Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Fibroblast growth factor (FGF)...
FGFR2-related syndromic...
Syndrome management
Pharmacologic blockade of...
Conclusions and future aspects
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(12):1479-1488. doi:10.7150/ijbs.22373 This issue Cite
Review
Fibroblast Growth Factor Receptor 2 (FGFR2) Mutation Related Syndromic Craniosynostosis
Saïd C. Azoury1, Sashank Reddy2, Vivek Shukla3 ![]() , Chu-Xia Deng4
, Chu-Xia Deng4 ![]()
1. Department of Surgery, The Johns Hopkins Hospital, Baltimore, MD, USA;
2. Department of Plastic and Reconstructive Surgery, The Johns Hopkins Hospital, Baltimore, MD, USA;
3. TGIB, NCI, National Institutes of Health, Bethesda, MD, USA;
4. Faculty of Health Sciences, University of Macau, Macau SAR, China.
Received 2017-8-14; Accepted 2017-10-1; Published 2017-11-2
Abstract

Craniosynostosis results from the premature fusion of cranial sutures, with an incidence of 1 in 2,100-2,500 live births. The majority of cases are non-syndromic and involve single suture fusion, whereas syndromic cases often involve complex multiple suture fusion. The fibroblast growth factor receptor 2 (FGFR2) gene is perhaps the most extensively studied gene that is mutated in various craniosynostotic syndromes including Crouzon, Apert, Pfeiffer, Antley-Bixler, Beare-Stevenson cutis gyrata, Jackson-Weiss, Bent Bone Dysplasia, and Seathre-Chotzen-like syndromes. The majority of these mutations are missense mutations that result in constitutive activation of the receptor and downstream molecular pathways. Treatment involves a multidisciplinary approach with ultimate surgical fixation of the cranial deformity to prevent further sequelae. Understanding the molecular mechanisms has allowed for the investigation of different therapeutic agents that can potentially be used to prevent the disorders. Further research efforts are need to better understand screening and effective methods of early intervention and prevention. Herein, the authors provide a comprehensive update on FGFR2-related syndromic craniosynostosis.
Keywords: Fibroblast Growth Factor Receptor, Mutations, Syndromic, Craniosynostosis.
Introduction
Craniosynostosis results from the premature fusion of cranial sutures, with an incidence of 1 in 2,100-2,500 live births (1-4). In normal development, cranial growth occurs perpendicular to suture lines leading to a well-proportioned head with a longer anteroposterior than bitemporal dimension (5). Sutures typically fuse in an orderly fashion with the first suture - metopic - closing within the first year of life and the final sutures squamosal and lambdoidal not closing until the fourth or fifth decade of life. Premature fusion of one or more sutures precludes growth of cranial bones perpendicular to the affected sutures and leads to compensatory growth in a parallel direction, resulting in a cranial dysmorphic head shape (6,7).
The vast majority of craniosynostoses involve single suture fusion, though multiple sutures can be affected (5). Depending on which sutures prematurely fused, patients will have characteristic phenotypes. There are four major sutures including sagittal, coronal, metopic and labdoid, as well as minor sutures including frontonasal, temporosquamosal, and frontosphenoidal. The sagittal suture is involved in 40-58% of cases, followed by unilateral or bilateral coronal (20-30%), metopic (5-15%) and lambdoid (2-4% of cases) suture fusion (3, 5, 8). Sagittal suture fusion results in an increase of the anterior-posterior diameter resulting in dolichocephaly or scaphocephaly (5). Unilateral coronal synostosis results in anterior plagiocephaly whereas bilateral coronal fusion results in brachycephaly. Premature metopic suture fusion is associated with triangular shaped forehead, trigonocephaly. Lambdoid synostosis is usually unilateral, resulting in asymmetric posterior plagiocephaly, whereas bilateral lambdoid synostosis results in flattened and widened occipital region (5). Kleeblattschadel or cloverleaf skull is a term given for a trilobed skull resulting from synostosis of lambdoidal and coronal sutures.
Cases of craniosynostoses are thought to result from a complex interaction of environment, epigenetics, and genetics (9, 10). Evidence from a population-based study suggests that multiple births, maternal age greater than 35, birth weight >4000 g, and male sex are risk factors for craniosynostosis (11). Other potential risk factors include advanced paternal age, higher parental education, maternal exposure to valproate acid, preterm delivery, and in-utero growth constraint (1,12-14). The vast majority of these cases are non-syndromic and 80-90% of cases involve a single suture line (15). On the contrary, approximately 10-20% of craniosynostosis cases are syndromic, mostly with autosomal dominant inheritance, incomplete penetrance, and variable expressivity (6). The syndromic synostoses, such as Crouzon, Apert, and Pfeiffer, may involve multiple sutures and extracranial phenotypes are mostly of the hands and feet (16). Occasionally complex or multisuture craniosynostosis occurs in patients without an identifiable syndrome (17). Although there have been reports of prenatal diagnoses of craniosynostosis, the majority are diagnosed postnatally including those associated with syndromes.
Recently, whole exome and genome sequencing has enabled identification of hundreds of mutations implicated in craniosynostosis. Genes most commonly mutated in familial craniosynostosis include FGFR2, FGFR3, twist homologue 1 (TWIST1) and ephrin-B1 (EFNB1) (2,18-21) Of these, FGFR variants have been most extensively characterized and serve as a basis for understanding the molecular underpinnings of craniosynostosis. Herein, the authors provide a comprehensive update on FGFR2-related syndromic craniosynostosis.
Fibroblast growth factor (FGF) receptors (FGFRs)
The FGF family, comprised of at least 22 known FGF ligands, binds to five related receptor tyrosine kinases, resulting in receptor monomer dimerization, activation of kinase domains and phosphorylation of the receptor (22-24). It is well known that FGF/FGFR signaling plays important roles in numerous biological processes, including bone development and homeostasis by controlling differentiation of mesenchymal and neuroectoermal cells (25-27). Mutations in three members of the FGFR family (FGFR1, FGFR2, and FGFR3) result in unregulated FGF signaling and premature suture closure. Much of the current knowledge regarding the role of FGF/FGFR in cranial growth is derived from mouse models. With the exception of FGFR5, members of the FGFR gene family encode a receptor tyrosine kinase (RTK), which has an extracellular ligand-binding portion composed of immunoglobulin-like domains (IgI, IgII, and IgIII), a transmembrane region, and intracellular tyrosine kinase domains (TK1 and TK2) (Figure 1) (3, 28-32).
Schematic representation of FGFR signaling pathway. A typical FGFR gene encodes a tyrosine kinase (TK) receptor. This receptor has an extracellular ligand-binding sites, which are comprised of immunoglobulin-like domains (IgI, IgII and IgIII), transmembrane region and divided intracellular tyrosine kinase domains, TK1 and TK2. The signaling pathway mainly operates through at least 3 distinct pathways. Initiation of RAS/MAPK pathways starts with formation of FRS2 complex and regulates cell proliferation and differentiation. The PI3K/AKT pathway controls cells survival and fate determination after getting initiated by FRS2 complex formation. Activation of PKC pathway started with binding of PLCγ to the activated FGFR and regulates morphogenesis and migration of cells.
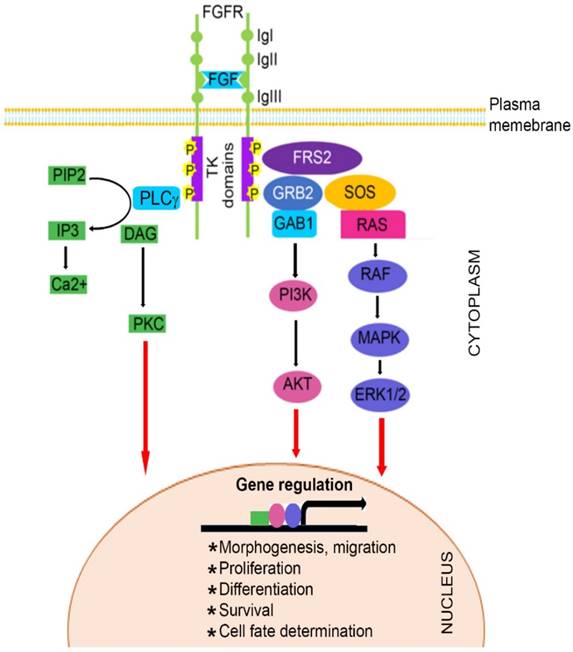
Within the IgII domain, a positively charged conserved region serves as the binding site for heparin sulphate (26). FGF binding occurs at IgII and IgIII as well as the link between these Ig-like domains. Alternative splicing in the IgIII domain results in various isoforms of the receptor with differing affinities for FGF ligands. The N-terminal part of the IgIII domain is encoded by exon IIIa and the C-terminal part is encoded by either exon IIIb or IIIc (26).
Ligand binding activates downstream pathways including phosphoinositide 3-kinase (PI3K)/Akt, protein kinase C, phospholipase C, and mitogen-activated protein kinase [20, 27, 28]. Downstream of RAS, the extracellular signal-related kinases 1 and 2 (ERK1/2) are key proteins mediating mitogen-activated protein kinase (MAPK) in FGFR2 signaling (31,33). Recent evidence suggests that reduced levels of ERF, which encodes an inhibitory ETS transcription factor directly bound to ERK1/2, results in complex multisuture craniosynostosis in humans and mice (32,34). This suggests that FGFR2 mediated ERK-MAPK activity regulates coronal suture formation (34).
Craniosynostotic disorders caused by mutations in FGFR2 gene are perhaps the most extensively studied (35). Both minor and major sutures fusion occurs in most patients with FGFR2-related craniofacial dyostosis (36). Abundant FGFR2 expression is found in the cartilage of the cranial base (25,37-39). FGFR2 in cranial sutures is largely expressed in differentiating osteoblasts and osteoprogenitor cells. Via downstream pathways, FGF/FGFR2 signaling mediates osteoprogenitor proliferation, differentiation, and apoptosis (25,40). Early differentiation is thought to be a leading factor involved in premature suture fusion (3). As will be discussed, autosomal dominant FGFR2 mutations enhance receptor signaling through dimerization independent of ligand binding with resultant constitutive activation (41,42). Most pathogenic variants of FGFR2 are missense variants, with less frequent reports of deletions, insertions, and splice site mutations (28). Mutations in FGFR2 less commonly result in non-syndromic craniosynostoses. The discussion in this review will be limited to FGFR2-related syndromic craniosynostosis.
FGFR2-related syndromic craniosynostosis
Heterozygous activating mutations of FGFR2 result in several craniosynostosis syndromes, such as Crouzon, Apert, Pfeiffer, Antley-Bixler, Beare-Stevenson cutis gyrata, Jackson-Weiss, Bent Bone Dysplasia, and Seathre-Chotzen-like syndromes (43-47) (Table 1). Whereas the majority of mutations in FGFR2 localize to two exons encoding the extracellular IgIII domain (IIIa/exon 8, and IIIc/exon 10), mutations have been identified in exons coding the tyrosine kinase region (exons 3, 5, 14, 15, 16 and 17), and exon 11 that encodes the transmembrane domain, as well as the IgI and IgII domains (48-51) (Table 1).
FGFR2- Associated Craniosynostotic Syndromes
| Syndrome | Inheritance | Mutations | Craniosynostosis Findings | Extracranial Phenotypes | Ref. |
|---|---|---|---|---|---|
| Crouzon Syndrome | Autosomal dominant | Cys278Phe, Trp289Gly, Tyr290Gly, Ser267Pro, Tyr328Cys, Gly338Arg, Tyr340His, Cys342Tyr, Cys342Arg, Cys342Phe, Cys342Ser, Cys342Trp, Ala344Gly, Asn549Thr, Ser347Cys, Ser354Cys | Bicoronal synostosis, pansynostosis late | Crouzonoid face (flattened forehead, proptosis, hypertelorism, beaked nose, midface hypoplasia), normal hands | [35, 51,54-56,61] |
| Pfeiffer Syndrome | Autosomal dominant | Ala314Ser, Asp321Ala, Thr342Pro, Cys278Phe, Cys342Tyr, Trp290Cys, Tyr340Cys, Cys342Tyr, Cys342Arg, Cys342Ser, Cys342Trp, Ser351Arg, Val359Phe | Type I: Bicoronal synostosisType II: Kleeblattschädel (cloverleaf skull) and extreme proptosisType III: Bicoronal synostosis, No cloverleaf skull | Nasal abnormalities, proptosis, hypertelorism, frontal bossing, broad thumbs and great toes, hydrocephalus with type II | [35,52, 55-57,61, 104] |
| Apert Syndrome | Autosomal dominant | Ser252Trp, Pro253Arg, Ser252Phe | Bicoronal synostosis | Midface hypoplasia, macrocephaly, downslated palpebral fissues, radial deviation of thumb, severe syndactyly of hands and feet | [3, 63-65] |
| Antley-Bixler Syndrome | Autosomal recessive/ dominant forms | Ser351CysTyr290Cys | Multisuture synostosis | Midface hypoplasia, radiohumeral synostosis, joint contractures and arachnodactyly | [77-82] |
| Beare-Stevenson cutis gyrata syndrome | Autosomal dominant | Tyr375Cys, Ser372Cys | Kleeblattschädel deformity | Cutis gyrata (furrowed skin), ear defects, anogenital deformities | [83-94] |
| Jackson-Weiss Syndrome | Autosomal dominant | Ala344Gly, Glu289Pro, Ser347Cys, Cys342Arg,Cys342Ser | Variable | Facial dysmorphism and foot malformations (hands typically normal) | [10, 35, 95-99] |
| Bent Bone Dysplasia syndrome | Autosomal dominant | Tyr381Asp, Met391Arg | Bicoronal synostosis | Dysmorphic oral maxillofacial features as well as hypoplastic pubis, clavicles, osteopenia, and bent long bones | [40, 99,100] |
| Seathre-Chotzen-like syndrome | Autosomal dominant | Gln289ProVV269-70del | Heterogeneous patterns of craniosynostosis: uni- or bicoronal, sagittal, or metopic synostosis | Syndactyly of hands and feet, ptosis, hearing loss, hypertelorism, and spine abnormalities | [35,101-103] |
Both Crouzon and Pfieffer are typically inherited in an autosomal dominant fashion and have similar molecular mechanisms. Crouzon is usually the mildest of the syndromes. Bicoronal synostosis is most common in Crouzon syndrome; however, pansynostosis may also be a late presenting feature in some cases. The crouzonoid facial appearance, which consists of flattened forehead, proptosis, hypertelorism, beaked nose, and midface hypoplasia without major limb deformities characterizes the disorder (26, 52) (Table 1).
The flattened forehead is secondary to bicoronal synostosis. In Pfieffer syndrome, the craniofacial severity is variable and extracranial manifestations include big toes and broad radially deviated thumbs (53). Various clinical subtypes of Pfieffer syndrome have been described, type 1 being compatible with life, and types 2 and 3 associated with more severe complex synostosis and higher associated mortality (54). The craniofacial features of the Pfieffer syndromes differ somewhat: type 1 typically has moderate-to-severe midface hypoplasia, type 2 has cloverleaf skull and extreme proptosis, and type 3 is characterized by turribrachycephaly (a high and prominent forehead) as well as proptosis (28). The FGFR2 mutations in Crouzon and Pfeiffer syndrome overlap, most often occurring in extracellular IgIII domain encoded by exons 8 (IIIa) or 10 (IIIc) (46). Both syndromes may be caused by the Cys278Phe and Cys342Tyr missense mutations (55-57). Severe cases of Pfeiffer syndrome are caused by a small subset of substitutions encoding Trp290Cys, Tyr340Cys, Cys342Arg or Ser351Cys (58-60). The Trp290Cys mutation has also been linked to tracheal cartilaginous sleeves in children with syndromic craniosynostosis (61). Recently, Ke and colleagues presented a unique report on a young boy with Crouzon syndrome who had bicoronal craniosynostosis, proptosis, and maxillary hypoplasia and identified a heterozygous Ser267Pro mutation in exon 8 (55). Other mutations identified in patients with features resembling the Crouzon/Pfeiffer syndrome include Leu617Phe mutation in the TK domain and Gly271Val in the IgIIIa domain of FGFR2 (62). A unique mutation leading to Ala315Ser in FGFR2 has also been identified in in a family with a mild phenotype of Crouzon craniofacial dyostosis but without overt craniosynostosis (63). A report from Japan also identified a novel FGFR2 Asn549Thr mutation in a patient with Crouzon syndrome (10).
Apert syndrome is characterized by bicoronal synostosis, midface hypoplasia, and complex syndactyly involving both hands and feet (Table 1). The abnormal skull morphology is a result of maldevelopment of brain calvarias and cranial base (64). This fusion of the digits differentiates Apert syndrome from other FGFR2-related syndromes. Apert syndrome mutations have been shown to follow autosomal dominant inheritance (3,65,66). Apert syndrome is typically caused by specific missense mutations of the highly-conserved linker region between IgII and IgIII domains of FGFR2, most often either Ser252Trp or Pro253Arg (16,43,67-69). Cleft palate can also occur in this disorder, most commonly in those with Ser252Trp mutations (43). Other studies corroborate the findings of Ser252Trp and Pro253Arg mutations in majority of patients with FGFR2 related Apert syndrome, although Ser252Trp variant is more common, and Ser252Phe mutations have also been reported (44,70-72). Interestingly, unlike the Ser252Trp variant, the Pro253Arg mutation results in generalized increased affinity of FGFs to FGFR2 (28,73). Such receptor mutations exhibit selective decrease in the dissociation kinetics of FGF2 but not of other FGF ligands examined (74). Recently, Andreou and colleagues reported a young girl with Pro253Arg FGFR2 mutation-associated Apert syndrome who also had early-onset low-grade papillary carcinoma (75). Studies by Lomri et al. on Apert syndrome provided evidence to suggest that FGFR2 mutations resulted in greater numbers of precursor cells entering the osteogenic pathway, which lead to premature calvarial ossification and bone matrix formation (76). FGFR2 mutations may result in premature suture fusion through upregulation of epidermal growth factor (EGF) and platelet derived growth factor (PDGF) α signaling (77).
Antley-Bixler is a rare syndrome associated with multisuture synostosis as well as a broad spectrum of other congenital anomalies including midface hypoplasia, radiohumeral synostosis, joint contractures and arachnodactyly (Table 1) (78-80). Prognosis is poor and mortality in the newborn is high, largely from airway issues. Although the syndrome has been described for several decades now, the molecular basis is evolving (81). Antley-Bixler syndrome was thought to have autosomal recessive inheritance, but more recently evidence has emerged of a dominant de novo mutation in FGFR2 resulting in a Ser351Cys amino acid substitution in the IgIII domain (82,83). These findings suggest that there may be both an autosomal recessive and dominant form.
Beare-Stevenson cutis gyrata syndrome is characterized by several anomalies in addition to the skin condition of redundant and convoluted folds, including craniosynostosis, ear defects, anogenital anomalies (Table 1) (84-86). The syndrome follows an autosomal dominant inheritance pattern. Although the syndrome is exceedingly rare with only a few cases reported, the majority of the patients with craniosynostosis had severe Kleeblattschadel deformity (87-90). Beare-Stevenson is caused by de novo heterozygous mutations in FGFR2, including Tyr375Cys mutation in the transmembrane domain, and Ser372Cys mutation in the carboxy-terminal end of the linker region between IgIII and transmembrane domains (90-94). These activating point mutations account for the majority of Beare-Stevenson syndrome cases. A de novo heterozygous 63-bp deletion in the FGFR2 was also noted in a boy presenting with Beare-Stevenson, and the authors proposed that such a deletion would alter the splicing of isoform IIIc (95). However, in the same report, no mutations were found in a few patients with the syndrome, suggesting genetic heterogeneity.
Jackson-Weiss syndrome is another rare craniosynostostic disorder, with variable phenotypes, inherited in an autosomal dominant fashion (Table 1). Patients with this syndrome also demonstrate facial dysmorphism and foot malformations including tarsal/metatarsal fusion, although the hands are normal in appearance (96). Further investigation of one of the families described in the original report of Amish kindreds led to the identification of Ala344Gly mutation in the conserved region of IgIII domain of FGFR2 (97). Similarly, other reports have confirmed missense mutations in FGFR2 exons IIIa and IIIc in Jackson-Weiss, including a Glu289Pro mutation in exon IIIa (98,99). A novel FGFR2 Ser347Cys mutation was identified in a Japanese patient with Jackson-Weiss syndrome (10).
De novo FGFR2 mutations have been identified in Bent Bone Dysplasia (BBDS), a rare occurring perinatal lethal skeletal dysplasia of autosomal dominant inheritance. BBDS is characterized by craniosynostosis, dysmorphic oral maxillofacial features as well as hypoplastic pubis, clavicles, osteopenia, and bent long bones (100,101). One study demonstrated that four individuals affected with this disorder were heterozygous for missense mutations that resulted in the addition of a polar amino acid to the hydrophobic transmembrane region of the receptor (100). In BBDS, unique mutations reduce receptor levels at the plasma membrane and decrease responsiveness to FGF2, therefore attenuating canonical signaling. Heterozygosity for the de novo missense mutations Tyr381Asp and Met391Arg has been reported (100). More recently, in addition to canonical signaling at the plasma membrane, FGFR2 has been shown to regulate bone formation within the nucleolus (40). These aforementioned mutations also enhance nucleolar occupancy of FGFR2 at the ribosomal DNA (rDNA) promoter (40).
Seathre-Chotzen is yet another autosomal dominant craniosynostotic condition most commonly associated with mutations in the TWIST gene (Table 1). However, Paznekas and colleagues reported on a single case with a val-val deletion in FGFR2 (102). Several years later, a Gln289Pro mutation in FGFR2 was detected in a family with Saethre-Chotzen syndrome (103). The spectrum of craniosynostosis observed may be either uni- or bicoronal, with sagittal suture fusion exceedingly rare and metopic suture fusion found in few reports (104). Other features of the disorder include short stature, including syndactyly of hands and feet, hypertelorism, ptosis, and abnormalities of the spine.
An unspecified craniosynostosis syndrome, craniofacial-skeletal-dermatological dysplasia, has been associated with a Trp290Cys mutation in the extracellular domain of FGFR2 (105). This syndrome is characterized by complex multiple suture craniosynostosis, hydrocephalus, corneal scleralization, atresia of auditory meatus, choanal stenosis, broad thumbs and great toes, scoliosis, acanthosis nigricans, and mental retardation.
Syndrome management
Management of syndromic craniosynostosis is optimized by a multidisciplinary approach involving plastic and reconstructive surgeons, neurosurgeons, neurologists, pediatricians, and genetic counselors. Various imaging modalities are used to better facilitate prenatal diagnosis including ultrasound, magnetic resonance imaging (MRI) and genetic testing (106). Ultrasound may be used to look for associated limb anomalies and craniofacial dysmorphologies that may point to a particular syndrome. 3-D ultrasound and MRI should be used to further assess findings suspicious for a craniosynostosis syndrome on two-dimensional ultrasound (53,107). MRI is particularly helpful in evaluating associated central nervous system anomalies.
Given the autosomal dominant inheritance pattern of FGFR2-related craniosynostosis syndromes, genetic counseling is critical once the diagnosis is confirmed. Follow-up should be scheduled regularly to monitor for signs and symptoms of increased intracranial pressure. Once a pathogenic variant has been identified, prenatal testing is possible, although the predictive value is poor. Non-invasive means such as the use of cell-free fetal DNA present in the maternal blood to assess for known pathogenic gene mutations is possible in the first trimester (108). Amniocentesis and chorionic villus sampling are invasive procedures traditionally use for prenatal screening. Testing for pathogenic variants of genes such as FGFR2 is indicated in high-risk pregnancies with family histories significant for craniosynostosis, but routine fetal ultrasound is sufficient for low-risk pregnancies (106).
Treatment is typically geared towards surgical correction of the clinical manifestations, the prevention of future deformation, while reducing the risk of increased intracranial pressure. Surgery is done emergently in cases of increased intracranial pressure. Otherwise, the optimal timing for fixation remains controversial, with some surgeons preferring early intervention to allow for expansion of a constricted brain and corneal protection, and others delaying surgery to allow for more stability of the skull in order to decrease the need for revisional surgery (106). The goal of surgery is to improve skull, orbit, and facial deformities. Whereas experience has shown that single suture craniosynostosis can often be managed successfully with a single surgical procedure, complex/multiple suture craniosynostosis often necessitates a staged approach (17). Surgical correction methods include open calvarial reconstruction, distraction, and less invasive techniques such as regional cranioectomy, liberation of occipital bone flap and spring-mediated cranial vault remodeling (106,109). Increasing experience at the Children's Hospital of Philadelphia, a high-volume center for syndromic craniosynostosis surgery, has favored posterior cranial vault distraction over fronto-orbital advancement as the initial cranial remodeling procedure (106). Posterior cranial vault distraction has several advantages including superior cranial volume expansion, improvements in anterior and posterior cranial morphology, with favorable morbidity profile (106). Early data in patients with Apert syndrome suggests that the overall number of major craniofacial procedures within the first 5 years of life is decreased following early posterior cranial vault distraction (106).
Pharmacologic blockade of craniosynostotic pathways
There is a developing interest in inhibiting the molecular pathways involved in the development of craniosynostosis in the aforementioned syndromes. A soluble form of FGFR2 with Ser252Trp mutation inhibits osteoblastic differentiation caused by gain-of-function mutations in FGFR2 in an Apert mouse model, and partially prevents craniosynostosis (110-112). Further attenuation of the signaling pathways has also been shown to prevent premature suture fusion (113). Uncoupling of the docking protein FRS2 and activated FGFR2 (Figure 1) leads to normal skull development in a murine model of Crouzon-like craniosynostosis (113). Using a murine model, Xu and colleagues showed that an activating mutation in FGFR2 results in increased trabecular bone formation and decreased bone resorption through a Wnt/β-catenin-dependent anabolic effect (114). Pharmacologic inhibition of Wnt/β-catenin signaling partially reverses this process (114). These early studies provide support for developing agents that inhibit the pathways that lead to craniosynostosis. Also, knowing that patients previously diagnosed with FGFR2-related Crouzon syndrome had ERF-related craniosynostosis, ERF may serve as an target for therapeutic modulation (32,115). In a mouse model of craniosynostosis, Shukla and colleagues demonstrated that a small hairpin RNA targeting the dominant mutant form of FGFR2 prevents craniosynostosis in mice (116,117). Pharmacologic blockade of mitogen-activated protein (MAP) kinase 1 and 2 (MEK1/2)/ERK pathway (Figure 1) by U0126 in mutant mice significantly inhibits craniosynostosis (116). These authors proposed that small-molecules could potentially be used for both prevention and therapy.
Compared to the investigations underway for FGFR-related malignancies, less progress has been made with regard to therapeutic strategies for FGFR-related skeletal and craniosynostotic disorders. Tyrosine kinase inhibition is one such promising strategy in mutated FGFR-related craniosynostotic conditions (118). PD173074, an FGFR tyrosine kinase inhibitor, has been used in a model of FGFR2 related Crouzon syndrome, demonstrating coronal suture patency and no evidence of craniosynostosis (119). Wang et al. observed ligand-independent phosphorylation of FGFR2 in a model of Beare-Stevenson cutis gyrate syndrome and p38 MAPK pathway activation (120). The use of p38 kinase inhibitor suppressed premature suture closure in mutated mice (120). Recently, a tyrosine kinase inhibitor (Figure 1), ARQ 087, has been shown to rescue the major pathological effects of activating mutations in FGFR1, FGFR2, and FGFR3 in experimental models of chondrodysplasias and craniosynostosis (121).
Conclusions and future aspects
The FGFR2 is perhaps the most extensively studied gene that is mutated in various craniosynostotic syndromes including Crouzon, Apert, Pfeiffer, Antley-Bixler, Beare-Stevenson cutis gyrata, Jackson-Weiss, Bent Bone Dysplasia, and Seathre-Chotzen-like syndromes. The majority of these mutations are missense mutations that result in constitutive activation of the receptor and downstream molecular pathways. Treatment involves a multidisciplinary approach with ultimate surgical fixation of the cranial deformity to prevent further sequelae. Understanding the molecular mechanisms has allowed for the investigation of different therapeutic agents that can potentially be used to prevent the disorders. Genetic studies are recommended for not only individuals with known syndromic features, but also for family members with uncommon or normal phenotypes to improve counseling outreach.
One of the more fascinating questions in the biology of the FGFR2 gene is why mutations in this single gene can give rise to such a wide range of disorders with such a broad spectrum of phenotypes. As seen in the preceding sections, some disorders involve single suture synostosis, whereas others affect multiple sutures. Some disorders are principally confined to the craniofacial skeleton, whereas others have prominent extracranial dysmorphologies. Given the extensive study of FGFR2 mutations in clinical samples, this gene could prove an interesting test case for systematically mapping genotype-phenotype relationships in a highly conserved pathway. It will be interesting to discover whether various mutations in FGFR2 and its pathway members have subtly different activities, and how these activities give rise to the observed spectrum of phenotypes. Another possibility is that modifier genes alter the phenotypic response in patients to a common array of gain or loss of function mutations. The accumulation of patient sequencing data and more studies in animal models will be important to sorting out these possibilities.
Further research efforts are need to better understand screening and effective methods of early intervention and prevention. Also, knowing that some individuals with mutations may be phenotypically normal and have syndromic offspring, a better understanding of the genetic, epigenetic, and environment interplay is critical. When deciding surgical options for patients with craniosynostosis, more data is necessary to guide optimal timing of intervention as well as the best method of cranial remodeling to allow for minimal morbidity and less need for future interventions.
Acknowledgements
The authors would like to thank the Department of Surgery and Plastic and Reconstructive Surgery at the Johns Hopkins hospital, as well as the National Institutes of Health (Bethesda, MD). This review was supported by the Chair Professor Grant (CPG2017-00026-FHS) granted to C.D by University of Macau.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lajeunie E, Le Merrer M, Bonaiti-Pellie C, Marchac D, Renier D. Genetic study of nonsyndromic coronal craniosynostosis. Am J Med Genet. 1995;55(4):500-4
2. Wilkie AOM, Byren JC, Hurst JA, Jayamohan J, Johnson D, Knight SJL. et al. Prevalence and Complications of Single-Gene and Chromosomal Disorders in Craniosynostosis. Pediatrics [Internet]. 2010;126(2):e391-400 Available from: http://pediatrics.aappublications.org/cgi/doi/10.1542/peds.2009-3491
3. Johnson D, Wilkie AOM. Craniosynostosis. Eur J Hum Genet [Internet]. 2011;19(4):369-76 Available from: http://www.nature.com/doifinder/10.1038/ejhg.2010.235
4. Wilkie AOM. Craniosynostosis: Genes and mechanisms. Hum Mol Genet. 1997;6(10 REV. ISS.):1647-56
5. Ciurea A V, Toader C. Genetics of craniosynostosis: review of the literature. J Med Life [Internet]. 2008;2(1):5-17 Available from: papers://a61f8494-ea99-4e99-9b25-0ee136663c66/Paper/p47
6. Badve CA, Mallikarjunappa MK, Iyer RS, Ishak GE, Khanna PC. Craniosynostosis: Imaging review and primer on computed tomography. Pediatric Radiology. 2013:728-42
7. Burke MJ, Winston KR, Williams S. Normal sutural fusion and the etiology of single sutural craniosynostosis: The microspicule hypothesis. Pediatr Neurosurg. 1955;22(5):241-7
8. Kimonis V, Gold JA, Hoffman TL, Panchal J, Boyadjiev SA. Genetics of Craniosynostosis. Semin Pediatr Neurol. 2007;14(3):150-61
9. Magge SN, Snyder K, Sajja A, DeFreitas TA, Hofherr SE, Broth RE. et al. Identical Twins Discordant for Metopic Craniosynostosis: Evidence of Epigenetic Influences. J Craniofac Surg. United States. 2017Jan;28(1):14-6
10. Ohishi A, Nishimura G, Kato F, Ono H, Maruwaka K, Ago M. et al. Mutation analysis of FGFR1-3 in 11 Japanese patients with syndromic craniosynostoses. Am J Med Genet Part A. 2017;173(1):157-62
11. Boulet SL, Rasmussen SA, Honein MA. A population-based study of craniosynostosis in metropolitan Atlanta, 1989-2003. Am J Med Genet Part A. 2008;146(8):984-91
12. Singh RP, Dhariwal D, Bhujel N, Shaikh Z, Davies P, Nishikawa H. et al. Role of parental risk factors in the aetiology of isolated non-syndromic metopic craniosynostosis. Br J Oral Maxillofac Surg. 2010;48(6):438-42
13. Sanchez-Lara PA, Carmichael SL, Graham JM, Lammer EJ, Shaw GM, Ma C. et al. Fetal constraint as a potential risk factor for craniosynostosis. Am J Med Genet Part A. 2010;152(2):394-400
14. Barik M, Bajpai M, Das RR, Panda SS. Study of environmental and genetic factors in children with craniosynostosis: A case-control study. J Pediatr Neurosci [Internet]. 2013;8(2):89-92 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3783739&tool=pmcentrez&rendertype=abstract
15. Boyadjiev SA. Genetic analysis of non-syndromic craniosynostosis. Orthod Craniofacial Res. 2007;10(3):129-37
16. Ko JM. Genetic syndromes associated with craniosynostosis. J Korean Neurosurg Soc. 2016;59(3):187-91
17. Czerwinski M, Kolar JC, Fearon JA. Complex Craniosynostosis. Plast Reconstr Surg [Internet]. 2011;128(4):955-61 Available from: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00006534-201110000-00029
18. Sharma VP, Fenwick AL, Brockop MS, McGowan SJ, Goos JAC, Hoogeboom AJM. et al. Mutations in TCF12, encoding a basic helix-loop-helix partner of TWIST1, are a frequent cause of coronal craniosynostosis. Nat Genet [Internet]. 2013;45(3):304-7 Available from: http://www.nature.com/doifinder/10.1038/ng.2531
19. Marie PJ, Kaabeche K, Guenou H. Roles of FGFR2 and twist in human craniosynostosis: Insights from genetic mutations in cranial osteoblasts. Front Oral Biol. 2008;12:144-59
20. Seto ML, Hing A V, Chang J, Hu M, Kapp-Simon KA, Patel PK. et al. Isolated sagittal and coronal craniosynostosis associated with TWIST box mutations. Am J Med Genet Part A. 2007;143(7):678-86
21. Johnson D, Wall SA, Mann S, Wilkie AO. A novel mutation, Ala315Ser, in FGFR2: a gene-environment interaction leading to craniosynostosis? Eur J Hum Genet [Internet]. 2000;8(8):571-7 Available from: http://www.nature.com/doifinder/10.1038/sj.ejhg.5200499
22. Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine and Growth Factor Reviews. 2005:139-49
23. Itoh N, Ornitz DM. Functional evolutionary history of the mouse Fgf gene family. Developmental Dynamics. 2008:18-27
24. Klint P, Claesson-Welsh L. Signal transduction by fibroblast growth factor receptors. Front Biosci [Internet]. 1999;4(7):D165-77 Available from: http://www.ncbi.nlm.nih.gov/pubmed/9989949
25. Su N, Jin M, Chen L. Role of FGF/FGFR signaling in skeletal development and homeostasis: learning from mouse models. Bone Res [Internet]. 2014;2:14003. Available from: http://www.nature.com/articles/boneres20143
26. Moosa S, Wollnik B. Altered FGF signalling in congenital craniofacial and skeletal disorders. Seminars in Cell and Developmental Biology. 2016:115-25
27. Lei H, Deng C-X. Fibroblast growth factor receptor 2 signaling in breast cancer. Int J Biol Sci. In press
28. Robin NH. FGFR-Related Craniosynostosis Syndromes. GeneReviews. 2007:1-30
29. Marie PJ, Coffin JD, Hurley MM. FGF and FGFR signaling in chondrodysplasias and craniosynostosis. J Cell Biochem. 2005;96(5):888-96
30. Sleeman M, Fraser J, McDonald M, Yuan S, White D, Grandison P. et al. Identification of a new fibroblast growth factor receptor, FGFR5. Gene. 2001;271(2):171-82
31. Miraoui H, Oudina K, Petite H, Tanimoto Y, Moriyama K, Marie PJ. Fibroblast growth factor receptor 2 promotes osteogenic differentiation in mesenchymal cells via ERK1/2 and protein kinase C signaling. J Biol Chem. 2009;284(8):4897-904
32. Twigg SRF, Vorgia E, McGowan SJ, Peraki I, Fenwick AL, Sharma VP. et al. Reduced dosage of ERF causes complex craniosynostosis in humans and mice and links ERK1/2 signaling to regulation of osteogenesis. Nat Genet [Internet]. 2013;45(3):308-13 Available from: http://www.nature.com/doifinder/10.1038/ng.2539
33. Plotnikov A, Chuderland D, Karamansha Y, Livnah O, Seger R. Nuclear Extracellular Signal-Regulated Kinase 1 and 2 Translocation Is Mediated by Casein Kinase 2 and Accelerated by Autophosphorylation. Mol Cell Biol [Internet]. 2011;31(17):3515-30 Available from: http://mcb.asm.org/cgi/doi/10.1128/MCB.05424-11
34. Pfaff MJ, Xue K, Li L, Horowitz MC, Steinbacher DM, Eswarakumar JVP. FGFR2c-mediated ERK-MAPK activity regulates coronal suture development. Dev Biol. 2016;415(2):242-50
35. Chen L, Deng CX. Roles of FGF signaling in skeletal development and human genetic diseases. Front Biosci. 2005;10:1961-76
36. Runyan CM, Xu W, Alperovich M, Massie JP, Paek G, Cohen BA. et al. Minor suture fusion in syndromic craniosynostosis. Plast Reconstr Surg. United States. 2017 May
37. Rice DPC, Rice R, Thesleff I. Fgfr mRNA isoforms in craniofacial bone development. Bone. 2003;33(1):14-27
38. Iseki S, Wilkie AO, Morriss-Kay GM. Fgfr1 and Fgfr2 have distinct differentiation- and proliferation-related roles in the developing mouse skull vault. Development [Internet]. 1999;126(24):5611-20 Available from: http://www.ncbi.nlm.nih.gov/pubmed/10572038
39. Lazarus JE, Hegde A, Andrade AC, Nilsson O, Baron J. Fibroblast growth factor expression in the postnatal growth plate. Bone. 2007;40(3):577-86
40. Neben CL, Idoni B, Salva JE, Tuzon CT, Rice JC, Krakow D. et al. Bent bone dysplasia syndrome reveals nucleolar activity for FGFR2 in ribosomal DNA transcription. Hum Mol Genet. 2014;23(21):5659-71
41. Mangasarian K, Li Y, Mansukhani A, Basilico C. Mutation associated with crouzon syndrome causes ligand-independent dimerization and activation of FGF receptor-2. J Cell Physiol. 1997;172(1):117-25
42. Robertson SC, Meyer a N, Hart KC, Galvin BD, Webster MK, Donoghue DJ. Activating mutations in the extracellular domain of the fibroblast growth factor receptor 2 function by disruption of the disulfide bond in the third immunoglobulin-like domain. Proc Natl Acad Sci U S A. 1998;95(April):4567-72
43. Slaney SF, Oldridge M, Hurst J a, Moriss-Kay GM, Hall CM, Poole MD. et al. Differential effects of FGFR2 mutations on syndactyly and cleft palate in Apert syndrome. Am J Hum Genet. 1996;58(5):923-32
44. Wilkie AOM, Slaney SF, Oldridge M, Poole MD, Ashworth GJ, Hockley AD. et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet [Internet]. 1995;9(2):165-72 Available from: http://www.nature.com/doifinder/10.1038/ng0295-165
45. Schell U, Hehr A, Feldman GJ, Robin NH, Zackai EH, De Die-smulders C. et al. Mutations in FGFR1 and FGFR2 cause familial and sporadic pfeiffer syndrome. Hum Mol Genet. 1995;4(3):323-8
46. Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet [Internet]. 1994;8(1):98-103 Available from: http://www.nature.com/doifinder/10.1038/ng0994-98
47. Passos-Bueno MR, Wilcox WR, Jabs EW, Sertie AL, Alonso LG, Kitoh H. Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat. 1999;14(2):115-25
48. Suh YJ, Bae HS, Choi JY, Lee JH, Kim MJ, Kim S. et al. A novel FGFR2 mutation in tyrosine kinase II domain, L617F, in Crouzon syndrome. J Cell Biochem. 2014;115(1):102-10
49. Zankl A, Jaeger G, Bonafé L, Boltshauser E, Superti-Furga A. Novel mutation in the tyrosine kinase domain of FGFR2 in a patient with Pfeiffer syndrome. Am J Med Genet Part A [Internet]. 2004;131A(3):299-300 Available from: http://doi.wiley.com/10.1002/ajmg.a.30366
50. McGillivray G, Savarirayan R, Cox TC, Stojkoski C, McNeil R, Bankier A. et al. Familial scaphocephaly syndrome caused by a novel mutation in the FGFR2 tyrosine kinase domain. J Med Genet. England. 2005Aug;42(8):656-62
51. Pulleyn LJ, Reardon W, Wilkes D, Rutland P, Jones BM, Hayward R. et al. Spectrum of craniosynostosis phenotypes associated with novel mutations at the fibroblast growth factor receptor 2 locus. Eur J Hum Genet [Internet]. 1996;4(5):283-91 Available from: https://www.ncbi.nlm.nih.gov/pubmed/8946174
52. Hoefkens MF, Vermeij-Keers C, Vaandrager JM. Crouzon syndrome: phenotypic signs and symptoms of the postnatally expressed subtype. J Craniofac Surg. 2004;15(2):233-240 -242
53. Giancotti A, D'Ambrosio V, Marchionni E, Squarcella A, Aliberti C, La Torre R. et al. Pfeiffer syndrome: literature review of prenatal sonographic findings and genetic diagnosis. J Matern Fetal Neonatal Med. England. 2017Sep;30(18):2225-31
54. Cohen MM. Pfeiffer syndrome update, clinical subtypes, and guidelines for differential diagnosis. Am J Med Genet. 1993;45(3):300-7
55. Ke R, Yang X, Tianyi C, Ge M, Lei J, Mu X. The C342R Mutation in FGFR2 Causes Crouzon Syndrome With Elbow Deformity. J Craniofac Surg [Internet]. 2015;26(2):584-6 Available from: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00001665-201503000-00070
56. Kress W, Collmann H, Büsse M, Halliger-Keller B, Mueller CR. Clustering of FGFR2 gene mutations inpatients with Pfeiffer and Crouzon syndromes (FGFR2-associated craniosynostoses). Cytogenet Cell Genet. 2000;91(1-4):134-7
57. Chen C-P, Lin S-P, Liu Y-P, Chern S-R, Chen S-W, Lai S-T. et al. Pfeiffer syndrome with FGFR2 C342R mutation presenting extreme proptosis, craniosynostosis, hearing loss, ventriculomegaly, broad great toes and thumbs, maxillary hypoplasia, and laryngomalacia. Taiwan J Obstet Gynecol. China (Republic : 1949- ). 2017Jun;56(3):412-4
58. Lajeunie E, Heuertz S, El Ghouzzi V, Martinovic J, Renier D, Le Merrer M. et al. Mutation screening in patients with syndromic craniosynostoses indicates that a limited number of recurrent FGFR2 mutations accounts for severe forms of Pfeiffer syndrome. Eur J Hum Genet [Internet]. 2006;14(3):289-98 Available from: http://www.nature.com/doifinder/10.1038/sj.ejhg.5201558
59. Chen CP, Lin SP, Su YN, Chien SC, Tsai FJ, Wang W. Craniosynostosis and congenital tracheal anomalies in an infant with Pfeiffer syndrome carrying the W290C FGFR2 mutation. Genet Couns. 2008;19(2):165-72
60. Chokdeemboon C, Mahatumarat C, Rojvachiranonda N, Tongkobpetch S, Suphapeetiporn K, Shotelersuk V. FGFR1 and FGFR2 Mutations in Pfeiffer Syndrome. J Craniofac Surg [Internet]. 2013;24(1):150-2 Available from: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00001665-201301000-00035
61. Wenger TL, Dahl J, Bhoj EJ, Rosen A, McDonald-McGinn D, Zackai E. et al. Tracheal cartilaginous sleeves in children with syndromic craniosynostosis. Genet Med [Internet]. 2016(January):1-7 Available from: http://www.nature.com/doifinder/10.1038/gim.2016.60
62. Goos JAC, van den Ouweland AMW, Swagemakers SMA, Verkerk AJMH, Hoogeboom AJM, van Veelen MLC. et al. A novel mutation in FGFR2. Am J Med Genet Part A. 2015;167(1):123-7
63. Graul-Neumann LM, Klopocki E, Adolphs N, Mensah MA, Kress W. Mutation c.943G>T (p.Ala315Ser) in FGFR2 Causing a Mild Phenotype of Crouzon Craniofacial Dysostosis in a Three-Generation Family. Mol Syndromol. 2017;8(2):93-7
64. Luo F, Xie Y, Xu W, Huang J, Zhou S, Wang Z. et al. Deformed Skull Morphology Is Caused by the Combined Effects of the Maldevelopment of Calvarias, Cranial Base and Brain in FGFR2-P253R Mice Mimicking Human Apert Syndrome. Int J Biol Sci. Australia. 2017;13(1):32-45
65. Glaser RL, Jiang W, Boyadjiev SA, Tran AK, Zachary AA, Van Maldergem L. et al. Paternal Origin of FGFR2 Mutations in Sporadic Cases of Crouzon Syndrome and Pfeiffer Syndrome. Am J Hum Genet [Internet]. 2000;66(3):768-77 Available from: http://linkinghub.elsevier.com/retrieve/pii/S0002929707640075
66. Mantilla-Capacho JM, Arnaud L, Díaz-Rodriguez M, Barros-Núñez P. Apert syndrome with preaxial polydactyly showing the typical mutation Ser252Trp in the FGFR2 gene. Genet Couns. 2005;16(4):403-6
67. Ferreira JC, Carter SM, Bernstein PS, Jabs EW, Glickstein JS, Marion RW. et al. Second-trimester molecular prenatal diagnosis of sporadic Apert syndrome following suspicious ultrasound findings. Ultrasound Obstet Gynecol. 1999;14(6):426-30
68. Ibarra-Arce A, Ortiz de Zarate-Alarcon G, Flores-Pena LG, Martinez-Hernandez F, Romero-Valdovinos M, Olivo-Diaz A. Mutations in the FGFR2 gene in Mexican patients with Apert syndrome. Genet Mol Res. Brazil. 2015Mar;14(1):2341-6
69. Kunwar F, Tewari S, Bakshi SR. Apert syndrome with S252W FGFR2 mutation and characterization using Phenomizer: An Indian case report. J Oral Biol Craniofacial Res. 2017;7(1):67-71
70. Lajeunie E, Cameron R, El Ghouzzi V, de Parseval N, Journeau P, Gonzales M. et al. Clinical variability in patients with Apert's syndrome. J Neurosurg [Internet]. 1999;90(3):443-7 Available from: http://thejns.org/doi/10.3171/jns.1999.90.3.0443
71. Oldridge M, Lunt PW, Zackai EH, McDonald-McGinn DM, Muenke M, Moloney DM. et al. Genotype-phenotype correlation for nucleotide substitutions in the IgII-IgIII linker of FGFR2. Hum Mol Genet. 1997;6(1):137-43
72. Oldridge M, Zackai EH, McDonald-McGinn DM, Iseki S, Morriss-Kay GM, Twigg SRF. et al. De Novo Alu-Element Insertions in FGFR2 Identify a Distinct Pathological Basis for Apert Syndrome. Am J Hum Genet [Internet]. 1999;64(2):446-61 Available from: http://linkinghub.elsevier.com/retrieve/pii/S0002929707617509
73. Ibrahimi O a, Chiu ES, McCarthy JG, Mohammadi M. Understanding the molecular basis of Apert syndrome. Plast Reconstr Surg. 2005;115(1):264-70
74. Andersen J, Burns HD, Enriquez-Harris P, Wilkie AOM, Heath JK. Apert syndrome mutations in fibroblast growth factor receptor 2 exhibit increased affinity for FGF ligand. Hum Mol Genet. 1998;7(9):1475-83
75. Andreou A, Lamy A, Layet V, Cailliez D, Gobet F, Pfister C. et al. Early-onset low-grade papillary carcinoma of the bladder associated with Apert syndrome and a germline FGFR2 mutation (Pro253Arg). Am J Med Genet A. United States. 2006Oct;140(20):2245-7
76. Lomri A, Lemonnier J, Hott M, de Parseval N, Lajeunie E, Munnich A. et al. Increased calvaria cell differentiation and bone matrix formation induced by fibroblast growth factor receptor 2 mutations in Apert syndrome. J Clin Invest [Internet]. 1998;101(6):1310-7 Available from: http://www.ncbi.nlm.nih.gov/cgi-bin/Entrez/referer?http://www.ncbi.nlm.nih.gov/htbin-post/Omim/getmim?field=medline_uid&search=9502772
77. Miraoui H, Ringe J, Häupl T, Marie PJ. Increased EFG-and PDGFα-receptor signaling by mutant FGF-receptor 2 contributes to osteoblast dysfunction in Apert craniosynostosis. Hum Mol Genet. 2010;19(9):1678-89
78. Rumball KM, Pang E, Letts RM. Musculoskeletal manifestations of the Antley-Bixler syndrome. J Pediatr Orthop B. 1999;8(2):139-43
79. McGlaughlin KL, Witherow H, Dunaway DJ, David DJ, Anderson PJ. Spectrum of Antley-Bixler Syndrome. J Craniofac Surg [Internet]. 2010;21(5):1560-4 Available from: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00001665-201009000-00055
80. R. A. Trapezoidocephaly, midfacial hypoplasia and cartilage abnormalities with multiple synostoses and skeletal fractures, MALFORMATION SYNDROMES [Internet]. Birth Defects: Original Article Series. 1975:397-401 Available from: http://ovidsp.ovid.com/ovidweb.cgi?T=JS&PAGE=reference&D=emcl2&NEWS=N&AN=0976112090
81. Robinson LK, Powers NG, Dunklee P, Sherman S, Jones KL. The Antley-Bixler syndrome. J Pediatr. 1982;101(2):201-5
82. Chun K, Siegel-Bartelt J, Chitayat D, Phillips J, Ray PN. FGFR2 mutation associated with clinical manifestations consistent with Antley-Bixler syndrome. Am J Med Genet. 1998;77(3):219-24
83. Reardon W, Smith A, Honour JW, Hindmarsh P, Das D, Rumsby G. et al. Evidence for digenic inheritance in some cases of Antley-Bixler syndrome? J Med Genet [Internet]. 2000;37(1):26-32 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1734444&tool=pmcentrez&rendertype=abstract
84. Beare JM, Dodge JA, Nevin NC. Cutis gyratum, acanthosis nigricans and other congenital anomalies. A new syndrome. Br J Dermatol. England. 1969Apr;81(4):241-7
85. Stevenson RE, Ferlauto GJ, Taylor HA. Cutis gyratum and acanthosis nigricans associated with other anomalies: a distinctive syndrome. J Pediatr. United States. 1978Jun;92(6):950-2
86. Hall BD, Cadle RG, Golabi M. Beare-Stevenson cutis gyrata syndrome. Am J Med Genet. 1992;44:82-9
87. Bratanič B, Praprotnik M, Novosel-Sever M. Congenital craniofacial dysostosis and cutis gyratum: The Beare-Stevenson syndrome. Eur J Pediatr. 1994;153(3):184-6
88. Cohen MM. Craniosynostosis update 1987. Am J Med Genet Suppl. 1988;4:99-148
89. Fonseca RF, Costa-Lima MA, Pereira ET, Castilla EE, Orioli IM. Beare-Stevenson cutis gyrata syndrome: A new case of a c.1124C→G (Y375C) mutation in the FGFR2 gene. Mol Med Rep. 2008;1(5):753-5
90. Przylepa KA, Paznekas W, Zhang M, Golabi M, Bias W, Bamshad MJ. et al. Fibroblast growth factor receptor 2 mutations in Beare-Stevenson cutis gyrata syndrome. Nat Genet [Internet]. 1996;13(4):492-4 Available from: http://www.nature.com/doifinder/10.1038/ng0896-492
91. Wang T-J, Huang C-B, Tsai F-J, Wu J-Y, Lai R-B, Hsiao M. Mutation in the FGFR2 gene in a Taiwanese patient with Beare-Stevenson cutis gyrata syndrome. Clin Genet. 2002;61:218-21
92. Vargas RAP, Maegawa GHB, Taucher SC, Leite JCL, Sanz P, Cifuentes J. et al. Beare-Stevenson syndrome: Two South American patients with FGFR2 analysis. Am J Med Genet A [Internet]. 2003;121A(1):41-6 Available from: http://www.ncbi.nlm.nih.gov/pubmed/12900900
93. Fonseca R, Costa-Lima MA, Cosentino V, Orioli IM. Second case of Beare-Stevenson syndrome with an FGFR2 Ser372Cys mutation. Am J Med Genet Part A. 2008;146(5):658-60
94. Wenger TL, Bhoj EJ, Wetmore RF, Mennuti MT, Bartlett SP, Mollen TJ. et al. Beare-Stevenson syndrome: Two new patients, including a novel finding of tracheal cartilaginous sleeve. Am J Med Genet Part A. 2015;167(4):852-7
95. Slavotinek A, Crawford H, Golabi M, Tao C, Perry H, Oberoi S. et al. Novel FGFR2 deletion in a patient with Beare-Stevenson-like syndrome. Am J Med Genet A. United States. 2009Aug;149A(8):1814-7
96. Jackson CE, Weiss L, Reynolds WA, Forman TF, Peterson JA. Craniosynostosis, midfacial hypoplasia and foot abnormalities: an autosomal dominant phenotype in a large Amish kindred. J Pediatr. United States. 1976Jun;88(6):963-8
97. Jabs EW, Li X, Scott AF, Meyers G, Chen W, Eccles M. et al. Jackson-Weiss-Syndrome and Crouzon-Syndrome Are Allelic with Mutations in Fibroblast Growth-Factor Receptor-2. Nat Genet. 1994;8(3):275-9
98. Tartaglia M, Di Rocco C, Lajeunie E, Valeri S, Velardi F, Battaglia PA. Jackson-Weiss syndrome: Identification of two novel FGFR2 missense mutations shared with Crouzon and Pfeiffer craniosynostotic disorders. Hum Genet. 1997;101(1):47-50
99. Meyers GA, Day D, Goldberg R, Daentl DL, Przylepa KA, Abrams LJ. et al. FGFR2 exon IIIa and IIIc mutations in Crouzon, Jackson-Weiss, and Pfeiffer syndromes: evidence for missense changes, insertions, and a deletion due to alternative RNA splicing. Am J Hum Genet. United States. 1996Mar;58(3):491-8
100. Merrill AE, Sarukhanov A, Krejci P, Idoni B, Camacho N, Estrada KD. et al. Bent bone dysplasia-FGFR2 type, a distinct skeletal disorder, has deficient canonical FGF signaling. Am J Hum Genet. 2012;90(3):550-7
101. Handa A, Okajima Y, Izumi N, Yamanaka M, Kurihara Y. Bent bone dysplasia (BBD)-FGFR2 type: the radiologic manifestations in early gestation. Pediatr Radiol. 2016;46(2):296-9
102. Paznekas WA, Cunningham ML, Howard TD, Korf BR, Lipson MH, Grix AW. et al. Genetic Heterogeneity of Saethre-Chotzen Syndrome, Due to TWIST and FGFR Mutations. Am J Hum Genet [Internet]. 1998;62(6):1370-80 Available from: http://linkinghub.elsevier.com/retrieve/pii/S0002929707627785
103. Lopes Burrone De Freitas ÉC, Dantas Nascimento SR, Palandi De Mello M, Gil-Da-Silva-Lopes VL. Q289P mutation in FGFR2 gene causes saethre-chotzen syndrome: Some considerations about familial heterogeneity. Cleft Palate-Craniofacial J. 2006;43(2):142-7
104. Johnson D, Horsley SW, Moloney DM, Oldridge M, Twigg SR, Walsh S. et al. A comprehensive screen for TWIST mutations in patients with craniosynostosis identifies a new microdeletion syndrome of chromosome band 7p21.1. Am J Hum Genet [Internet]. 1998;63(5):1282-93 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1377539&tool=pmcentrez&rendertype=abstract
105. Shotelersuk V, Ittiwut C, Srivuthana S, Mahatumarat C, Lerdlum S, Wacharasindhu S. Distinct craniofacial-skeletal-dermatological dysplasia in a patient with W290C mutation in FGFR2. Am J Med Genet. 2002;113(1):4-8
106. Taylor JA, Bartlett SP. What's New in Syndromic Craniosynostosis Surgery? Plast Reconstr Surg. United States. 2017Jul;140(1):82e-93e
107. Giancotti A, D'Ambrosio V, De Filippis A, Aliberti C, Pasquali G, Bernardo S. et al. Comparison of ultrasound and magnetic resonance imaging in the prenatal diagnosis of Apert syndrome: report of a case. Childs Nerv Syst [Internet]. 2014;30(8):1445-8 Available from: https://www.ncbi.nlm.nih.gov/pubmed/24566675
108. Benn P. Non-Invasive Prenatal Testing Using Cell Free DNA in Maternal Plasma: Recent Developments and Future Prospects. J Clin Med [Internet]. 2014;3(2):537-65 Available from: http://www.mdpi.com/2077-0383/3/2/537/
109. Governale LS. Craniosynostosis. Pediatric Neurology. 2015:394-401
110. Tanimoto Y, Yokozeki M, Hiura K, Matsumoto K, Nakanishi H, Matsumoto T. et al. A soluble form of fibroblast growth factor receptor 2 (FGFR2) with S252W mutation acts as an efficient inhibitor for the enhanced osteoblastic differentiation caused by FGFR2 activation in Apert syndrome. J Biol Chem. United States. 2004Oct;279(44):45926-34
111. Morita J, Nakamura M, Kobayashi Y, Deng CX, Funato N, Moriyama K. Soluble form of FGFR2 with S252W partially prevents craniosynostosis of the apert mouse model. Dev Dyn. 2014;243(4):560-7
112. Yokota M, Kobayashi Y, Morita J, Suzuki H, Hashimoto Y, Sasaki Y. et al. Therapeutic effect of nanogel-based delivery of soluble FGFR2 with S252W mutation on craniosynostosis. PLoS One. 2014:9 (7)
113. Eswarakumar VP, Ozcan F, Lew ED, Bae JH, Tomé F, Booth CJ. et al. Attenuation of signaling pathways stimulated by pathologically activated FGF-receptor 2 mutants prevents craniosynostosis. Proc Natl Acad Sci U S A [Internet]. 2006;103(49):18603-8 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1693709&tool=pmcentrez&rendertype=abstract
114. Xu W, Luo F, Wang Q, Tan Q, Huang J, Zhou S. et al. Inducible activation of FGFR2 in adult mice promotes bone formation after bone marrow ablation. J Bone Miner Res. United States. 2017 Jun
115. Kan S, Elanko N, Johnson D, Cornejo-Roldan L, Cook J, Reich EW. et al. Genomic Screening of Fibroblast Growth-Factor Receptor 2 Reveals a Wide Spectrum of Mutations in Patients with Syndromic Craniosynostosis. Am J Hum Genet [Internet]. 2002;70(2):472-86 Available from: http://linkinghub.elsevier.com/retrieve/pii/S0002929707639603
116. Shukla V, Coumoul X, Wang R-H, Kim H-S, Deng C-X. RNA interference and inhibition of MEK-ERK signaling prevent abnormal skeletal phenotypes in a mouse model of craniosynostosis. Nat Genet [Internet]. 2007;39(9):1145-50 Available from: http://www.nature.com/doifinder/10.1038/ng2096
117. Shukla V, Coumoul X, Deng CX. RNAi-based conditional gene knockdown in mice using a U6 promoter driven vector. Int J Biol Sci. 2007;3(2):91-9
118. Carter EP, Fearon AE, Grose RP. Careless talk costs lives: fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. England. 2015Apr;25(4):221-33
119. Perlyn CA, Morriss-Kay G, Darvann T, Tenenbaum M, Ornitz DM. A model for the pharmacological treatment of Crouzon syndrome. Neurosurgery. 2006;59(1):210-5
120. Wang Y, Zhou X, Oberoi K, Phelps R, Couwenhoven R, Sun M. et al. p38 inhibition ameliorates skin and skull abnormalities in Fgfr2 Beare-Stevenson mice. J Clin Invest. 2012;122(6):2153-64
121. Balek L, Gudernova I, Vesela I, Hampl M, Oralova V, Kunova Bosakova M. et al. ARQ 087 inhibits FGFR signaling and rescues aberrant cell proliferation and differentiation in experimental models of craniosynostoses and chondrodysplasias caused by activating mutations in FGFR1, FGFR2 and FGFR3. Bone. United States. 2017Aug;105:57-66
Author contact
![]() Corresponding author: Vivek Shukla, PhD, Senior Research Fellow, Thoracic and Gastrointestinal Oncology Branch, National Cancer Institute, National Institutes of Health, 10 Center Drive, Room 3W5848, Bethesda, MD 20892 P: 301-594-8961 Email: vivek.shuklagov And Chuxia Deng, Ph.D., Dean and Chair Professor, E12, Room 4041, Faculty of Health Sciences, University of Macau, Macau SAR, China Phone: (853) 8822 4997 Fax: 8822 2314 Email: cxdengmo
Corresponding author: Vivek Shukla, PhD, Senior Research Fellow, Thoracic and Gastrointestinal Oncology Branch, National Cancer Institute, National Institutes of Health, 10 Center Drive, Room 3W5848, Bethesda, MD 20892 P: 301-594-8961 Email: vivek.shuklagov And Chuxia Deng, Ph.D., Dean and Chair Professor, E12, Room 4041, Faculty of Health Sciences, University of Macau, Macau SAR, China Phone: (853) 8822 4997 Fax: 8822 2314 Email: cxdengmo