International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(1):69-77. doi:10.7150/ijbs.22259 This issue Cite
Research Paper
Aryl Hydrocarbon Receptor Activation Modulates Intestinal Epithelial Barrier Function by Maintaining Tight Junction Integrity
Min Yu1#, Qimeng Wang1#, Yuanhang Ma1, Liangzi Li1, Kun Yu1, Zhicao Zhang1, Guoqing Chen1, Xiangsheng Li1, Weidong Xiao1, Pengyuan Xu2, Hua Yang1 ![]()
1. Department of General Surgery, Xinqiao Hospital, Third Military Medical University, Chongqing, China,
2. Department of Gastrointestinal Surgery, the Second Affiliated Hospital of Kunming Medical University, Kunming, China
#These authors contributed equally to this work.
Received 2017-8-7; Accepted 2017-12-14; Published 2018-1-11
Citation:
Yu M, Wang Q, Ma Y, Li L, Yu K, Zhang Z, Chen G, Li X, Xiao W, Xu P, Yang H. Aryl Hydrocarbon Receptor Activation Modulates Intestinal Epithelial Barrier Function by Maintaining Tight Junction Integrity. Int J Biol Sci 2018; 14(1):69-77. doi:10.7150/ijbs.22259. https://www.ijbs.com/v14p0069.htm
Other stylesAbstract

Activation of Aryl hydrocarbon receptor (AhR) is involved in the control of intestinal mucosal homeostasis. Intestinal barrier dysfunction contributes to the development of many intestinal diseases, such as inflammatory bowel disease (IBD). In this study, we investigated the mechanisms of AhR activation in the maintenance of intestinal barrier function. Adult C57BL/6 mice were treated with dextran sulphate sodium (DSS) for 7 days, with or without 6-Formylindolo(3,2-b)carbazole (FICZ), a ligand of AhR. We found that AhR activation by FICZ attenuated the decreased TJ protein expression in the colonic mucosa of the DSS-induced mice. Further, the increase of both MLC phosphorylation and MLCK expression in the mice with DSS-induced colitis was also significantly inhibited by FICZ induced AhR activation. For in vitro experiments, Caco-2 cells were treated with tumour necrosis factor alpha (TNF-α)/interferon gamma (IFN-γ) for 48 h, with or without FICZ. AhR activation prevented TNF-α/IFN-γ-induced decrease in TER and morphological disruption of the TJs in Caco-2 monolayers. It also inhibited TNF-α/IFN-γ-induced increase in MLCK expression and MLC phosphorylation by suppression of NF-κB p65 signaling pathway. Thus, AhR-activating factors might have potential as therapeutic agents for the treatment of patients with IBD.
Keywords: Aryl hydrocarbon receptor, inflammatory bowel disease, intestinal barrier function, tight junction, myosin light chain
Introduction
Intestinal epithelial cells (IECs) with their intact tight junctions (TJs) create a barrier between the external environment and the mammalian host. The intestinal epithelial TJ barrier acts as an important line of defence against toxins and pathogenic organisms present in the intestinal lumen[1, 2]. Disruption of this barrier induces the increased permeation of toxic luminal antigens into the lamina propria, triggering an inflammatory cascade and promoting intestinal inflammation, as well as the development of a variety of intestinal diseases, including inflammatory bowel disease (IBD) [3]. Notably, intestinal TJ barrier function was reported to be defective in patients with IBD [4-6]. And therapeutic enhancement of the intestinal TJ barrier ameliorated the development of intestinal inflammation [7].
Aryl hydrocarbon receptor (AhR) has been recently reported to inhibit inflammatory processes in the gut and to have a protective effect against IBD [8, 9]. AhR is a ligand-activated transcription factor that belongs to the basic helix-loop-helix (bHLH)/PerARNT-Sim (PAS) superfamily of proteins [10, 11]. In the absence of a ligand, AhR is retained in the cytosol and binds to the chaperone proteins heat shock protein 90 (Hsp90), AhR-associated protein 9 (ARA9) and p23 [12, 13]. Upon ligand binding, AhR translocates to the nucleus and binds to its dimerization partner, AhR nuclear translocator (ARNT), which is another bHLH/PAS-domain transcription factor. The AhR-ARNT complex then activates the transcription of a variety of target genes that contain dioxin response elements (DREs) or xenobiotic response elements (XREs), including cytochrome P450 (CYP)1A1 [14-16].
6-Formylindolo(3,2-b)carbazole (FICZ), an ultraviolet photoproduct of L-tryptophan, is a high-affinity endogenous ligand for AhR [17-20]. Recent data have suggested that AhR helps to maintain intestinal immune homeostasis [8, 9]. Activation of AhR by FICZ results in down-regulation of the expression of inflammatory cytokines, up-regulation of the expression of IL-22, and inhibition of inflammation in the gut [11, 21]. In the intestine, activation of this receptor is essential for the maintenance of intestinal intraepithelial lymphocytes (IELs), a subset of T cells present in the intestinal epithelium that are involved in the maintenance of intestinal epithelial integrity and localized immune quiescence [22].
However, current studies of the role of AhR in IBD have been largely focused on immune cells, and little is known about the potential role of AhR activation in IECs, in which this receptor is also highly expressed. Our previous work showed that AhR activation by FICZ downregulated intestinal epithelial cells(IEC)-derived IL-7 expression in mice with DSS-induced colitis and inhibit inflammation in the gastrointestinal tract of mice [23], suggesting that AhR activation in IECs may also play an important role in IBD.
In the present study, we focused on the effect of AhR activation on intestinal epithelial barrier function. We found that AhR activation by FICZ attenuated disruption of intestinal epithelial barrier function both in vivo and in vitro. In addition, our data suggested that AhR activation ameliorated tumour necrosis factor alpha (TNF-α)/interferon gamma (IFN-γ)-induced alterations in the expression and localization of TJ proteins. We also demonstrated that AhR activation suppressed TNF-α/IFN-γ-induced activation of the MLCK-pMLC signalling pathway by inhibiting NF-κB p65 activation.
Materials and Methods
Animal model
C57BL/6 mice were purchased from the Experiment Animal Center (Third Military Medical University, Chongqing, P.R. China) for use in the in vivo experiments, performed as previously described. Acute colitis was induced by administration of 3% dextran sulphate sodium (DSS; 36-50 kDa, MP Biomedicals, Solon, OH), which was dissolved in the animals' drinking water, for 7 days. Control mice were provided with distilled water. FICZ (1 µg/mouse) was administered by intraperitoneal injection daily, beginning on the third day of DSS administration. All of the animal experiments were approved by the University Committee on the Use and Care of Animals of the Third Military Medical University.
Immunohistochemical (IHC) staining
Segments of colon were harvested and fixed in 4% paraformaldehyde. Then, 5 µm-thick, paraffin-embedded sections of colon were prepared and incubated in 0.3% hydrogen peroxide in methanol for 20 min at room temperature to inhibit endogenous peroxidase activity. For antigen retrieval, the sections were treated with citrate buffer (pH 6.0) and heated three times in a microwave oven for 5 min each time. The sections were then blocked with 5% bovine serum albumin (BSA) at room temperature for 30 min and were subsequently incubated with the following primary antibodies overnight at 4 °C: ZO-1 (1:50, Proteintech), Occludin (1:50, Proteintech), Claudin-1 (1:50, Proteintech), MLCK (1:50, Proteintech) and pMLC (1:100, Abcam). Next, the sections were incubated with the respective secondary antibodiesat room temperature for 40 min. After being washed with phosphate-buffered serum (PBS), the sections were incubated with an avidin-biotin complex according to the manufacturer's instructions of a Boster ABC Kit (Boster, Wuhan, China). Peroxidase activity was assessed using diaminobenzidine (DAB) as a chromogen. Sections were counterstained with haematoxylin, and histological examination was then performed under a Leica TCS SP light microscope (Leica, Heidelberg, Germany).
Intestinal Epithelial Cell Isolation
Intestinal epithelial cell isolation was used for western blotting and quantitative real-time polymerase chain reaction (QRT-PCR) assays. Briefly, the whole small intestine was removed and placed in tissue culture medium (RPMI 1640, with 10% foetal calf serum).The section was cut into 5-mm pieces, washed in an isolation buffer containing 190 mg ethylenediaminetetraacetic acid and 80 mg DDT dissolved in phosphate-buffered saline (PBS) 500ml, and incubated in the same buffer with continuous brisk stirring at 37°C for 30 min. The supernatant was filtered rapidly through a glass wool column. After centrifugation, the pellets were purified in 40% isotonic Percoll (GE Healthcare Biosciences), and the cells recovered in the suspension were collected for RNA or protein extraction.
Intestinal permeability assay
The detection of intestinal permeability was performed as previously described [24, 25]. In brief, colonic tissues were rapidly harvested and cut longitudinally. The mucosal membranes were mounted in modified Ussing chambers (Physiologic Instruments, San Diego, CA), with an exposed tissue area of 0.3 cm2. Transepithelial electrical resistance (TER) in ohm cm2 was determined according to the spontaneous potential difference and short circuit current.
RNA isolation and quantitative RT-PCR analysis
CYP1A gene expression was determined by real-time Q-PCR. Total RNA was extracted from the colons using Trizol reagent (Takara Co., Ltd, Dalian, China), according to the manufacturer's instructions. The total RNA was reverse transcribed into complementary DNA (cDNA) using a SuperScript FirstStrand Synthesis System RT-PCR Kit (Invitrogen, Carlsbad, CA). The sequences of the primers used were as follows (gene symbol: forward, reverse): CYP1A: CAATGAGTTTGGGGAGGTTACTG, CCCTTCTCAAATGTCCTGTAGTG; and β-actin: CTTCTTTGCAGCTCCTTCGTT, AGGAGTCCTTCTGACCCATTC. PCR was performed under the following conditions: 30 sec at 94˚C, 1 min at 60˚C, and 30 sec at 68˚C for 40 cycles, followed by 7 min at 68˚C. Relative gene expression was determined by calculating the ratio between the Ct values for the target gene and β-actin.
Cell culturing and TNF-α and IFN-γ treatment
Caco-2 cells were cultured in Eagle's Minimum Essential Medium supplemented with 10% heat-inactivated foetal bovine serum (Gemini Bioproducts) and 1% non-essential amino acids (Invitrogen). Cells were plated on transwell filters with a pore size of 0.4 μm (Corning, USA). TNF-α (20 ng/ml) and IFN-γ (10 ng/ml) were added basolaterally to monolayers with or without 100 nM FICZ.
TER measurement
Caco-2 monolayers were grown on 0.33 cm2 Transwell supports (Millipore). TER was measured with a Millicell-ERS voltohmmeter (Millipore) as described previously [26]. TER measurements were calculated in ohms cm2 after subtracting the blank value for the membrane insert. The TER values were normalized to the initial values and were expressed as percentages of the initial resistance values.
Western blot analysis
Whole proteins were extracted from the Caco-2 monolayers using a previously described method. Protein concentrations were determined using BCA assay reagent (Beyotime). Samples (25 μg protein) were electrophoresed in an 8 or 10% polyacrylamide gel, and the proteins were then electrophoretically transferred to a polyvinylidene fluoride membrane. The membrane was blocked with 5% BSA for 2 h at room temperature and was then incubated with the following primary antibodies overnight at 4℃: ZO-1 (1:500, Proteintech), Occludin (1:5000, Epitomics), Claudin-1 (1:1,000, Abcam), CYP1A1 (1:500, Proteintech), MLCK (1:1,000, Abcam), pMLC (1:1,000, Abcam), NF-κB p65 (1:500, Santa cruz), p-NF-κB p65 (1:1000, Cell signaling technology) and GAPDH (1:1,000, Goodhere Biotechnology). The membrane was subsequently incubated with a secondary antibody at room temperature for 1 h. Membrane imaging was performed using ECL western blotting detection reagent (Millipore) according to the manufacturer's instructions, and images were captured using an Image Station 4000R (Kodak).
Immunofluorescence and confocal microscopy
Caco-2 monolayers were fixed with 4% paraformaldehyde for 15 min and permeabilized with 0.2% Triton X-100 for 10 min. Then, the cells were blocked by incubation with PBS containing 3% BSA for 1 h at room temperature. Next, they were incubated with primary antibodies, including ZO-1 (1:50, BD), Occludin (1:50, R&D), Claudin-1 (1:50, Abcam) and NF-κB p65 (1:50, Santa cruz), at 4℃ overnight and then with secondary antibodies (diluted 1:400) for 1 h in the dark. Subsequently, the cells were washed and mounted using ProLongR Gold Antifade Reagent with DAPI (Invitrogen). Fluorescence was visualized by laser scanning fluorescence microscopy (Leica TCS SP5; Leica).
Statistical analyses
The results were expressed as the mean ± standard deviation (SD). Analysis was performed using SPSS software (Statistical Package for the Social Sciences). Analysis of variance (ANOVA) was used for comparisons among 3 or more groups, and Student's t-test was used for comparisons between 2 groups. Differences were considered statistically significant at a p<0.05. If not otherwise stated, all experiments included three independent replicates and were performed in triplicate to ensure for reproducibility.
Results
AhR activation ameliorates the disruption of intestinal barrier function in a mouse model of DSS-induced colitis
C57BL/6J wild-type (WT) mice were treated with 3% DSS for 7 days, and FICZ was administered to the mice for 5 days beginning at 2 days after the start of DSS administration. CYP1A1 expression was detected for the indication of AhR activation (Fig.1A). Histological examination of colonic tissues in the different groups revealed that the mice treated with FICZ developed less severe mucosal inflammatory infiltration compared with DSS-induced mice (Fig.1B).
Intestinal permeability and epithelial TJ barrier function have critical roles in the pathogenesis of IBD [3, 4]. To investigate the role of FICZ in colonic paracellular permeability, the TER of mouse colonic tissues mounted in Ussing chambers was measured. The colonic TER was significantly reduced in the DSS group compared with the control group, suggesting that intestinal permeability was increased in the DSS mice. Interestingly, FICZ prevented the decline in TER induced by DSS (Fig.1C).
We next examined the effect of FICZ on the DSS-induced disruption of TJs. Markedly reduced expressions of ZO-1, Occludin and Claudin-1 were observed in the DSS mice compared with the control mice. However, FICZ prevented the DSS-induced reduction in TJ protein expression (Fig. 2A). FICZ induced remarkably increase of CYP1A1 indicated that AhR was activated.
IHC staining also revealed that DSS treatment resulted in the depletion of ZO-1, Occludin and Claudin-1 at the epithelial junctions (Fig. 2B). FICZ administration attenuated this DSS-induced depletion of TJ proteins at the intercellular junctions.
These data indicate that AhR activation by FICZ ameliorates the DSS-induced disruption of intestinal barrier function by decreasing paracellular permeability and maintaining TJ barrier integrity.
Figure 1
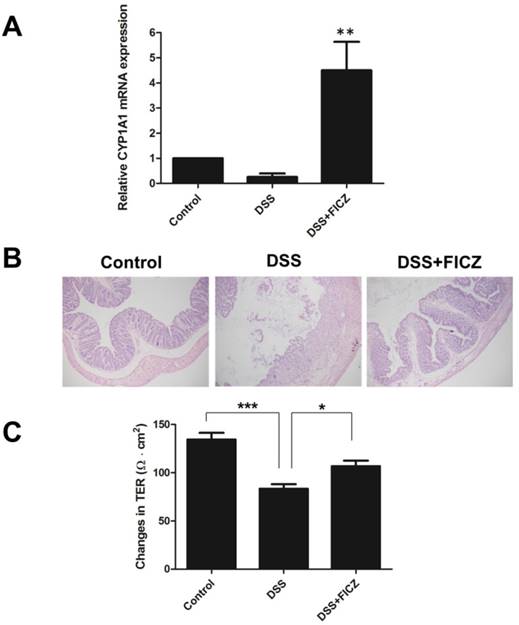
FICZ attenuates the loss of intestinal barrier function during DSS-induced colitis. C57BL/6J WT mice were treated with 3% DSS for 7 days, and FICZ was administered to the mice for 5 days beginning at 2 days after the start of DSS administration. (A) The CYP1A1 mRNA level in colon homogenates. The data are presented as the mean ± SD (n = 5). **P<0.01 versus DSS group. (B) Colons from the control, DSS-treated and DSS+FICZ-treated groups were stained with haematoxylin-eosin. Magnification: ×100. (C) TER was measured to assess the permeability of the colonic epithelium on day 7 of the experiment. The data are presented as the mean ± SD (n = 5). *P<0.05 versus control group, ***P<0.001 versus DSS+FICZ group.
AhR activation attenuates increased paracellular permeability of the intestinal epithelium induced by TNF-α/IFN-γ
To further assess the impact of AhR activation on intestinal barrier function, a well-established in vitro intestinal epithelial model system was used. Caco-2 monolayers were treated with 20 ng/ml TNF-α and 10 ng/ml IFN-γ for 48 h, respectively, with or without FICZ. As shown in Fig. 3A, TNF-α/IFN-γ induced significant decrease in the TER of the Caco-2 monolayers within 48 h. However, the decrease was prevented by FICZ.
AhR activation ameliorates TNF-α/IFN-γ-induced alterations in the expression and localization of TJ proteins
It has been reported that both the reduced expression and altered subcellular localization of TJ proteins contribute to intestinal epithelial barrier dysfunction [27-29].
We next examined the effect of AhR activation on the expression of TJ proteins in Caco-2 monolayers. However, in the Caco-2 monolayers, western blot analysis revealed that ZO-1, Occludin, and Claudin-1 expression was not significantly altered following stimulation with TNF-α/IFN-γ, FICZ, or both compounds (Fig. 3B).
Subsequently, immunofluorescence staining was performed to examine the localization of TJ proteins in the Caco-2 monolayers. As shown in Figure 3C, in the control Caco-2 monolayers, ZO-1 staining showed the presence of continuous belt-like structures. FICZ did not affect the ZO-1 distribution in the absence of TNF-α/IFN-γ. Following treatment with TNF-α/IFN-γ, ZO-1 localization at the cellular junctions became discontinuous, with a decreased intensity and areas of a gap-like appearance. Similar results were obtained for Occludin (Fig. 3D) and Claudin-1 (Fig. 3E). Moreover, the cytoplasmic staining of Occludin and Claudin-1 was seen in Caco-2 monolayers upon TNF-α/IFN-γ stimulation. However, co-administration of FICZ ameliorated these TNF-α/IFN-γ-induced changes.
Figure 2
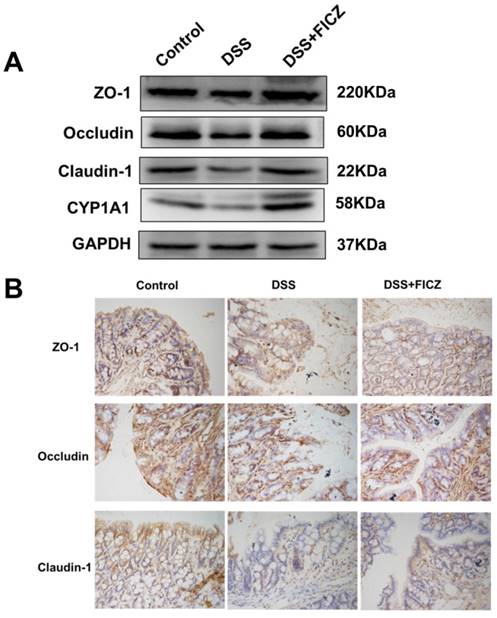
FICZ prevents DSS-induced disruption of TJ. (A) Expressions of TJ proteins (ZO-1, Occludin and Claudin-1) and CYP1A1 in the colonic mucosa were evaluated by western blotting. (B) Immunohistochemical analysis of TJ proteins in the colonic mucosa. Magnification: ×400.
AhR activation suppress TNF-α/IFN-γ-induced activation of the MLCK-pMLC signalling pathway
It is widely recognized that myosin light chain phosphorylation by MLCK plays an important role in the regulation of intestinal barrier function and TJ structure [30-32].
To further investigate the effect of AhR activation on the MLCK-pMLC signalling pathway, we examined MLCK expression and MLC phosphorylation. In the Caco-2 monolayers, treatment with FICZ alone had no effect on activation of the MLCK-pMLC signalling pathway. By 48 h of TNF-α/IFN-γ treatment, significant increases in MLCK protein expression and MLC phosphorylation were observed. FICZ treatment remarkably attenuated the increases in MLCK protein expression and MLC phosphorylation induced by TNF-α/IFN-γ (Fig. 4A). In addition, the MLCK-pMLC signalling pathway was also activated in the mice with DSS-induced colitis and administration of FICZ inhibited the activation of the MLCK-pMLC signalling pathway (Fig. 4B and 4C).
AhR activation suppress NF-κB p65 signaling pathway induced by TNF-α/IFN-γ
Previous study has reported that TNF-α-activated NF-κB p65 signaling could induce upregulation of the MLCK promoter activity and MLCK transcription and the subsequent increase in TJ permeability [30, 33, 34]. To investigate whether FICZ-induced AhR activation could block the activation of NF-kB p65, protein levels of total and phospho-p65 were detected by western blot analysis. As shown in Fig. 5A, FICZ alone did not affect basal p65 activity. TNF-α/IFN-γ treatment for 30 min induced a significant increase in levels of phosphorylated p65. Pre-incubation with 100 nM FICZ for 60 min prior to TNF-α/IFN-γ exposure for 30 min, remarkably attenuated TNF-α/IFN-γ induced increased phosphorylation of p65 (Fig. 5A).
Figure 3
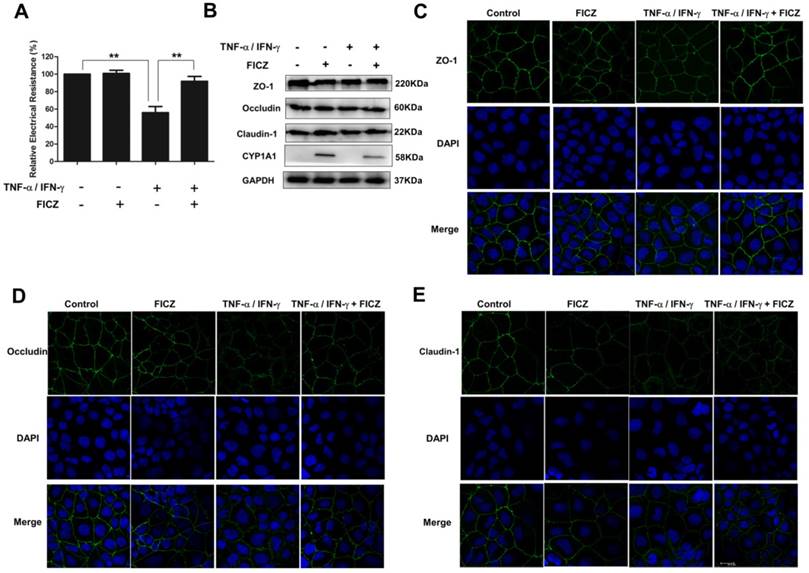
The effects of FICZ and TNF-α/IFN-γ on the expression and localization of tight junction proteins in vitro. Caco-2 cell monolayers were treated with 20 ng/ml TNF-α and 10 ng/ml IFN-γ for 48 h, respectively, with or without FICZ. (A) TER was detected. The results are expressed as the mean ± SD (n = 3). **P<0.01. (B) Expression of TJ proteins was assessed by western blotting. The results are representative of three independent experiments. The subcellular localization of ZO-1 (C, green), Occludin (D, green) and Claudin-1 (E, green) was determined in Caco-2 cells treated as described above by immunofluorescence staining. Scale bar, 20 μM.
Consistent with this, immunofluorescence of NF-kB p65 showed that p65 accumulation within the nucleus in Caco-2 monolayers treated with TNF-α/IFN-γ for 30 min. However, pre-incubation with FICZ decreased the nuclear accumulation of p65 elicited by TNF-α/IFN-γ (Fig. 5B).
These results suggested that FICZ-induced AhR activation could protect against TNF-α/IFN-γ induced increase in intestinal TJ permeability through the suppression of NF-kB p65 signaling.
Discussion
AhR has been shown to contribute to the maintenance of intestinal mucosal homeostasis. AhR expression is down-regulated in patients with IBD [21]. In addition, down-regulation of AhR in the SW480 human epithelial colon cancer cell line has been demonstrated to enhance the lipopolysaccharide-induced inflammatory response [9]. In contrast, enhancement of AhR activation has been reported to inhibit inflammatory pathways in the gut [9, 21, 35, 36].
In our study, administration of FICZ, a powerful activator of AhR, was shown to suppress the pathogenesis of DSS-induced colitis and to ameliorate the DSS-induced disruption of intestinal epithelial barrier function.
As intestinal epithelial barrier function is regulated by apical TJs, we measured TJ protein levels in colonic tissues. Our results revealed that ZO-1, Occludin and Claudin-1 were present at reduced levels in DSS-induced colitis. However, FICZ prevented the changes in TJ protein expression and distribution, thus explaining the dramatically improved barrier function.
IFN-γ and TNF-α have been extensively characterized as important proinflammatory cytokines involved in the pathogenesis of IBD. These cytokines have been reported to impair intestinal TJ barrier function through different mechanisms [1-3, 6, 33, 37, 38]. Biological therapies targeting TNF-α have become a very important part of IBD treatment.
Figure 4
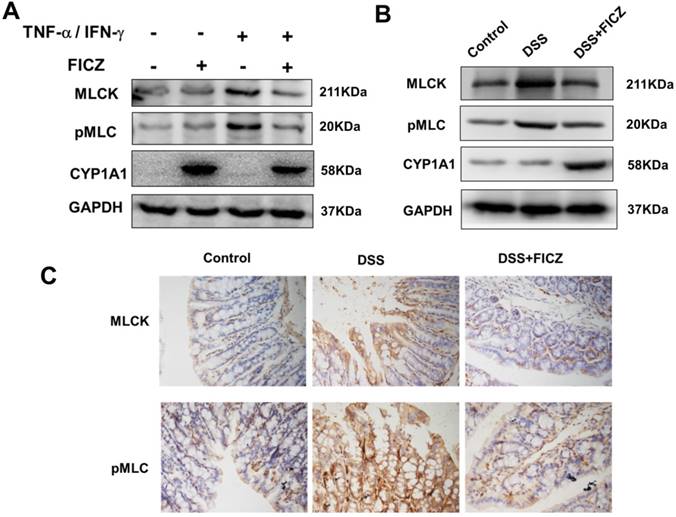
FICZ inhibits the increases in MLC phosphorylation and MLCK expression. (A) Caco-2 cell monolayers were treated with 20 ng/ml TNF-α and 10 ng/ml IFN-γ for 48 h, respectively, with or without FICZ. MLCK and pMLC expression was assessed by western blotting. (B) Expression of MLCK and pMLC in the colonic mucosa was evaluated by western blotting. The results are representative of three independent experiments. (C) Immunohistochemical analysis of MLCK and pMLC in the colonic mucosa. Magnification: ×400.
Therefore, we used Caco-2 cell monolayers as model of intestinal epithelial monolayers. These monolayers were exposed to TNF-α/IFN-γ to disrupt monolayer barrier function. Caco-2 cells exhibited significant decreases in TER after TNF-α/IFN-γ stimulation for 48 h. However, FICZ treatment significantly alleviated these TNF-α/IFN-γ-induced reductions of barrier function. These results demonstrate that FICZ antagonizes the intestinal epithelial monolayer barrier disruption induced by TNF-α/IFN-γ.
The expression of TJ proteins was then measured in Caco-2 monolayers. The expression of TJ proteins was not significantly altered by treatment with TNF-α/IFN-γ with or without FICZ. However, immunofluorescence analysis of TJ proteins in the Caco-2 monolayers revealed that TNF-α/IFN-γ induced a significant change in TJ distribution and that FICZ attenuated this destructive effect. These results suggest that AhR activation ameliorates TJ barrier dysfunction by regulating both the expression and distribution of TJ proteins.
Previous studies conducted by our laboratory and others have suggested that the MLCK-pMLC signalling pathway plays a central role in the regulation of TJ permeability in the intestinal epithelium. MLCK protein and gene expression was increased by TNF-α/IFN-γ, and corresponded with increased MLC phosphorylation [30, 39, 40]. MLCK expression and MLC phosphorylation was also increased in the colon tissues of patients with active Crohn's disease [41]. In the present study, we also examined the role of AhR activation in regulation of the MLCK-pMLC signalling pathway. FICZ was found to inhibit the TNF-α/IFN-γ-induced increases in MLCK and pMLC protein expression. These results suggest that AhR activation may play a role in protecting intestinal epithelial barrier function against disruption induced by TNF-α/IFN-γ by suppressing activation of the MLCK-pMLC signalling pathway.
Previous studies have demonstrated that TNF-α-induced activation of MLCK-pMLC signaling was mediated by NF-κB activation [30, 33, 34]. Activated NF-κB p50/p65 led to a stepwise increase in MLCK transcription, which induced increase in MLCK protein expression and activity, followed by MLCK-mediated opening of the intestinal epithelial TJ barrier.
Figure 5
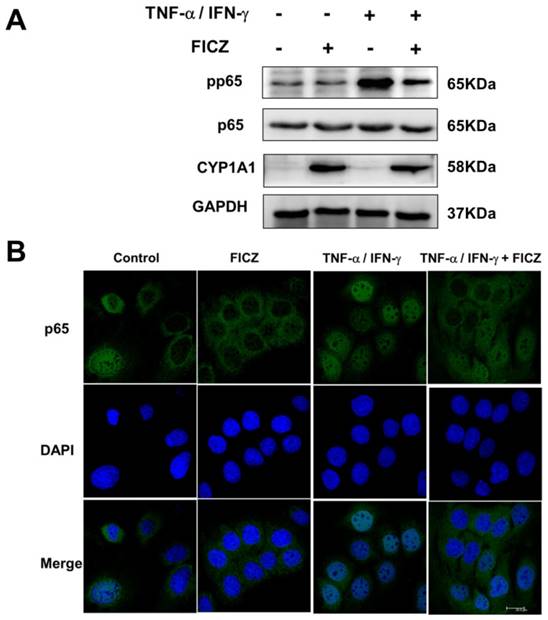
FICZ suppress TNF-α/IFN-γ induced activation of NF-κB p65. Caco-2 monolayers were exposed to 20 ng/ml TNF-α and 10 ng/ml IFN-γ for 30 min with or without FICZ. (A) The phosphorylation level of NF-κB p65 was assessed by western blotting. (B) Caco-2 monolayers were stained for NF-κB p65 by immunofluorescence. Scale bar, 20 μM.
It has been shown that there was a considerable cross-talk between AhR-signaling and NF-κB transcription factors [9, 42-46]. AhR-agonists such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) and β-naphthoflavone (BNF) were reported to suppressed NF-κB signaling induced by TNF-α, LPS and crystalline silica [9, 44, 46]. In addition, Ovrevik et al. found that depletion of AhR resulted in increased TNF-α-induced phosphorylation of p65 in human bronchial epithelial cells [45]. In the present study, we also found that remarkable activation of NF-kB p65 induced by TNF-α/IFN-γ. Pre-treatment with FICZ significantly suppressed NF-kB p65 activation induced by TNF-α/IFN-γ.
In conclusion, our study revealed the molecular mechanisms for the ability of AhR activation to against the intestinal barrier disrupted by TNF-α/IFN-γ. This protective effect may provide a potential approach to restoring TJ barrier function and AhR may be a novel therapeutic target in intestinal disease such as IBD.
Supplementary Material
Supplementary figures.
Acknowledgements
This research was supported by grants from the National Natural Science Foundation of China (NSFC 81501661 to M.Y., NSFC 81330013 to H.Y., NSFC 81770524 and 81470803 to W.D.X., and NSFC 81400602 to G.Q.C), Innovative Research Team of Ministry of Education of China (IRT 13050 to H.Y.) and this work was also supported by the Program of Yunnan academican and expert workstation (2015-2).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Nusrat A, Turner JR, Madara JL. Molecular physiology and pathophysiology of tight junctions. IV. Regulation of tight junctions by extracellular stimuli: nutrients, cytokines, and immune cells. Am J Physiol Gastrointest Liver Physiol. 2000;279:G851-7
2. Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799-809
3. Turner JR. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am J Pathol. 2006;169:1901-9
4. Bruewer M, Samarin S, Nusrat A. Inflammatory bowel disease and the apical junctional complex. Ann N Y Acad Sci. 2006;1072:242-52
5. Meddings J. The significance of the gut barrier in disease. Gut. 2008;57:438-40
6. Suenaert P, Bulteel V, Lemmens L, Noman M, Geypens B, Van Assche G. et al. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn's disease. Am J Gastroenterol. 2002;97:2000-4
7. Mennigen R, Nolte K, Rijcken E, Utech M, Loeffler B, Senninger N. et al. Probiotic mixture VSL#3 protects the epithelial barrier by maintaining tight junction protein expression and preventing apoptosis in a murine model of colitis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1140-9
8. Arsenescu R, Arsenescu V, Zhong J, Nasser M, Melinte R, Dingle RW. et al. Role of the xenobiotic receptor in inflammatory bowel disease. Inflamm Bowel Dis. 2011;17:1149-62
9. Furumatsu K, Nishiumi S, Kawano Y, Ooi M, Yoshie T, Shiomi Y. et al. A role of the aryl hydrocarbon receptor in attenuation of colitis. Dig Dis Sci. 2011;56:2532-44
10. Gu YZ, Hogenesch JB, Bradfield CA. The PAS superfamily: sensors of environmental and developmental signals. Annu Rev Pharmacol Toxicol. 2000;40:519-61
11. Monteleone I, Pallone F, Monteleone G. Aryl hydrocarbon receptor and colitis. Semin Immunopathol. 2013;35:671-5
12. Esser C, Rannug A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol Rev. 2015;67:259-79
13. Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. Exactly the same but different: promiscuity and diversity in the molecular mechanisms of action of the aryl hydrocarbon (dioxin) receptor. Toxicol Sci. 2011;124:1-22
14. Beischlag TV, Luis Morales J, Hollingshead BD, Perdew GH. The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr. 2008;18:207-50
15. Hu W, Sorrentino C, Denison MS, Kolaja K, Fielden MR. Induction of cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Mol Pharmacol. 2007;71:1475-86
16. Koch DC, Jang HS, O'Donnell EF, Punj S, Kopparapu PR, Bisson WH. et al. Anti-androgen flutamide suppresses hepatocellular carcinoma cell proliferation via the aryl hydrocarbon receptor mediated induction of transforming growth factor-beta1. Oncogene. 2015;34:6092-104
17. Rannug U, Rannug A, Sjoberg U, Li H, Westerholm R, Bergman J. Structure elucidation of two tryptophan-derived, high affinity Ah receptor ligands. Chem Biol. 1995;2:841-5
18. Farmahin R, Crump D, Kennedy SW. Sensitivity of avian species to the aryl hydrocarbon receptor ligand 6-formylindolo [3,2-b] carbazole (FICZ). Chem Biol Interact. 2014;221:61-9
19. Mukai M, Tischkau SA. Effects of tryptophan photoproducts in the circadian timing system: searching for a physiological role for aryl hydrocarbon receptor. Toxicol Sci. 2007;95:172-81
20. Oberg M, Bergander L, Hakansson H, Rannug U, Rannug A. Identification of the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole, in cell culture medium, as a factor that controls the background aryl hydrocarbon receptor activity. Toxicol Sci. 2005;85:935-43
21. Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L. et al. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 2011;141:237-48 48 e1
22. Li Y, Innocentin S, Withers DR, Roberts NA, Gallagher AR, Grigorieva EF. et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 2011;147:629-40
23. Ji T, Xu C, Sun L, Yu M, Peng K, Qiu Y. et al. Aryl Hydrocarbon Receptor Activation Down-Regulates IL-7 and Reduces Inflammation in a Mouse Model of DSS-Induced Colitis. Dig Dis Sci. 2015;60:1958-66
24. Qiu Y, Yu M, Yang Y, Sheng H, Wang W, Sun L. et al. Disturbance of intraepithelial lymphocytes in a murine model of acute intestinal ischemia/reperfusion. J Mol Histol. 2014;45:217-27
25. Han B, Sheng B, Zhang Z, Pu A, Yin J, Wang Q. et al. Aryl Hydrocarbon Receptor Activation in Intestinal Obstruction Ameliorates Intestinal Barrier Dysfunction Via Suppression of MLCK-MLC Phosphorylation Pathway. Shock. 2016;46:319-28
26. Yu M, Yang S, Qiu Y, Chen G, Wang W, Xu C. et al. Par-3 modulates intestinal epithelial barrier function through regulating intracellular trafficking of occludin and myosin light chain phosphorylation. J Gastroenterol. 2015;50:1103-13
27. Bruewer M, Utech M, Ivanov AI, Hopkins AM, Parkos CA, Nusrat A. Interferon-gamma induces internalization of epithelial tight junction proteins via a macropinocytosis-like process. FASEB J. 2005;19:923-33
28. Schumann M, Gunzel D, Buergel N, Richter JF, Troeger H, May C. et al. Cell polarity-determining proteins Par-3 and PP-1 are involved in epithelial tight junction defects in coeliac disease. Gut. 2012;61:220-8
29. Hopkins AM, Walsh SV, Verkade P, Boquet P, Nusrat A. Constitutive activation of Rho proteins by CNF-1 influences tight junction structure and epithelial barrier function. J Cell Sci. 2003;116:725-42
30. Cunningham KE, Turner JR. Myosin light chain kinase: pulling the strings of epithelial tight junction function. Ann N Y Acad Sci. 2012;1258:34-42
31. Su L, Nalle SC, Shen L, Turner ES, Singh G, Breskin LA. et al. TNFR2 activates MLCK-dependent tight junction dysregulation to cause apoptosis-mediated barrier loss and experimental colitis. Gastroenterology. 2013;145:407-15
32. Cao M, Wang P, Sun C, He W, Wang F. Amelioration of IFN-gamma and TNF-alpha-induced intestinal epithelial barrier dysfunction by berberine via suppression of MLCK-MLC phosphorylation signaling pathway. PLoS One. 2013;8:e61944
33. Ye D, Ma I, Ma TY. Molecular mechanism of tumor necrosis factor-alpha modulation of intestinal epithelial tight junction barrier. Am J Physiol Gastrointest Liver Physiol. 2006;290:G496-504
34. Chen SW, Zhu J, Zuo S, Zhang JL, Chen ZY, Chen GW. et al. Protective effect of hydrogen sulfide on TNF-alpha and IFN-gamma-induced injury of intestinal epithelial barrier function in Caco-2 monolayers. Inflamm Res. 2015;64:789-97
35. Takamura T, Harama D, Fukumoto S, Nakamura Y, Shimokawa N, Ishimaru K. et al. Lactobacillus bulgaricus OLL1181 activates the aryl hydrocarbon receptor pathway and inhibits colitis. Immunol Cell Biol. 2011;89:817-22
36. Huang Z, Jiang Y, Yang Y, Shao J, Sun X, Chen J. et al. 3,3'-Diindolylmethane alleviates oxazolone-induced colitis through Th2/Th17 suppression and Treg induction. Mol Immunol. 2013;53:335-44
37. Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-{alpha} modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005;288:G422-30
38. Yang S, Yu M, Sun L, Xiao W, Yang X, Zhang C. et al. Interferon-gamma-induced intestinal epithelial barrier dysfunction by NF-kappaB/HIF-1alpha pathway. J Interferon Cytokine Res. 2014;34:195-203
39. Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409-19
40. Graham WV, Wang F, Clayburgh DR, Cheng JX, Yoon B, Wang Y. et al. Tumor necrosis factor-induced long myosin light chain kinase transcription is regulated by differentiation-dependent signaling events. Characterization of the human long myosin light chain kinase promoter. J Biol Chem. 2006;281:26205-15
41. Blair SA, Kane SV, Clayburgh DR, Turner JR. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest. 2006;86:191-201
42. Tian Y. Ah receptor and NF-kappaB interplay on the stage of epigenome. Biochem Pharmacol. 2009;77:670-80
43. Tian Y, Rabson AB, Gallo MA. Ah receptor and NF-kappaB interactions: mechanisms and physiological implications. Chem Biol Interact. 2002;141:97-115
44. Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA. Ah receptor and NF-kappaB interactions, a potential mechanism for dioxin toxicity. J Biol Chem. 1999;274:510-5
45. Ovrevik J, Lag M, Lecureur V, Gilot D, Lagadic-Gossmann D, Refsnes M. et al. AhR and Arnt differentially regulate NF-kappaB signaling and chemokine responses in human bronchial epithelial cells. Cell Commun Signal. 2014;12:48
46. Beamer CA, Seaver BP, Shepherd DM. Aryl hydrocarbon receptor (AhR) regulates silica-induced inflammation but not fibrosis. Toxicol Sci. 2012;126:554-68
Author contact
![]() Corresponding author: Hua Yang, M.D., Ph.D., Department of General Surgery, Xinqiao Hospital, Chongqing 400037, China, Phone: (86) 23-687-55705 (Office), E-mail: huayangedu.cn
Corresponding author: Hua Yang, M.D., Ph.D., Department of General Surgery, Xinqiao Hospital, Chongqing 400037, China, Phone: (86) 23-687-55705 (Office), E-mail: huayangedu.cn
Citation styles
APA
Yu, M., Wang, Q., Ma, Y., Li, L., Yu, K., Zhang, Z., Chen, G., Li, X., Xiao, W., Xu, P., Yang, H. (2018). Aryl Hydrocarbon Receptor Activation Modulates Intestinal Epithelial Barrier Function by Maintaining Tight Junction Integrity. International Journal of Biological Sciences, 14(1), 69-77. https://doi.org/10.7150/ijbs.22259.
ACS
Yu, M.; Wang, Q.; Ma, Y.; Li, L.; Yu, K.; Zhang, Z.; Chen, G.; Li, X.; Xiao, W.; Xu, P.; Yang, H. Aryl Hydrocarbon Receptor Activation Modulates Intestinal Epithelial Barrier Function by Maintaining Tight Junction Integrity. Int. J. Biol. Sci. 2018, 14 (1), 69-77. DOI: 10.7150/ijbs.22259.
NLM
Yu M, Wang Q, Ma Y, Li L, Yu K, Zhang Z, Chen G, Li X, Xiao W, Xu P, Yang H. Aryl Hydrocarbon Receptor Activation Modulates Intestinal Epithelial Barrier Function by Maintaining Tight Junction Integrity. Int J Biol Sci 2018; 14(1):69-77. doi:10.7150/ijbs.22259. https://www.ijbs.com/v14p0069.htm
CSE
Yu M, Wang Q, Ma Y, Li L, Yu K, Zhang Z, Chen G, Li X, Xiao W, Xu P, Yang H. 2018. Aryl Hydrocarbon Receptor Activation Modulates Intestinal Epithelial Barrier Function by Maintaining Tight Junction Integrity. Int J Biol Sci. 14(1):69-77.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.