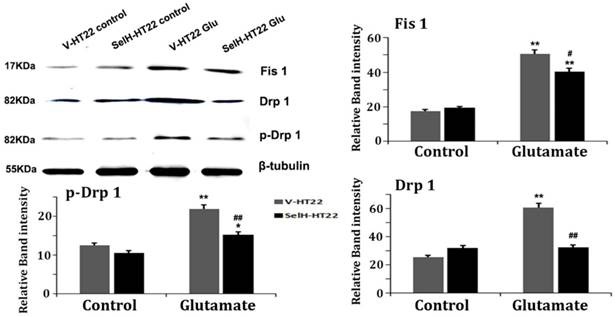
Glutamate enhanced expression of mitochondrial dynamics related proteins were diminished in SelH overexpressing cells. V-HT22 and SelH-HT22 cells were exposed to glutamate for 24 hrs and the expression of mitochondrial fission proteins Fis1 and Drp1, and pDRP1, were quantified by immunoblots. Glutamate significantly increased the expression of Fis1, Drp1 and p-Drp1 in V-HT22 cells compared to cells unexposed to glutamate. Glutamate-dependent increases in expression of Fis1, Drp1 and p-Drp1 were blocked by SelH overexpression in HT22. **p<0.01 vs. non-glutamate control and #p<0.05, ##p<0.01 vs. V-HT22 cell with similar treatment.
Figure 8
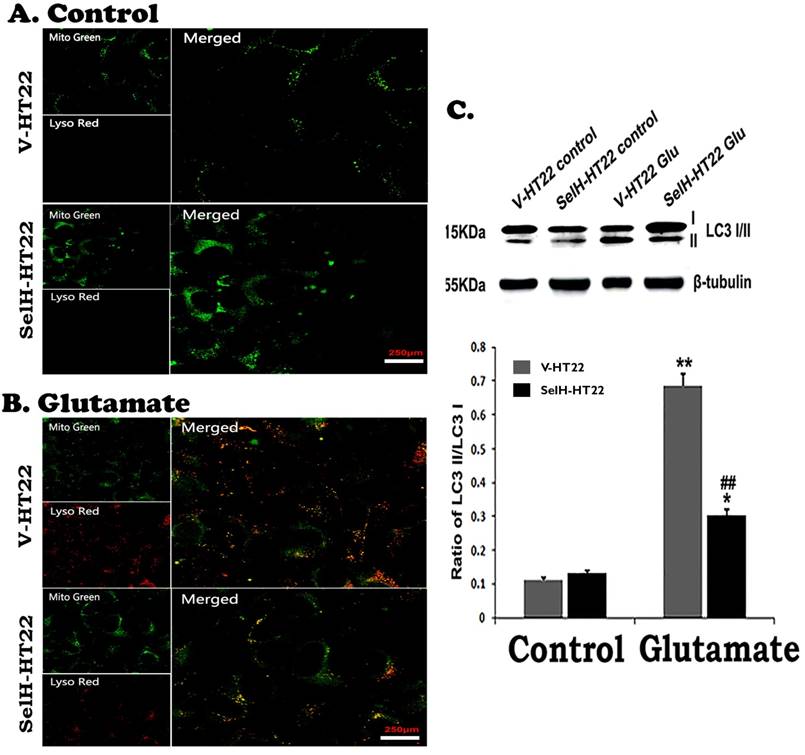
Glutamate stimulated activation of mitophagy marker protein was reversed by SelH expression. (A) Representative immunostaining images from control V-HT22 and SelH-HT22. (B) Representative immunostaining images from glutamate treated V-HT22 and SelH-HT22. (C) Western blot analysis of LC3-I/II. V-HT22 and SelH-HT22 cells were exposed to glutamate for 24 hrs and the expression of autophagy markers was measured. Mitochondria (green stain) are visible in cells devoid of glutamate treatment. Glutamate exposure significantly increased the immunointensity of LysoTracker Red suggesting increased number of lysosomes. LysoTracker Red colocalized with MitoTracker green (orange color) suggesting ongoing mitophagy. Protein blot shows differential increase in the LC3-II level after glutamate exposure in V-HT22, which was significantly attenuated in SelH-HT22 cells. **p<0.01 vs. non-glutamate control and ##p<0.01 vs. V-HT22 cell with the same treatment. Scale bar=250 µm.
Overexpression of SelH in HT22 significantly constrained the expression levels of these pro-apoptotic enzymes. Our results on caspase-3 is at variance with the early studies [53] who reported that glutamate did not cause an increase in cleaved caspase-3 and caspase inhibitor failed to protect HT22 cells from glutamate toxicity. The authors concluded that glutamate cytotoxicity was not mediated by caspase-3, but by the AIF nuclear translocation and calpain activation in HT22 cells. However, recent studies [49,54] demonstrated that glutamate increased cleaved caspase-3, Bax and inhibited Bcl-2 in HT22 cells. Our results are in line with these recent studies. It is not known why the results obtained in recent year are different from those observed on decades ago. The purity and specificity of the antibodies and inhibitors may have been much improved in recent years.
Overexpression of SelH in HT22 significantly constrained the expression levels of these pro-apoptotic enzymes. Our results on caspase-3 is at variance with the early studies [51] who reported that glutamate did not cause an increase in cleaved caspase-3 and caspase inhibitor failed to protect HT22 cells from glutamate toxicity. The authors concluded that glutamate cytotoxicity was not mediated by caspase-3, but by the AIF nuclear translocation and calpain activation in HT22 cells. However, recent studies [52,36] demonstrated that glutamate increased cleaved caspase-3, Bax and inhibited Bcl-2 in HT22 cells. Our results are in line with these recent studies. It is not known why the results obtained in recent year are different from those observed on decades ago. The purity and specificity of the antibodies and inhibitors may have been much improved in recent years.
Selenium supplementation protects against glutamate-induced excitotoxic insult in cell culture by de novo protein synthesis [55]. Selenium pretreatment significantly ameliorated ROS production caused by glutamate toxicity and hypoxia by increasing antioxidant enzyme activities [17,56,57,58]. Based on these findings we hypothesized that overexpression of SelH protein could protect neurons from cell death in a glutamate excitotoxicity model. Consistent with these findings we demonstrate in this report that SelH counteracts glutamate toxicity in HT22 neurons in a manner similar to protection by elemental selenium.
Mitochondrial dynamics is regulated by mechanisms that involve the fission and fusion of mitochondria. Both fission and fusion play prominent roles in sustaining a healthy mitochondrial network and contribute to disease-related processes such as apoptosis and mitophagy. Mitochondrial fission is mediated by dynamin related protein 1 (Drp1) and fission 1 (Fis1). Fis1 is a mitochondrial receptor for Drp1, a predominantly cytosolic protein recruited to the mitochondria during fission [59]. In vivo experiments suggests that selenium accumulate mainly in the mitochondria and in selenoproteins such as glutathione peroxidase. Restriction of selenium or mutations in selenoproteins decrease the activity of these enzymes and exacerbate neuronal cell death, whereas, selenium treatment or enhanced expression of selenoenzymes ameliorates neurodegenerative conditions [60,61]. Existing evidence suggest that glutamate promotes mitochondrial dynamic imbalance by activating Drp1 and Fis1 in HT22 cells while selenium treatment protected the mitochondria by inhibiting the increase in p-Drp1 and Fis1 [26]. Consistent with these reports the present study shows a significant reduction in the cellular contents of the three mitochondrial fission markers in SelH-transfected cells relative to vector-transfected cells and suggests that SelH rescues cells from glutamate insult in part by restoring normal balance to the process of mitochondrial fission.
The central role of mitochondrial autophagy is to selectively target degradation of defective mitochondria in autophagosomes. Evidence exists to show that expression of pro-fission proteins such as Fis1 reduces mitochondrial number by activating mitophagy and apoptosis [62]. One consequence of glutamate excitotoxicity is the activation of mitochondrial fission, fragmentation, and condensation, which were clearly evidenced in this study. During mitophagy, defective mitochondria engulfed into autophagosomes recruit microtubule-associated protein 1 light chain LC3-II (LC3-II) and are delivered to the lysosomes for degradation [63]. We considered the activation of mitophagy after glutamate exposure in V-HT22 cells by the increase in conversion of LC3-I to LC3-II. Moreover, we showed that SelH overexpression decreased the conversion of LC3-I to LC3-II under glutamate tension. More importantly, we observed a significant increase in lysosome to mitochondria ratio in the vector transfected cells, which suggested enhanced level of mitophagic clearance of fragmented and damaged mitochondria in those cells.
In the present study, we have shown that high concentration of glutamate negatively impacts the mitochondria by altering the mitochondrial structure and dysregulation of mitochondria dynamics, which are associated with increase in ROS production and subsequent activation of autophagy in HT22 cells. These glutamate-induced effects are prevented by cellular overexpression of SelH protein. As such we conclude that SelH exerts its neuroprotective effects by reducing glutamate-induced ROS production, restoration of mitochondrial dynamic balance, and modulation of autophagy. Therefore, upregulating SelH gene could provide proof of concept for an efficient strategy to mitigate the toxic effects of glutamate that often lead to neuronal cell death in the event of trauma and other neurologic disease conditions. Selenium supplements, such as synthetic inorganic sodium selenite, selenase and selenate, organic L-selenomethionine, and organic selenium rich yeast, increase selenoprotein levels. Future studies linking the selenium dietary supplementation to the severity of cerebral ischemic brain damage is highly recommended.
Acknowledgements
This work is supported by Natural Science Foundation of China (81560208, 81260184) and West China Top Class Discipline Project in Basic Medical Sciences, Ningxia Medical University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Burk RF, Hill KE. Selenoprotein P: an extracellular protein with unique physical characteristics and a role in selenium homeostasis. Annu Rev Nutr. 2005;25:215-235
2. Labunsky VM, Hatfield DL, Gladyshev VN. Selenoproteins: molecular pathways and physiological roles. Physiol Rev. 2014;94:739-777
3. Maulik N, Yoshida T, Das DK. Regulation of cardiomyocyte apoptosis in ischemic reperfused mouse heart by glutathione peroxidase. Mol Cell Biochem. 1999;196:13-21
4. Voetsch B, Jin RC, Bierl C, Benke KS, Kenet G, Simioni P, Ottaviano F, Damasceno BP, Annichino-Bizacchi JM, Handy DE. et al. Promoter polymorphisms in the plasma glutathione peroxidase (GPx-3) gene: a novel risk factor for arterial ischemic stroke among young adults and children. Stroke. 2007;38:41-49
5. Hatfield DL, Yoo MH, Carlson BA, Gladyshev VN. Selenoproteins that function in cancer and promotion. Biochim Biophys Acta. 2009;1790:1541-1545
6. Brigelius-Flohe R. Selenium compounds and selenoproteins in cancer. Chem Biodivers. 2008;5:389-395
7. Lovell MA, Xiong S, Lyubartseva G, Markesbery WR. Organoselenium (Sel-Plex diet) decreases amyloid burden and RNA and DNA oxidative damage in APP/PS1 mice. Free Radic Biol Med. 2009;46(11):1527-1533
8. Nakayama A, Hill KE, Austin LM, Motley AK, Burk RF. All regions of mouse brain are dependent on selenoprotein P for maintenance of selenium. J Nutr. 2007;137(3):690-693
9. Panee J, Stoytcheva ZR, Liu W, Berry MJ. Selenoprotein H is a redox-sensing high mobility group family DNA-binding protein that up-regulates genes involved in glutathione synthesis and phase II detoxification. J Biol Chem. 2007;282(33):23759-23765
10. Ben Jilani KE, Panee J, He Q, Berry MJ, Li PA. Overexpression of selenoprotein H reduces Ht22 neuronal cell death after UVB irradiation by preventing superoxide formation. Int J Biol Sci. 2007;3(4):198-204
11. Mendelev N, Witherspoon S, Li PA. Overexpression of human selenoprotein H in neuronal cells ameliorates ultraviolet irradiation-induced damage by modulating cell signaling pathways. Exp Neurol. 2009;220(2):328-334
12. Mendelev N, Mehta SL, Witherspoon S, He Q, Sexton JZ, Li PA. Upregulation of human selenoprotein H in murine hippocampal neuronal cells promotes mitochondrial biogenesis and functional performance. Mitochondrion. 2011;11(1):76-82
13. Wu RT, Cao L, Chen BP, Cheng WH. Selenoprotein H suppresses cellular senescence through genome maintenance and redox regulation. J Biol Chem. 2014;289(49):34378-34388
14. Wirth EK, Conrad M, Winterer J, Wozny C, Carlson BA, Roth S, Schmitz D, Bornkamm GW, Coppola V, Tessarollo L. Neuronal selenoprotein expression is required for interneuron development and prevents seizures and neurodegeneration. FASEB J. 2010;24(3):844-852
15. Venardos K, Ashton K, Headrick J, Perkins A. Effects of dietary selenium on post-ischemic expression of antioxidant mRNA. Mol Cell Biochem. 2005;270(1-2):131-138
16. Furling D, Ghribi O, Lahsaini A, Mirault ME, Massicotte G. Impairment of synaptic transmission by transient hypoxia in hippocampal slices: improved recovery in glutathione peroxidase transgenic mice. Proc Natl Acad Sci U S A. 2000;97(8):4351-4356
17. Ansari MA, Ahmad AS, Ahmad M, Salim S, Yousuf S, Ishrat T, Islam F. Selenium protects cerebral ischemia in rat brain mitochondria. Biol Trace Elem Res. 2004;101(1):73-86
18. Almeida A, Allen KL, Bates TE, Clark JB. Effect of reperfusion following cerebral ischaemia on the activity of the mitochondrial respiratory chain in the gerbil brain. J Neurochem. 1995;65(4):1698-1703
19. Bowling AC, Beal MF. Bioenergetic and oxidative stress in neurodegenerative diseases. Life Sci. 1995;56(14):1151-1171
20. Piantadosi CA, Zhang J. Mitochondrial generation of reactive oxygen species after brain ischemia in the rat. Stroke. 1996;27(2):327-331 discussion 332
21. Lancelot E, Beal MF. Glutamate toxicity in chronic neurodegenerative disease. Prog Brain Res. 1998;116:331-347
22. Sattler R, Tymianski M. Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol Neurobiol. 2001;24(1-3):107-129
23. Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2(6):1547-1558
24. Pereira CF, Oliveira CR. Oxidative glutamate toxicity involves mitochondrial dysfunction and perturbation of intracellular Ca2+ homeostasis. Neurosci Res. 2000;37(3):227-236
25. Wu S, Zhou F, Zhang Z, Xing D. Mitochondrial oxidative stress causes mitochondrial Fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J. 2011;278(6):941-954
26. Kumari S, Mehta SL, Li PA. Glutamate induces mitochondrial dynamic imbalance and autophagy activation: preventive effects of selenium. PLoS One. 2012;7(6):e39382
27. Hoffmann PR, Berry MJ. The influence of selenium on immune responses. Mol Nutr Food Res. 2008;52(11):1273-1280
28. Wang C, Wang H, Luo J, Hu Y, Wei L, Duan M, He H. Selenium deficiency impairs host innate immune response and induces susceptibility to Listeria monocytogenes infection. BMC Immunol. 2009;10:55
29. Benstoem C, Goetzenich A, Kraemer S, Borosch S, Manzanares W, Hardy G, Stoppe C. Selenium and its supplementation in cardiovascular disease-what do we know? Nutrients. 2015;7(5):3094-3118
30. Brigo F, Storti M, Lochner P, Tezzon F, Nardone R. Selenium supplementation for primary prevention of cardiovascular disease: proof of no effectiveness. Nutr Metab Cardiovasc Dis. 2014;24(1):e2-3
31. Rees K, Hartley L, Day C, Flowers N, Clarke A, Stranges S. Selenium supplementation for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013:CD009671
32. Cox AG, Tsomides A, Kim AJ, Saunders D, Hwang KL, Evason KJ, Heidel J, Brown KK, Yuan M, Lien EC. Selenoprotein H is an essential regulator of redox homeostasis that cooperates with p53 in development and tumorigenesis. Proc Natl Acad Sci U S A. 2016;113(38):E5562-5571
33. Xie W, Yang M, Chan J, Sun T, Mucci LA, Penney KL, Lee GS, Kantoff PW. Association of genetic variations of selenoprotein genes, plasma selenium levels, and prostate cancer aggressiveness at diagnosis. Prostate. 2016;76(7):691-699
34. Kohrle J, Jakob F, Contempre B, Dumont JE. Selenium, the thyroid, and the endocrine system. Endocr Rev. 2005;26(7):944-984
35. Cardoso BR, Roberts BR, Bush AI, Hare DJ. Selenium, selenoproteins and neurodegenerative diseases. Metallomics. 2015;7(8):1213-1228
36. Dominiak A, Wilkaniec A, Wroczynski P, Adamczyk A. Selenium in the Therapy of Neurological Diseases. Where is it Going? Curr Neuropharmacol. 2016;14(3):282-299
37. Pitts MW, Byrns CN, Ogawa-Wong AN, Kremer P, Berry MJ. Selenoproteins in nervous system development and function. Biol Trace Elem Res. 2014;161(3):231-245
38. Novoselov SV, Kryukov GV, Xu XM, Carlson BA, Hatfield DL, Gladyshev VN. Selenoprotein H is a nucleolar thioredoxin-like protein with a unique expression pattern. J Biol Chem. 2007;282(16):11960-11968
39. Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J Neurosci. 2006;26(41):10514-10523
40. Gliyazova NS, Huh EY, Ibeanu GC. A novel phenoxy thiophene sulphonamide molecule protects against glutamate evoked oxidative injury in a neuronal cell model. BMC Neurosci. 2013;14:93
41. Alfadda AA, Sallam RM. Reactive oxygen species in health and disease. J Biomed Biotechnol. 2012:936486
42. Meldrum BS. The role of glutamate in epilepsy and other CNS disorders. Neurology. 1994;44(11 Suppl 8):S14-23
43. Fukui M, Song JH, Choi J, Choi HJ, Zhu BT. Mechanism of glutamate-induced neurotoxicity in HT22 mouse hippocampal cells. Eur J Pharmacol. 2009;617(1-3):1-11
44. Chen J, Small-Howard A, Yin A, Berry MJ. The responses of Ht22 cells to oxidative stress induced by buthionine sulfoximine (BSO). BMC Neurosci. 2005;6:10
45. Tan S, Sagara Y, Liu Y, Maher P, Schubert D. The regulation of reactive oxygen species production during programmed cell death. J Cell Biol. 1998;141(6):1423-1432
46. Ha JS, Park SS. Glutamate-induced oxidative stress, but not cell death, is largely dependent upon extracellular calcium in mouse neuronal HT22 cells. Neurosci Lett. 2006;393(2-3):165-169
47. Mergenthaler P, Dirnagl U, Meisel A. Pathophysiology of stroke: lessons from animal models. Metab Brain Dis. 2004;19(3-4):151-167
48. Kang Y, Tiziani S, Park G, Kaul M, Paternostro G. Cellular protection using Flt3 and PI3Kalpha inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat Commun. 2014;5:3672
49. Lee DS, Ko W, Kim DC, Kim YC, Jeong GS. Cudarflavone B provides neuroprotection against glutamate-induced mouse hippocampal HT22 cell damage through the Nrf2 and PI3K/Akt signaling pathways. Molecules. 2014;19(8):10818-10831
50. Hacker G. The morphology of apoptosis. Cell Tissue Res. 2000;301(1):5-17
51. Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam AR, Kashani HH. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24-49
52. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495-516
53. Zhang Y, Bhavnani BR. Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci. 2006:15 7:49
54. Yue L, Zhao L, Liu H, Li X, Wang B, Guo H, Gao L, Feng D, Qu Y. Adiponectin Protects against glutamate-Induced excitotoxicity via Activating SIRT1-Dependent PGC-1α Expression in HT22 Hippocampal Neurons. Oxid Med Cell Longev. 2016;11:30
55. Savaskan NE, Brauer AU, Kuhbacher M, Eyupoglu IY, Kyriakopoulos A, Ninnemann O, Behne D, Nitsch R. Selenium deficiency increases susceptibility to glutamate-induced excitotoxicity. FASEB J. 2003;17(1):112-114
56. Bordoni A, Biagi PL, Angeloni C, Leoncini E, Danesi F, Hrelia S. Susceptibility to hypoxia/reoxygenation of aged rat cardiomyocytes and its modulation by selenium supplementation. J Agric Food Chem. 2005;53(2):490-494
57. Zhou YJ, Zhang SP, Liu CW, Cai YQ. The protection of selenium on ROS mediated-apoptosis by mitochondria dysfunction in cadmium-induced LLC-PK(1) cells. Toxicol In Vitro. 2009;23(2):288-294
58. Sarada SK, Himadri P, Ruma D, Sharma SK, Pauline T, Mrinalini. Selenium protects the hypoxia induced apoptosis in neuroblastoma cells through upregulation of Bcl-2. Brain Res. 2008;1209:29-39
59. Loson OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell. 2013;24(5):659-667
60. Crack PJ, Taylor JM, Flentjar NJ, de Haan J, Hertzog P, Iannello RC, Kola I. Increased infarct size and exacerbated apoptosis in the glutathione peroxidase-1 (Gpx-1) knockout mouse brain in response to ischemia/reperfusion injury. J Neurochem. 2001;78(6):1389-1399
61. Hoehn B, Yenari MA, Sapolsky RM, Steinberg GK. Glutathione peroxidase overexpression inhibits cytochrome C release and proapoptotic mediators to protect neurons from experimental stroke. Stroke. 2003;34(10):2489-2494
62. Gomes LC, Scorrano L. High levels of Fis1, a pro-fission mitochondrial protein, trigger autophagy. Biochim Biophys Acta. 2008;1777(7-8):860-866
63. Baumann K. Autophagy: Mitophagy receptors unravelled. Nat Rev Mol Cell Biol. 2015;16(10):580
Author contact
![]() Corresponding authors: Dr. P. Andy Li, Email: pliedu or Dr. Jian-Zhong Zhang, Email: zhangjzedu.cn
Corresponding authors: Dr. P. Andy Li, Email: pliedu or Dr. Jian-Zhong Zhang, Email: zhangjzedu.cn
Citation styles
APA
Ma, Y.M., Guo, Y.Z., Ibeanu, G., Wang, L.Y., Dong, J.D., Wang, J., Jing, L., Zhang, J.Z., Li, P.A. (2017). Overexpression of selenoprotein H prevents mitochondrial dynamic imbalance induced by glutamate exposure. International Journal of Biological Sciences, 13(11), 1458-1469. https://doi.org/10.7150/ijbs.21300.
ACS
Ma, Y.M.; Guo, Y.Z.; Ibeanu, G.; Wang, L.Y.; Dong, J.D.; Wang, J.; Jing, L.; Zhang, J.Z.; Li, P.A. Overexpression of selenoprotein H prevents mitochondrial dynamic imbalance induced by glutamate exposure. Int. J. Biol. Sci. 2017, 13 (11), 1458-1469. DOI: 10.7150/ijbs.21300.
NLM
Ma YM, Guo YZ, Ibeanu G, Wang LY, Dong JD, Wang J, Jing L, Zhang JZ, Li PA. Overexpression of selenoprotein H prevents mitochondrial dynamic imbalance induced by glutamate exposure. Int J Biol Sci 2017; 13(11):1458-1469. doi:10.7150/ijbs.21300. https://www.ijbs.com/v13p1458.htm
CSE
Ma YM, Guo YZ, Ibeanu G, Wang LY, Dong JD, Wang J, Jing L, Zhang JZ, Li PA. 2017. Overexpression of selenoprotein H prevents mitochondrial dynamic imbalance induced by glutamate exposure. Int J Biol Sci. 13(11):1458-1469.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.