Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(1):10-20. doi:10.7150/ijbs.22768 This issue Cite
Research Paper
Disruption of Gen1 Causes Congenital Anomalies of the Kidney and Urinary Tract in Mice
Herui Wang1,2,5*, Chi Zhang1,2,6*, Xiaowen Wang2,7, Yaru Lian1,3, Bin Guo1, Miao Han1,3, Xiaoe Zhang2, Xiaoting Zhu1, Sixian Xu1, Zengli Guo1, Yunli Bi2, Qian Shen2, Xiang Wang2, Jiaojiao Liu2, Yuan Zhuang1,4, Ting Ni1,3, Hong Xu2 ![]() , Xiaohui Wu1,2
, Xiaohui Wu1,2 ![]()
1. State Key Laboratory of Genetic Engineering and National Center for International Research of Development and Disease, Institute of Developmental Biology and Molecular Medicine, Collaborative Innovation Center of Genetics and Development, School of Life Sciences, Fudan University, Shanghai 200433, China;
2. Shanghai Kidney Development and Pediatric Kidney Disease Research Center, Children's Hospital of Fudan University, Shanghai 201102, China;
3. MOE Key Laboratory of Contemporary Anthropology, Fudan University, Shanghai 200433, China;
4. Department of Immunology, Duke University Medical Center, Durham, NC 27710, USA;
5. Neuro-Oncology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, Maryland, USA;
6. Department of Molecular, Cellular, and Developmental Biology, University of Colorado, Boulder, CO 80309, USA;
7. Wuhan Children's Hospital (Wuhan Maternal and Child Healthcare Hospital), Tongji Medical College, Huazhong University of Science & Technology, Wuhan 430000, China.
* These authors contributed equally to this work.
Received 2017-9-10; Accepted 2017-10-2; Published 2018-1-1
Abstract

Congenital anomalies of the kidney and urinary tract (CAKUT) are among the most common developmental defects in humans. Despite of several known CAKUT-related loci (HNF1B, PAX2, EYA1, etc.), the genetic etiology of CAKUT remains to be elucidated for most patients. In this study, we report that disruption of the Holliday Junction resolvase gene Gen1 leads to renal agenesis, duplex kidney, hydronephrosis, and vesicoureteral reflux (VUR) in mice. GEN1 interacts with SIX1 and enhances the transcriptional activity of SIX1/EYA1, a key regulatory complex of the GDNF morphogen. Gen1 mutation impairs Grem1 and Gdnf expression, resulting in excessive ureteric bud formation and defective ureteric bud branching during early kidney development. These results revealed an unidentified role of GEN1 in kidney development and suggested its contribution to CAKUT.
Introduction
Congenital anomalies of the kidney and urinary tract (CAKUT) occur in three to six per 1000 live births and serve as the leading cause of renal failure in infants and children (1-3). CAKUT cover a wide range of structural malformations of the kidney and/or the urinary tract, such as renal agenesis, duplex kidneys or ureters, vesicoureteric reflux, pelviureteric junction obstruction, and ureterovesical junction obstruction. Monogenic causes of CAKUT are supported by familial clustering as well as mouse genetic models (4). To date, more than twenty disease-causing genes have been identified for human CAKUT and mutations in 12 dominant CAKUT genes are reported to be responsible for over 6% of families with isolated CAKUT (5). However, most CAKUT patients still lack any molecular diagnosis (5-7).
CAKUT result from abnormal nephrogenesis. In mammals, kidney and urinary tract development is initiated upon the specification of metanephric mesenchyme (MM) in the nephrogenic cord (NC), followed by reciprocal interactions between MM and the adjacent nephric duct (ND) (8). A single ureteric bud (UB) is firstly induced on ND by signals from MM, then invades into and branches in MM to form the future collecting duct system. At the same time, MM cells are specialized by signals from UB tips to form future nephrons and fuse with the collecting duct (9). The formation and initial branching of UBs are critical for the development of the kidney and urinary tract (9). Blockage of either process could result in solitary kidney or renal agenesis, while extra UB formation usually leads to duplex kidney or ureters (3, 9).
During the early stages of kidney development, the secreted growth factor GDNF is expressed by MM to interact with its cognate receptor RET/GFRα-1 on ND to induce UB formation and branching (10, 11). GDNF expression is positively regulated by a group of transcription factors such as SIX1, SIX2, SIX4, PAX2, and EYA1. Disruption of any of these genes would block kidney development in mice (12-15). Meanwhile, GDNF expression are restricted by SLIT2/ROBO2 pathway that regulates the separation between ND and NC, such that supernumerary kidneys were observed in Robo2 mutants (16, 17). GDNF signal is transduced by the negative regulator Spry1, which acts downstream of Ret (18, 19). In addition to GDNF, Spry1 also modulates FGF signals. Deletion of Fgf10, which is expressed during the period of UB formation, caused less severe renal agenesis than Gdnf or Ret mutations. As expected, the renal phenotype in Fgf10 mutants could be rescued by heterozygous Spry1 deletions (19). BMPs provide another type of signals to UB development. In particular, BMP4 is expressed in stromal cells enveloping ND to inhibit UB branching but promote ureteric stalk elongation (20). BMP4 activity is locally restricted by GREMLIN1 (GREM1), a DAN domain protein that preferentially binds to BMP2/4 (21). GREM1-mediated reduction of BMP4 activity is essential to initiate UB outgrowth (22, 23). Loss of Grem1 causes arrested kidney development prior to UB invasion into MM (22).
The Rad2/XPG family member GEN1 was initially identified as a Holliday junction resolvase in mammalian cells (24). It has been shown that GEN1 is involved not only in DNA homologous recombination, but also in maintaining centrosome integrity (24-26). To explore the physiological role of Gen1, we analyzed mice carrying an insertion mutation in this gene. Here we report that disruption of Gen1 affects the expression of both Gdnf and Grem1 in mice, and caused multiple congenital anomalies of the kidney and urinary tract. These results revealed an unidentified role of GEN1 in kidney development and suggested its contribution to CAKUT.
Results
Gen1 mutants display multiple congenital defects in the kidney and urinary tract
We identified a Gen1 mutation (Gen1PB) in a large-scale mutagenesis project in mice (27, 28). This allele carries a piggyBac (PB) transposon insertion in the second intron (Fig. 1A). Real-time RT-PCR detected severely compromised gene expression in the mutant mice. Gen1 transcription in homozygous (Gen1PB/PB) and heterozygous (Gen1PB/+) embryos was reduced to 11.6% and 62.6% of that in the wild type, respectively (Fig. 1B).
Kidney and urinary tract defects in Gen1 mutants. (A) Genomic structure of the Gen1PB allele. A PB[Act-RFP] transposon was inserted in the second intron (solid lines) of Gen1. Open boxes, exons; black arrow, transcription direction; green arrows, PB termini; red arrow, RFP expression cassette. (B) Real-time PCR analysis of Gen1 expression in E12.5 embryos. Data is shown as the mean ± s.e.m. *** P < 0.001 determined by t test. (C) Percentages of newborn mice with normal number of kidneys (NK), unilateral renal agenesis (URA), or duplex kidneys (DK). Percentages of mice having accompanying hydronephrosis (HN) are indicated at the top of each column. (D-G) Representative images of the urinary system in newborn mice. URA (E), unilateral DK (F,G), severe HN and hydroureter (G) are presented. (H-O) Hematoxylin-eosin (H&E) staining of the paraffin sections of kidneys in newborn mice. A wild type kidney (H), URA (I), DK (J), and severe HN (K) are presented. Details within the boxes are enlarged in (L-O), respectively. DK is defined by double renal pelvises (arrowheads). Well differentiated glomeruli (arrows) could be observed in the wild type, URA, and DK. In contrast, tubular elements and glomeruli are almost missing in severe HN (O). Scale bars, 2 mm in D-G; 250 μm in H-K; 50 μm in L-O.
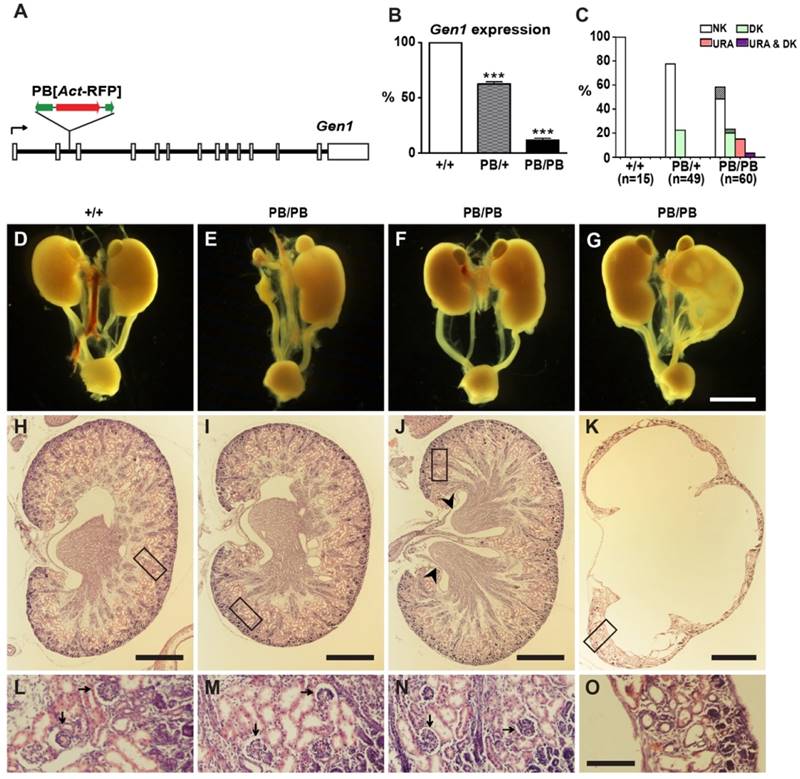
Gen1PB/PB mice are viable and fertile, but frequently developed kidney and urinary tract defects (Fig. 1C-O). We detected thirty-one abnormal individuals from sixty newborn pups by ultrasonic imaging. Varied and sometimes overlapped morphological defects were further revealed by following anatomical and histological analysis (Fig. 1C). We found unilateral duplex kidneys in fifteen mutants (Fig. 1F, J, N), bilateral duplex kidneys in one mutant, unilateral renal agenesis in eleven mutants (Fig. 1E, I, M), and unilateral hydronephrosis in eight mutants (Fig. 1G, K, O). The duplex kidneys in Gen1PB/PB mice are bi-papillary (Fig. 1J). Tubular elements and glomeruli were readily detected in solitary or duplex kidneys, but not in severe hydronephrotic kidneys. In addition to these morphological changes, we also observed two cases of unilateral vesicoureteric reflux (VUR) in nine Gen1PB/PB mice at weaning stages (Supplementary Fig. 1A and B). Gen1PB/+ mice have similar but weaker malformations (Fig. 1C). We only detected eleven unilateral duplex kidneys from forty-nine newborn Gen1PB/+ pups. No renal agenesis or hydronephrosis were observed. We also checked the renal function by measuring blood urea nitrogen (BUN) and creatinine concentration in the serum of four-month Gen1PB/PB mice. No significant differences were observed for both parameters (Supplementary Fig. 1C and D).
Other than the kidney and urinary tract defects, Gen1PB/PB mice have kinky tails as a result of delayed neural tube closure (Supplementary Fig. 2). All congenital defects were caused by the PB insertion. Generating revertant animals by removing PB in Gen1 could result in normal animals with no kidney and urinary tract defects or delayed neural tube closure (Supplementary Fig. 3).
Excessive UB formation accompanied by altered Grem1 and Gdnf expression in Gen1 mutants
The congenital kidney and urinary tract malformations observed in Gen1PB mutants were reminiscent of the defects in CAKUT patients. To identify the developmental abnormalities that may cause the kidney and urinary tract malformations, we followed the formation of ureteric buds (UB), a key event that leads to the development of the renal collecting system and renal vesicles (8). In mice, UB formation starts around embryonic day 10.5 (E10.5) in mice. At E11.0, a single UB is readily observed on each side in wild type embryos (Fig. 2A). We examined eleven Gen1PB/PB embryos at this stage (Table 1). Surprisingly, nine of the mutant embryos (81.8%) possessed an ectopic UB on one side (Fig. 2B). The others developed single UBs in a more anterior position to the end of the nephric duct on one or both sides (Fig. 2C). Gen1PB/+ mice exhibit less severe defects, as only three of twenty-one (14.3%) Gen1PB/+ embryos at E11.0 were detected to have an ectopic UB. Thus, disruption of Gen1 leads to excessive UB formation in mice.
Gen1 mutation leads to ectopic UBs accompanied by altered Grem1 and Gdnf expression. (A-C) Ectopic UBs detected in E11 embryos by c-Ret RNA in situ hybridization. (D-F) Gdnf RNA in situ hybridization in E10.5 embryos. Homozygous embryos (E,F) express less Gdnf than the wild type (D). (G) Real-time RT-PCR of Grem1 from E10.5 wild type (n=4) and homozygous mutant (n=6) kidney primordia. Dashed lines indicate UB. Scale bars, 0.2 mm. * P<0.05.
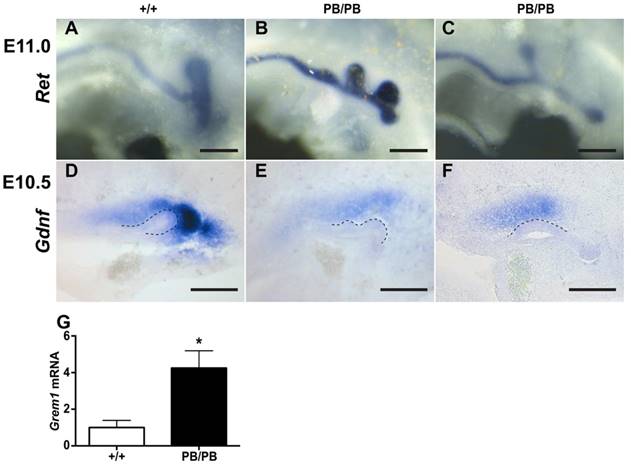
Defective UB formation cases in E11 Gen1 mutant embryos. The percentage of the embryos with indicated phenotype is shown in brackets. WT, wild type.
| Total embryos | Normal | Embryos with ectopic buds | Embryos with anterior buds | |
|---|---|---|---|---|
| WT | 8 | 8 (100%) | 0 | 0 |
| PB/+ | 21 | 18 (85.7%) | 3 (14.3%) | 0 |
| PB/PB | 11 | 0 | 9 (81.8%) | 2 (19.2%) |
Gen1 is ubiquitously expressed in the mesoderm of E10.5 embryos, including MM and UB regions (Supplementary Fig. 4). To explore if Gen1 mutation alters the signals controlling UB formation, we examined the expression of genes belong to the GDNF, FGF, SLIT, and BMP pathways in E10.5 kidney primordia (Supplementary Table 1) (8, 9). Among them, the BMP4 antagonist Grem1 is the only gene that was significantly upregulated, as revealed by RNA-seq. Real-time RT-PCR of E10.5 kidney primordia confirmed significant Grem1 upregulation in Gen1PB/PB embryos (Fig. 2G). In addition to Grem1, RNA-seq revealed significantly lower expression of GDNF pathway genes Eya1, Pax2, Pax8, Sall1, Six2, and Wt1 (Supplementary Table 1). We thus examined the expression of Gdnf in Gen1PB mutants by in situ hybridization, despite the fact that Gdnf expression alteration was not recognized by RNA-seq. We detected reduced Gdnf expression in six of eleven (54.5%) homozygous embryos (Fig. 2D-F).
Defective UB branching with decreased GNDF signal in Gen1 mutants
Excessive UB formation usually leads to duplex kidneys. To explore the reason for frequently observed renal agenesis in Gen1 mutants, we further examined the branching process after UB formation. In wild type mice, UB always branches in the metanephric mesenchyme (MM) to form a T-shaped bifurcation at E11.5 (Fig. 3A). In contrast, Gen1PB/PB embryos are often affected by UB branching failure at this stage (Table 2). In fact, we observed eleven branching failure events in forty-seven UBs from nineteen E11.5 homozygotes (Fig. 3B). Of eleven embryos without ectopic UBs, three had unilateral branching failures. In seven embryos with a unilateral ectopic UB, we observed three embryos with one unbranched UB and two with both on the side of two UBs, respectively (Fig. 3B). The only embryo with bilateral ectopic UBs also had branching failure in one of the UBs. UB branching defects were weaker in Gen1PB/+ mice. In twenty-five E11.5 heterozygous embryos, no branching failure was observed in eighteen embryos without ectopic UB, while one out of seven embryos with a unilateral ectopic UB had one UB failed to branch on that side. These results suggest that UB branching failure as the reason for renal agenesis in Gen1 mutants.
Ureteric branching defects in Gen1 mutants. (A) c-Ret RNA in situ hybridization at E11.5 showed normal branch in wild type embryos. (B) Both UBs at the same side failed to branch in some E11.5 Gen1PB/PB embryos. (C) Gdnf RNA in situ hybridization showed Gdnf expression around the UB branches in E11.5 wild type embryos. (D) No Gdnf expression was detected around the un-branched UBs in E11.5 Gen1PB/PB embryos. (E) Quantitative RT-PCR showed decreased Gdnf expression in MM of E11.5 Gen1PB/PB embryos. Dashed lines indicate UB. Scale bars, 0.2 mm.
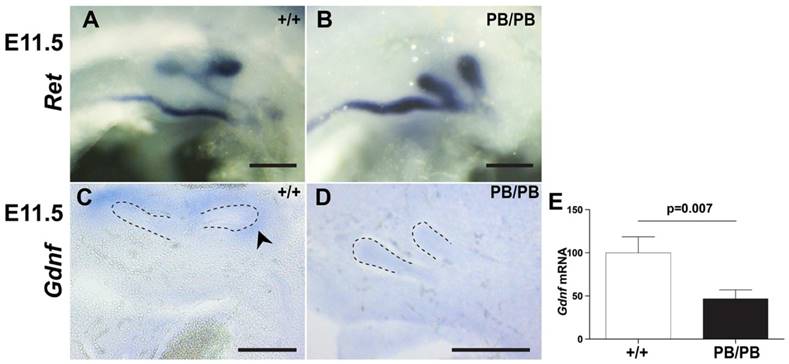
Defective UB branching cases in E11.5 Gen1 mutant embryos. The percentage of the embryos with indicated phenotype is shown in brackets. *, The only Gen1PB/+ embryo with one unbranched UB has two buds on that side.
| Total embryos | Normal | Embryos with one unbranched UB | Embryos with two unbranched UBs | |
|---|---|---|---|---|
| WT | 14 | 14 (100%) | 0 | 0 |
| PB/+ | 25 | 24 (96%) | 1 (4%)* | 0 |
| PB/PB | 19 | 10 (52.6%) | 7 (36.8%) | 2 (10.5%) |
In wild type embryos, presence of GDNF in the condensed mesenchyme surrounding the tips of UB is essential for branching (Fig. 3C) (8). In E11.5 Gen1PB/PB embryos, Gdnf expression was present surrounding the tips of all thirty branched UBs but was undetectable in any of the seven unbranched UBs (Fig. 3D). Quantitative RT-PCR also detected approximately 50% drop of Gdnf transcription in Gen1PB/PB MM than that in wild type embryos (Fig. 3E). These results suggest that lower Gdnf expression caused by the Gen1 mutation may critically contribute to UB branching defects in the mutants.
GEN1 binds with SIX1 and regulates the transcriptional activity of SIX1/EYA1 complex
We next explored the molecular mechanism through which Gen1 regulates Gdnf during UB formation and branching. As a HJ resolvase, GEN1 was originally identified from nuclear extracts (24). It has been shown that approximately 20% of GEN1 proteins exist in the nuclear fraction during interphase (29). Since several transcription factors have been identified as regulators of Gdnf expression during early kidney development (12-14, 30-34), we hypothesize that GEN1 may interact with the upstream transcription factors to regulate Gdnf expression and tested potential interactions among them by Co-IP. We found that GEN1 could interact with SIX1, but not with its molecular partner EYA1, nor with SIX2, SIX4, PAX2, or FOXC1 in HEK293T cells (Fig. 4A, B and supplementary Fig. 5). Thus, GEN1 may interact with SIX1 to affect Gdnf expression.
GEN1 binds with SIX1 and enhances transcriptional activity of the SIX1/EYA1 complex. (A) HA-tagged SIX1 can be pulled down by FLAG-tagged GEN1. (B) GFP-tagged GEN1 can be pulled down by FLAG-tagged SIX1. (C) Luciferase activity of a SIX1/EYA1 reporter. Supplemented by SIX1, EYA1, and GEN1, the luciferase became more active than under any other environment. * P < 0.05, ** P < 0.01, *** P < 0.001 determined by t test. (D,E) In situ hybridization at E10.5 revealed lower Six2 expression in homozygous (E) than in wild type embryos (D). Dashed lines indicate UB. Full-length blots are presented in Supplementary Figure 6. Scale bar, 0.1 mm.
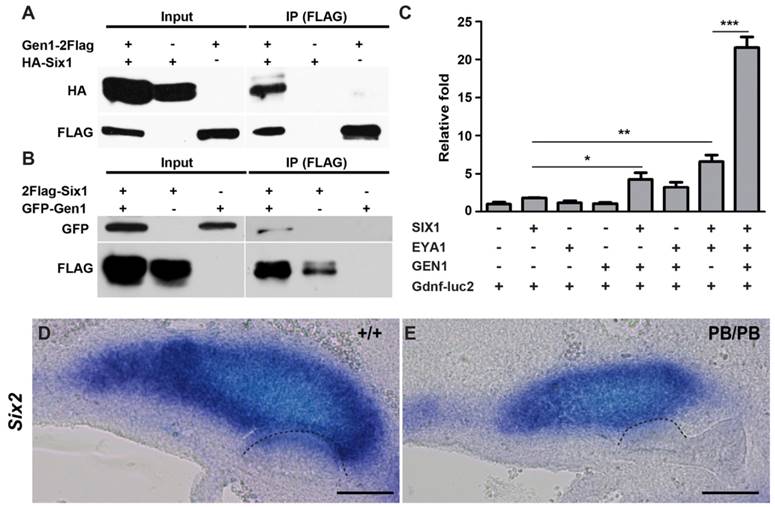
SIX1 was proposed to form a functional complex with EYA1 in regulating Gdnf during early kidney development (9). We thus examined if GEN1 could alter the transcription activity of SIX1/EYA1 complex. Consensus SIX1 binding sites have been identified in the intron of Gdnf (35). We found the expression of a luciferase reporter driven by a minimal promoter and consensus SIX1 binding sites (Gdnf-luc2) remained at the basal level in HEK293T cells when GEN1 was provided alone. The presence of both SIX1 and EYA1 increased luciferase expression by about six times, while GEN1, SIX1 and EYA1 together enhanced the transcription by approximately twenty folds (Fig. 4C). Metanephric mesenchyme expression of Six2 was also reduced in half (3/6) of the E10.5 Gen1PB/PB embryos (Fig. 4D, E). Taken together, these results suggested that GEN1 could enhance Gdnf expression by stimulating the transcriptional activity of the SIX1/EYA1 complex during early kidney development.
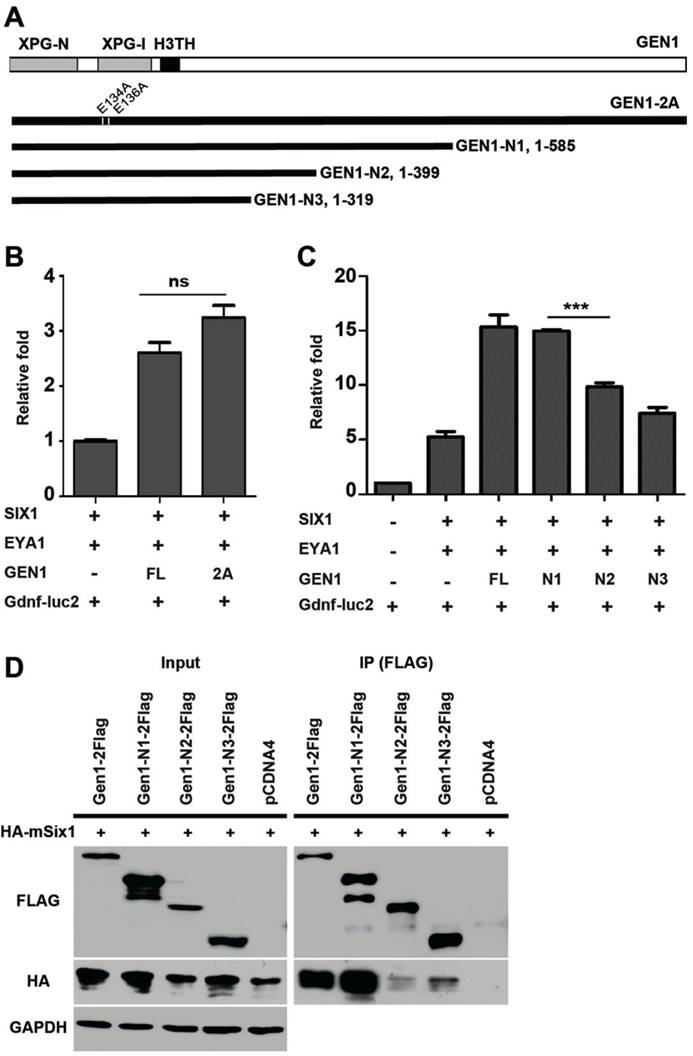
Previous studies have identified two conserved glutamic acid residues (E134/E136) required for GEN1 nuclease activity (24). We found the presence of a nuclease-inactive mutant, GEN1-2A (E134A/E136A), enhanced the expression of luciferase to similar levels as the wild type did (Fig. 5A, B). Thus, the resolvase activity of GEN1 is not required for GEN1 to promote SIX1/EYA1-mediated transcription. We also tested truncated GEN1 protein using the same assay. We found that a truncated GEN1 fragment (GEN1-N1, residues 1-585) that contains the potent resolvase fragment (24, 36) is able to enhance the activity of the SIX1/EYA1 complex, while two shorter fragments (GEN1-N2, residues 1-399; GEN1-N3, residues 1-319) could not (Fig. 5C). Co-IP experiment also proved impaired interaction between GEN1-N2/N3 and SIX1 (Fig. 5D). These data suggests that amino acids 400-585 are important for the transcriptional stimulation activity of GEN1.
Impaired interaction between mutant GEN1 and SIX1. (A) Schematic view of mutant GEN1 proteins. (B) Luciferase assay showed potent activity of GEN1-2A in enhancing the transcriptional activity of SIX1/EYA1 complex. (C) Luciferase assay showed impaired activity of truncated GEN1s in enhancing the transcriptional activity of SIX1/EYA1 complex. (D) Co-IP experiments showed impaired binding activity between truncated GEN1s and SIX1. Full-length blots are presented in Supplementary Figure 7. XPG-N, N-terminal nuclease domain; XPG-I, internal nuclease domain; H3TH, helix-hairpin-helix DNA binding domain. FL, full length; 2A, E134A/E136A.
Discussion
Initially purified as a mammalian Holliday junction resolvase, GEN1 was later reported necessary for centrosome integrity in mammalian cells (24-26, 29, 37). Here we found that Gen1 is required for kidney and urinary tract development and neural tube closure in mice. These are the first evidence showing that Gen1 is involved in developmental regulation. During UB formation, GEN1 not only affects the expression of BMP antagonist GREM1, but also stimulates the transcription of UB promoting signal GDNF, which may imply its role as a coordinator of both pathways to ensure normal nephrogenesis.
Intracellular localization was proposed as a critical factor for GEN1 activity. GEN1 was reported to be spatially regulated during cell cycle, such that its resolvase activity is restricted until mitosis when the nuclear membrane breaks down. With the strong nuclear exporting signal, it has been shown that approximately 80% of GEN1 proteins are presented in the cytoplasm (29). We have shown that GEN1 proteins could bind with the transcription factor SIX1 and enhance the transcriptional activity of the SIX1/EYA1 complex. Consistently, Gen1 mutations led to decreased expression of SIX1 downstream genes during kidney development. These results suggest that the apparently minor nuclear fraction of GEN1 may be critical for developmental regulation.
Not all Gen1PB/PB mice display kidney and urinary tract abnormalities. This may be due to the fact that Gen1PB is a partial loss-of-function allele, such that significant amount of potent GEN1 still exist in the homozygous mutant mice. In fact, incomplete penetrance is not uncommon for mutations that affect kidney development. Heterozygotes of Six1, Eya1, or Foxc1 knock out mutant mice all have varied kidney abnormalities (13, 15, 34). In addition, varied kidney and urinary tract abnormalities in Gen1PB/PB mice may reflect the complexity of the regulatory signals during early kidney development.
During UB formation, we have detected down-regulation of Gdnf signals but up-regulation of Grem1. Gdnf is usually considered as a UB promoting signal since complete ablation of Gdnf would lead to failed UB formation in mice (38). However, it needs to be pointed out that UB formation is sensitive to the dosage of Gdnf signals. In Six1-/- and Sall1-/- mutant embryos, Gdnf expression was partially decreased, which did not interfere with normal UB formation, but caused failure of the first UB branching (13, 39). On the other hand, GREM1-mediated BMP antagonism is essential to initiate UB formation (23). Grem1 knockout mice exhibited disrupted metanephric development at the stage of UB formation; while additional recombinant GREM1 protein could induce additional budding of the wild type nephric duct (23, 40). In Gen1PB/PB mutants, Gdnf expression was partially reduced (Fig. 2E and F), while Grem1 transcription was increased to four folds of wild type embryos (Fig. 2G). It is likely the decreased Gdnf signal did not affect UB formation in E10.5 Gen1PB/PB embryos, while the extra UB formation was due to the elevated Grem1 expression. Thus, varied Grem1 expression may lead to partially unobserved ectopic UB formation. During UB branching, ectopic UB in a single nephric duct may not always develop into a duplex kidney due to the lack of adequate GDNF signals so that less duplex kidney mutants were observed after birth. Unlike kidney defects, kinky tails were consistently observed in all Gen1 homozygotes. This suggests that a different threshold of GEN1 activity or a different mechanism is required for neural tube development.
The trunk of the initial UB differentiates into ureter, connecting the kidney with the bladder (41). Higher or lower UB formation on the nephric duct causes abnormal positioning of the ureter orifice in the trigone, leading to obstruction or reflux (42). We have observed single UBs at more anterior position in two Gen1PB/PB embryos (Fig. 2C). These abnormally positioned ureters may join the bladder at a position more lateral or anterior to the proper site in the trigone. This could shorten the intra-vesicular ureteral segment and disrupt the valve mechanism that prevents the urine backflow to the kidney, thus resulted in VUR. On the other hand, although we didn't observe proximal ureteral atresia or PUJO in Gen1 mutants (Fig.1), we could not exclude the possible contribution of potential ureteral atresia to the severe hydronephrotic phenotype. Similarly, our study did not exclude the possibility that Gen1 mutation may cause delayed kidney development. To distinguish this event, global quantification of tissue dynamics can be applied to monitor the developmental process of nephron number and branching at different stages in Gen1 mutant mice (43).
Our study raises the possibility of GEN1 as a disease gene of human CKAUT. To date, more than 90% of CAKUT patients still lack any molecular diagnosis (5-7). We have detected CAKUT phenotypes in both heterozygous and homozygous Gen1 mutant mice and shown that GEN1 proteins containing the N-terminal 399 amino acids could not function properly in regulating the transcriptional activity of SIX1/EYA1. At the same time, thirty-seven loss-of-function variants have been reported in the Exome Aggregation Consortium (ExAC) database (http://exac.broadinstitute.org), all of which are presented in heterozygosity and predicted to encode truncated GEN1 proteins with less than 400 residues in the N-terminus. Although incomplete penetrance, as implied by the mouse data, may bring complications in human genetic analysis, it is still worth to explore the genetic burden of GEN1 mutations in CAKUT patients.
Methods
Mice
All animal experiments were performed in accordance with protocols approved by the Animal Care and Use Committee of the Institute of Developmental Biology and Molecular Medicine (IDM) at Fudan University. The Gen1 mutant strain (081125049-HLA) was established and maintained on the FVB/N background. In the Gen1PB allele, the PB insertion was mapped in the second intron of Gen1 (Chr:12.11268138, Ensembl release 54).
PCR
Genotyping PCR was performed with a PB specific primer LB2 (5'-CTGAGATGTCCTAAATGCACAGCG-3') and two flanking genomic primers 081125049-L1A (5'-TAGTGTGGGCACATGCAAGC-3') and 081125049-R1A (5'-CCCCTTTGCCTGTTCTTAACCTC-3'). Real-time RT PCR was performed with SYBR Green (Agilent) according to the manufacturer's instruction. Gapdh was selected as the internal control for normalization. The primers used for RT-PCR were: Gen1-RT-F1 in exon 2: 5'-GCACAGACAGTGAAGAAAATG-3'; Gen1-RT-B1 in exon 3: 5'-TGCTTATGACATCAGCTTTCAG-3'; PCR product size 151 bp. Grem1-RT-F: 5'-CCTTTCAGTCTTGCTCCTTCTGC-3'; Grem1-RT-R: 5'-TTCTTCTTGGTGGGTGGCTGTAGC-3'; PCR product size 94 bp. Gapdh-RT-F1: 5'-TGTTCCTACCCCCAATGTGTCC-3'; Gaphd-RT-B1: 5'-GGAGTTGCTGTTGAAGTCGCAG-3'. PCR product size 169 bp.
Histology
Kidneys and the urinary tract were dissected and embedded with paraffin according to the standard protocol (31). Hematoxylin-eosin (H&E) staining was then performed on the sections of 7 μm as previously described (31).
RNA-seq
Total RNA from kidney primordia of E10.5 embryos was extracted by TRIzol (Invitrogen) and GlycoBlue (Ambion) coprecipitation. We combined two mice to obtain 0.5 μg of total RNA for either wild type or Gen1PB/PB samples. Strand-specific RNA-seq libraries starting with 0.5 μg of total RNA were constructed as previously described (44). Two biological replicates of RNA-seq for each group (wild type or mutants) were included to analyze differentially expressed genes. The RNA-seq libraries were sequenced on the Illumina HiSeq2000 platform (Genergy Biotechnology, Shanghai). FastQC was used to evaluate the quality of high-throughput sequencing data (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/). Paired-end reads were mapped to mm9 genome using TopHat2 with default parameters except -g -r and -mate-std-dev (45). In specificity, -g was set to 1 to ensure that there is only one alignment for a given read (45). Bowtie was used to get a good approximation of -r and -mate-std-dev, which is the expected inner distance between mate pairs and the standard deviation for the distribution on inner distances between mate pairs, respectively (46). The known transcripts supplied to TopHat2 were based on the Ensemble database and the NONCODE database (47). Cufflinks was used to assemble transcripts and estimates their abundances for each sample and Cuffdiff was used to get the differentially expressed genes (48).
RNA in situ hybridization
The coding sequences of Ret, Gdnf, and Six2 were cloned from E12.5 embryos through RT-PCR. Digoxigenin-labeled anti-sense probes were then produced by the DIG RNA Labeling kit (Sp6/T7) (Roche). Whole-mount in situ hybridization was performed as previously described (49). Frozen sections of 50 μm were then used for signal detection.
Co-immunoprecipitation (Co-IP) and luciferase assay
HEK293T cells were transfected with Lipofectamine 2000 (Invitrogen) for both co-immunoprecipitation and luciferase assay. Co-IP and immunoblot were performed as previously described (50). The antibodies used include mouse anti-HA (Covance), rabbit anti-HA (Abcam), mouse anti-FLAG (Sigma), and rabbit-anti-GFP (Cell signaling). For luciferase assay, each transfection was triplicated with 400 ng wild type or mutant Gen1, 100 ng pCX-EGFP, 200 ng HA-Six1, 200 ng HA-Eya1, and 200 ng Gdnf-luc2. Luciferase activity was scored 48 hours after transfection and calibrated by the percentage of GFP positive cells (determined by FACS). Each experiment was performed at least twice independently.
Statistics
All data are presented as mean ± SEM. The comparison of two groups was performed with unpaired two-tailed t-test. The threshold for significance was set at P < 0.05.
Data Availability
All data generated or analyzed during this study are included in this published article and its Supplementary Information files.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Drs. Min Han and Tian Xu for helpful discussion and suggestion, Yan Jin for sharing pNTAP-2Flag and pCTAP-2Flag plasmids before publication, Dr. Kai Lei for pcDNA4-HA plasmid, Dr. Aiwu Dong for the rabbit anti-HA antibody, Dr. Frank Costantini for Hoxb7/myr-Venus plasmid, Yanyan Nie, Xiaorong Huang, and Zhiyan Xia for technical assistance, Dr. Maria Lai and Dr. Lavan Khadan for comments on the manuscript. This study was supported by grants from the National Natural Science Foundation of China (81270763), Chinese Key Projects for Basic Research (2011CB944001), Chinese Hi-tech Research and Development Project (2014AA021104), and the Research Fund of the State Key Laboratory of Genetic Engineering, Fudan University.
Author Contributions
H.W. and C.Z. designed and performed experiments, interpreted data, and wrote the manuscript. X.W. (Xiaowen Wang) performed histology analysis. B.G., Z.G., Y.B., Q.S., X.W. (Xiang Wang), and J.L. participated in phenotypic analysis. Y.L., M.H., and T.N. carried out RNA-seq analysis. X.Z. (Xiaoe Zhang), X.Z. (Xiaoting Zhu) and S.X. participated in molecular biology experiments. Y.Z. gave intellectual input; H.X. supervised the collaborations. X.W. (Xiaohui Wu) conceived of and designed the studies, supervised the work, and wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Pohl M, Bhatnagar V, Mendoza SA, Nigam SK. Toward an etiological classification of developmental disorders of the kidney and upper urinary tract. Kidney international. 2002;61(1):10-9 Epub 2002/01/12. doi: 10.1046/j.1523-1755.2002.00086.x. PubMed PMID: 11786080
2. Scott JE, Renwick M. Antenatal diagnosis of congenital abnormalities in the urinary tract. Results from the Northern Region Fetal Abnormality Survey. British journal of urology. 1988;62(4):295-300 Epub 1988/10/01. PubMed PMID: 3191352
3. Weber S. Novel genetic aspects of congenital anomalies of kidney and urinary tract. Current opinion in pediatrics. 2012;24(2):212-8 Epub 2012/01/17. doi: 10.1097/MOP.0b013e32834fdbd4. PubMed PMID: 22245908
4. Nicolaou N, Pulit SL, Nijman IJ, Monroe GR, Feitz WF, Schreuder MF. et al. Prioritization and burden analysis of rare variants in 208 candidate genes suggest they do not play a major role in CAKUT. Kidney international. 2016;89(2):476-86 doi: 10.1038/ki.2015.319. PubMed PMID: 26489027
5. Hwang DY, Dworschak GC, Kohl S, Saisawat P, Vivante A, Hilger AC. et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney international. 2014;85(6):1429-33 doi: 10.1038/ki.2013.508. PubMed PMID: 24429398; PubMed Central PMCID: PMC4040148
6. Kohl S, Hwang DY, Dworschak GC, Hilger AC, Saisawat P, Vivante A. et al. Mild recessive mutations in six Fraser syndrome-related genes cause isolated congenital anomalies of the kidney and urinary tract. Journal of the American Society of Nephrology: JASN. 2014;25(9):1917-22 doi: 10.1681/ASN.2013101103. PubMed PMID: 24700879; PubMed Central PMCID: PMC4147986
7. Vivante A, Kohl S, Hwang DY, Dworschak GC, Hildebrandt F. Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr Nephrol. 2014;29(4):695-704 doi: 10.1007/s00467-013-2684-4. PubMed PMID: 24398540
8. Michos O. Kidney development: from ureteric bud formation to branching morphogenesis. Current opinion in genetics & development. 2009;19(5):484-90 Epub 2009/10/16. doi: 10.1016/j.gde.2009.09.003. PubMed PMID: 19828308; PubMed Central PMCID: PMC2783241
9. Davidson AJ. Mouse kidney development. StemBook. Cambridge (MA)2008.
10. Sanchez MP, Silos-Santiago I, Frisen J, He B, Lira SA, Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382(6586):70-3 Epub 1996/07/04. doi: 10.1038/382070a0. PubMed PMID: 8657306
11. Basson MA, Watson-Johnson J, Shakya R, Akbulut S, Hyink D, Costantini FD. et al. Branching morphogenesis of the ureteric epithelium during kidney development is coordinated by the opposing functions of GDNF and Sprouty1. Developmental biology. 2006;299(2):466-77 Epub 2006/10/07. doi: 10.1016/j.ydbio.2006.08.051. PubMed PMID: 17022962
12. Brodbeck S, Besenbeck B, Englert C. The transcription factor Six2 activates expression of the Gdnf gene as well as its own promoter. Mechanisms of development. 2004;121(10):1211-22 Epub 2004/08/26. doi: 10.1016/j.mod.2004.05.019. PubMed PMID: 15327782
13. Xu PX, Zheng W, Huang L, Maire P, Laclef C, Silvius D. Six1 is required for the early organogenesis of mammalian kidney. Development. 2003;130(14):3085-94 Epub 2003/06/05. PubMed PMID: 12783782
14. Kobayashi H, Kawakami K, Asashima M, Nishinakamura R. Six1 and Six4 are essential for Gdnf expression in the metanephric mesenchyme and ureteric bud formation, while Six1 deficiency alone causes mesonephric-tubule defects. Mechanisms of development. 2007;124(4):290-303 Epub 2007/02/16. doi: 10.1016/j.mod.2007.01.002. PubMed PMID: 17300925
15. Sajithlal G, Zou D, Silvius D, Xu PX. Eya 1 acts as a critical regulator for specifying the metanephric mesenchyme. Developmental biology. 2005;284(2):323-36 Epub 2005/07/16. doi: 10.1016/j.ydbio.2005.05.029. PubMed PMID: 16018995
16. Wainwright EN, Wilhelm D, Combes AN, Little MH, Koopman P. ROBO2 restricts the nephrogenic field and regulates Wolffian duct-nephrogenic cord separation. Developmental biology. 2015;404(2):88-102 doi: 10.1016/j.ydbio.2015.05.023. PubMed PMID: 26116176
17. Grieshammer U, Le M, Plump AS, Wang F, Tessier-Lavigne M, Martin GR. SLIT2-mediated ROBO2 signaling restricts kidney induction to a single site. Developmental cell. 2004;6(5):709-17 Epub 2004/05/08. PubMed PMID: 15130495
18. Michos O, Cebrian C, Hyink D, Grieshammer U, Williams L, D'Agati V. et al. Kidney development in the absence of Gdnf and Spry1 requires Fgf10. PLoS genetics. 2010;6(1):e1000809. Epub 2010/01/20. doi: 10.1371/journal.pgen.1000809. PubMed PMID: 20084103; PubMed Central PMCID: PMC2797609
19. Basson MA, Akbulut S, Watson-Johnson J, Simon R, Carroll TJ, Shakya R. et al. Sprouty1 is a critical regulator of GDNF/RET-mediated kidney induction. Developmental cell. 2005;8(2):229-39 doi: 10.1016/j.devcel.2004.12.004. PubMed PMID: 15691764
20. Miyazaki Y, Oshima K, Fogo A, Hogan BL, Ichikawa I. Bone morphogenetic protein 4 regulates the budding site and elongation of the mouse ureter. The Journal of clinical investigation. 2000;105(7):863-73 doi: 10.1172/JCI8256. PubMed PMID: 10749566; PubMed Central PMCID: PMC377476
21. Church RH, Krishnakumar A, Urbanek A, Geschwindner S, Meneely J, Bianchi A. et al. Gremlin1 preferentially binds to bone morphogenetic protein-2 (BMP-2) and BMP-4 over BMP-7. The Biochemical journal. 2015;466(1):55-68 doi: 10.1042/BJ20140771. PubMed PMID: 25378054
22. Michos O, Panman L, Vintersten K, Beier K, Zeller R, Zuniga A. Gremlin-mediated BMP antagonism induces the epithelial-mesenchymal feedback signaling controlling metanephric kidney and limb organogenesis. Development. 2004;131(14):3401-10 Epub 2004/06/18. doi: 10.1242/dev.01251. PubMed PMID: 15201225
23. Michos O, Goncalves A, Lopez-Rios J, Tiecke E, Naillat F, Beier K. et al. Reduction of BMP4 activity by gremlin 1 enables ureteric bud outgrowth and GDNF/WNT11 feedback signalling during kidney branching morphogenesis. Development. 2007;134(13):2397-405 Epub 2007/05/25. doi: 10.1242/dev.02861. PubMed PMID: 17522159
24. Ip SC, Rass U, Blanco MG, Flynn HR, Skehel JM, West SC. Identification of Holliday junction resolvases from humans and yeast. Nature. 2008;456(7220):357-61 Epub 2008/11/21. doi: 10.1038/nature07470. PubMed PMID: 19020614
25. Gao M, Rendtlew Danielsen J, Wei LZ, Zhou DP, Xu Q, Li MM. et al. A Novel Role of Human Holliday Junction Resolvase GEN1 in the Maintenance of Centrosome Integrity. PloS one. 2012;7(11):e49687. Epub 2012/11/21. doi: 10.1371/journal.pone.0049687. PubMed PMID: 23166748; PubMed Central PMCID: PMC3500319
26. Wang X, Wang H, Guo B, Zhang Y, Gong Y, Zhang C. et al. Gen1 and Eme1 Play Redundant Roles in DNA Repair and Meiotic Recombination in Mice. DNA and Cell Biology. 2016. Epub July 6. 2016
27. Sun LV, Jin K, Liu Y, Yang W, Xie X, Ye L. et al. PBmice: an integrated database system of piggyBac (PB) insertional mutations and their characterizations in mice. Nucleic acids research. 2008;36(Database issue):D729-34 Epub 2007/10/13. doi: gkm790 [pii] 10.1093/nar/gkm790. PubMed PMID: 17932058; PubMed Central PMCID: PMC2238892
28. Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;122(3):473-83 Epub 2005/08/13. doi: S0092-8674(05)00707-5 [pii] 10.1016/j.cell.2005.07.013. PubMed PMID: 16096065
29. Chan YW, West SC. Spatial control of the GEN1 Holliday junction resolvase ensures genome stability. Nature communications. 2014;5:4844. doi: 10.1038/ncomms5844. PubMed PMID: 25209024; PubMed Central PMCID: PMC4172962
30. Self M, Lagutin OV, Bowling B, Hendrix J, Cai Y, Dressler GR. et al. Six2 is required for suppression of nephrogenesis and progenitor renewal in the developing kidney. The EMBO journal. 2006;25(21):5214-28 Epub 2006/10/13. doi: 10.1038/sj.emboj.7601381. PubMed PMID: 17036046; PubMed Central PMCID: PMC1630416
31. Xu PX, Adams J, Peters H, Brown MC, Heaney S, Maas R. Eya1-deficient mice lack ears and kidneys and show abnormal apoptosis of organ primordia. Nature genetics. 1999;23(1):113-7 Epub 1999/09/02. doi: 10.1038/12722. PubMed PMID: 10471511
32. Brophy PD, Ostrom L, Lang KM, Dressler GR. Regulation of ureteric bud outgrowth by Pax2-dependent activation of the glial derived neurotrophic factor gene. Development. 2001;128(23):4747-56 Epub 2001/12/04. PubMed PMID: 11731455
33. Narlis M, Grote D, Gaitan Y, Boualia SK, Bouchard M. Pax2 and pax8 regulate branching morphogenesis and nephron differentiation in the developing kidney. Journal of the American Society of Nephrology: JASN. 2007;18(4):1121-9 Epub 2007/02/23. doi: 10.1681/ASN.2006070739. PubMed PMID: 17314325
34. Kume T, Deng K, Hogan BL. Murine forkhead/winged helix genes Foxc1 (Mf1) and Foxc2 (Mfh1) are required for the early organogenesis of the kidney and urinary tract. Development. 2000;127(7):1387-95 Epub 2000/03/08. PubMed PMID: 10704385
35. Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK. et al. Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature. 2003;426(6964):247-54 Epub 2003/11/25. doi: 10.1038/nature02083. PubMed PMID: 14628042
36. Bailly AP, Freeman A, Hall J, Declais AC, Alpi A, Lilley DM. et al. The Caenorhabditis elegans homolog of Gen1/Yen1 resolvases links DNA damage signaling to DNA double-strand break repair. PLoS genetics. 2010;6(7):e1001025. Epub 2010/07/28. doi: 10.1371/journal.pgen.1001025. PubMed PMID: 20661466; PubMed Central PMCID: PMC2908289
37. Garcia-Luis J, Machin F. Mus81-Mms4 and Yen1 resolve a novel anaphase bridge formed by noncanonical Holliday junctions. Nature communications. 2014;5:5652. doi: 10.1038/ncomms6652. PubMed PMID: 25466415
38. Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, Grinberg A. et al. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382(6586):73-6 doi: 10.1038/382073a0. PubMed PMID: 8657307
39. Kiefer SM, Robbins L, Stumpff KM, Lin C, Ma L, Rauchman M. Sall1-dependent signals affect Wnt signaling and ureter tip fate to initiate kidney development. Development. 2010;137(18):3099-106 Epub 2010/08/13. doi: 10.1242/dev.037812. PubMed PMID: 20702564; PubMed Central PMCID: PMC2926958
40. Nie X, Xu J, El-Hashash A, Xu PX. Six1 regulates Grem1 expression in the metanephric mesenchyme to initiate branching morphogenesis. Developmental biology. 2011;352(1):141-51 Epub 2011/02/02. doi: 10.1016/j.ydbio.2011.01.027. PubMed PMID: 21281623; PubMed Central PMCID: PMC3113441
41. Costantini F. Genetic controls and cellular behaviors in branching morphogenesis of the renal collecting system. Wiley Interdiscip Rev Dev Biol. 2012;1(5):693-713 doi: 10.1002/wdev.52. PubMed PMID: 22942910; PubMed Central PMCID: PMCPMC3430146
42. Mendelsohn C. Using mouse models to understand normal and abnormal urogenital tract development. Organogenesis. 2009;5(1):306-14 PubMed PMID: 19568352; PubMed Central PMCID: PMCPMC2659372
43. Short KM, Combes AN, Lefevre J, Ju AL, Georgas KM, Lamberton T. et al. Global quantification of tissue dynamics in the developing mouse kidney. Developmental cell. 2014;29(2):188-202 doi: 10.1016/j.devcel.2014.02.017. PubMed PMID: 24780737
44. Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S. et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic acids research. 2009;37(18):e123. doi: 10.1093/nar/gkp596. PubMed PMID: 19620212; PubMed Central PMCID: PMC2764448
45. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome biology. 2013;14(4):R36. doi: 10.1186/gb-2013-14-4-r36. PubMed PMID: 23618408; PubMed Central PMCID: PMC4053844
46. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome biology. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. PubMed PMID: 19261174; PubMed Central PMCID: PMC2690996
47. Xie C, Yuan J, Li H, Li M, Zhao G, Bu D. et al. NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic acids research. 2014;42(Database issue):D98-103 doi: 10.1093/nar/gkt1222. PubMed PMID: 24285305; PubMed Central PMCID: PMC3965073
48. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature protocols. 2012;7(3):562-78 doi: 10.1038/nprot.2012.016. PubMed PMID: 22383036; PubMed Central PMCID: PMC3334321
49. Correia KM, Conlon RA. Whole-mount in situ hybridization to mouse embryos. Methods. 2001;23(4):335-8 Epub 2001/04/24. doi: 10.1006/meth.2000.1145. PubMed PMID: 11316434
50. Zhang X, Lei K, Yuan X, Wu X, Zhuang Y, Xu T. et al. SUN1/2 and Syne/Nesprin-1/2 complexes connect centrosome to the nucleus during neurogenesis and neuronal migration in mice. Neuron. 2009;64(2):173-87 Epub 2009/10/31. doi: 10.1016/j.neuron.2009.08.018. PubMed PMID: 19874786; PubMed Central PMCID: PMC2788510
Author contact
![]() Corresponding authors: Xiaohui Wu: xiaohui_wuedu.cn; Hong Xu: hxuedu.cn
Corresponding authors: Xiaohui Wu: xiaohui_wuedu.cn; Hong Xu: hxuedu.cn