Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(1):87-99. doi:10.7150/ijbs.22498 This issue Cite
Research Paper
Comparison of microRNA Profiles between Bovine Mammary Glands Infected with Staphylococcus aureus and Escherichia coli
Zhuo-Ma Luoreng1,2*, Xing-Ping Wang1,2*, Chu-Gang Mei1, Lin-Sen Zan1 ![]()
1. College of Animal Science and Technology, National Beef Cattle Improvement Centre, Northwest A&F University, Yangling Shaanxi, China;
2. Key Laboratory of Zoology in Hunan Higher Education, College of Life Science, Hunan University of Arts and Science, Changde Hunan, China.
* These authors contributed equally to this work.
Received 2017-8-23; Accepted 2017-11-28; Published 2018-1-11
Abstract

MicroRNAs (miRNAs) play crucial roles in regulating innate and adaptive immunity in humans and animals. Infection with E. coli or S. aureus can cause inflammation of the mammary glands, which results in significant economic losses in dairy cattle. However, the regulatory mechanisms of miRNAs in response to E. coli or S. aureus infection in bovine mammary glands have not been thoroughly explored. To discover the differential expression of miRNA in bovine mammary gland challenged with E. coli or S. aureus, we performed miRNA sequencing on tissue samples. A total of 1838 miRNAs were identified, including 580 known-miRNAs (included in the miRbase database) and 1258 predicted novel miRNAs. The miRNA expression patterns indicated that, compared with control samples, 279 miRNAs and 305 miRNAs were differentially expressed miRNAs (DIE-miRNA) in S. aureus and E. coli infected tissues, respectively. Moreover, the results of comparison the DIE-miRNAs between the E. coli and S. aureus infected groups showed that 197 DIE-miRNAs are identical, 108 DIE-miRNAs are specific to the E. coli group, and 82 DIE-miRNAs are specific to the S. aureus group. Many DIE-miRNAs, such as bta-miR-144, bta-miR-451 and bta-miR-7863, might be the useful biomarkers of mastitis caused by E. coli and S. aureus. In addition, target genes of the DIE-miRNAs were predicted. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis indicated that these DIE-miRNAs are likely involved in many immune signaling pathways, including the Toll-like receptor signaling pathways, MAPK signaling pathway, cell adhesion molecules, TGF-β signaling pathway, leukocyte trans endothelial migration, cytokine-cytokine receptor interaction, and chemokine signaling pathways. This study has provided supportive evidence that miRNAs may serve as diagnostic biomarkers of mastitis in dairy cows, and suggests potentially of effective strategies to combat mastitis.
Keywords: miRNA, E. coli, S. aureus, mastitis, dairy cattle, deep sequencing
Introduction
Mastitis is considered to be the most prevalent and costly disease of dairy cow, which is caused by a variety of pathogens including the Gram-negative bacterium E. coli and the Gram-positive bacterium S. aureus [1, 2]. Most cases of E. coli mastitis present as an acute and severe infection, which can be either naturally cleared by the immune system or through antibiotic therapy if initiated within 10 days of the time of infection [3]. In contrast, S. aureus causes a persistent and chronic infection, which may range from mild to severe, with antibiotic treatment being largely ineffective [4], suggesting a different molecular pathogenesis.
Revealing the mechanisms of host immune and inflammatory responses to infection is particularly important for the prevention and treatment of mastitis. Previous studies have reported that the immune responses of bovine mammary epithelial cells (bMECs) infected with E. coli and S. aureus are significantly different [3, 5]. Furthermore, differential expression of inflammatory cytokines and other immune related proteins, in response to E. coli or S. aureus, were also detected in mammary tissues and milk [6-8].
MicroRNAs (miRNAs), endogenous and small non-coding RNA molecules, act as post-transcriptional regulators of gene expression by directly binding to the 3´ untranslated region (UTR) of target mRNA. Recently, it has been reported that miRNAs participate in multiple biological processes, such as development and immunity in humans and animals [9-14]. For example, it has been confirmed that miR-146 is a NF-κB-dependent gene, and suppresses the production of TNF-α and IL-1β in human and mice through targeting of the IRAK1 and TRAF6 genes. This suggests that miRNAs act as a key regulator of epithelial immune responses [15, 16]. Zhou et al. [17] reviewed that miRNAs may regulate epithelial immune responses in multiple steps of innate immune networks, including the production and secretion of inflammatory cytokines, chemokines, adhesion and co-stimulatory molecules. However, the roles of miRNAs in the modulation of immune and inflammatory responses to bacterial infection are not well understood in dairy cattle. Many studies have reported that the expression levels of many miRNAs detected by qPCR exhibit obvious changes in response to bovine mastitis or inflammatory pathway activation in bMECs. For example, it has been reported that the expressions of miRNAs (miR-223, miR-146a, miR-146b, miR-15a-3p, miR-15a-5p) were upregulated, whereas three miRNAs (miR-16a, -31, and -181a) were downregulated in infected versus healthy mammary tissue [11, 18, 19]. Although some reports have examined miRNAs and mastitis in dairy cows, the current understanding of the molecular mechanisms of the relationship is far from comprehensive. Thus, further investigation into the molecular mechanisms of the regulation of mastitis by miRNAs is necessary.
Increasingly, more robust techniques like high-throughput sequencing are being applied to detect the expression profile of known miRNAs, as well as to identify novel miRNAs. As a result, more information is becoming available to support our exploration of the expression and roles of miRNAs in the regulation of bovine mammary gland inflammation and their applications as biomarkers of mastitis.
It is well established that the cellular environment influences expression levels of intracellular genes, including miRNAs [20, 21]. However, few studies have been reported on transcriptomic differences in bovine mammary gland infected with S. aureus or E. coli. In the present study, in order to elucidate the differential expression miRNA (DIE-miRNA) and investigate the roles of miRNAs in host defenses against S. aureus or E. coli, 2 models of bovine mastitis were employed, using low dose exposures over an extended time frame to infect mammary glands of Chinese Holstein cows. Next, samples were sequenced and the miRNA profiles were compared. These data allowed for the detection of DIE-miRNA through the pairwise comparison of S. aureus, E. coli, and control groups. These findings provide a new understanding of the molecular mechanisms of mastitis in cows, and can guide further studies of pathogenesis of both S. aureus and E. coli type mastitis.
Materials and methods
Animals and ethics statement
A group of 2 year-old, half-sibling, Chinese Holstein cows in mid-lactation from rural regions of Yangling, Shannxi, China, were provided identical feed and management. All animals were allowed food and water ad libitum. Milk samples from each cow were tested for 3 weeks prior to the initiation of the trail to confirm that they contained <200,000 somatic cells/mL, and were free of mastitis. All studies involving dairy cattle were performed using humane procedures according to the provisions of the Animal Husbandry and Use Committee of Northwest A & F University.
Induction of mastitis and sample collection
S. aureus (ATCC 25923) or E. coli (ATCC 25922) (purchased from Shanghai Fuxiang Biotechnology Co., Ltd., China) were incubated on solid LB medium for 14 h at 37°C. A single colony was transferred to 2 mL of liquid LB medium and incubated for 7 h at 37°C. A 10 μL sample was transferred to 10 mL of liquid LB medium and incubated for 7 h (37°C). Serial dilutions were then made to prepare the desired inoculum dose of approximately 105 CFU/mL. The cows were randomly divided into three groups (uninfected control group, S. aureus infected, and E. coli infected). The teats of each animal were cleaned and disinfected with 75% ethanol, then were directly injected into the mammary glands with a 5 mL suspension of either sterile PBS or 105 CFU/mL S. aureus or E. coli through the teat canal, respectively. At 7 days, mammary tissues were surgically collected using aseptic technique. Pathological evaluation of H&E (hematoxylin-eosin) stained sections was conducted to ensure the successful induction of mastitis. These samples were used for RNA isolation, RNA-seq and qPCR verification.
RNA extraction
Total RNA was extracted from all samples using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocols. RNA purity was measured using the NanoPhotometer spectrophotometer (IMPLEN, CA, USA). RNA concentration and integrity were evaluated using the Qubit 2.0 Fluorometer (Life Technologies, CA, USA) and the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA), respectively.
Library preparation, clustering and sequencing for small RNA
All RNA samples within each group were sent to BIOMARKER Biotechnology Co., LTD (Beijing, China) for small-RNA library construction and sequencing. The libraries were constructed as previously described [22, 23]. Briefly, a total of 1.5 μg RNA per sample was used for the construction of sequencing libraries. Library quality was assessed using the Agilent Bioanalyzer 2100. Clustering of the index-coded samples was carried out using a cBot Cluster Generation System according to the manufacturer's recommendations. Library preparations were then sequenced on an Illumina Hiseq 2500 platform (Illumina, Santa Clara, CA, USA) and paired-end reads were generated.
Quality control and miRNAs identification
Raw data were first filtered using in-house perl scripts. During this process, clean reads were generated by removing low quality reads, as well as other reads consisting of the adapter and ploy-N reads from the raw data. Clean reads were obtained after being further trimmed by removing the inappropriate sequences with a length of < 18 nt or > 30 nt. Concurrently, Q20, Q30, GC-content and the sequence duplication level of the clean data were calculated for quality control of sequencing [22, 23]. All the following analyses were based on the resulting clean reads.
Using the Bowtie software, the clean reads were respectively aligned with the Silva, GtRNAdb, Rfam and Repbase databases to identify and filter out tRNA, rRNA, snRNA, snoRNA, as well as other ncRNA and repeats. The remaining reads were used to detect known and novel miRNAs as predicted using the miRDeep2 software package [24]. Briefly, all 3′-adapter trimmed sequences with a length of <18 bp and mismatch >1 were excluded from the analysis. The probability score of novel miRNAs were calculated. Those with a probability score of >80% were considered to be true positives. The secondary structure of novel miRNA was predicted using the Randfold software.
Quantification of miRNA expression levels and differential expression analysis
MiRNA expression levels were estimated for each sample as follows: sRNAs were compared with the bovine genome, and the read count for each miRNA was obtained from the mapping results. The expression level of each gene was calculated according to the frequency of clean reads. The data were subsequently normalized to the number of transcripts per million clean tags (TPM) [25], using the following formula:
TPM = (specific miRNA reads ×1,000,000) / total miRNA mapped reads
Differential expression analyses of two samples were conducted using the IDEG6 software. P-values were corrected to q-values [26]. The significantly differentially expressed miRNAs were identified by the criteria of q-value < 0.005, |log2 (fold-change)| ≥1 and FDR< 0.01.
Target gene functional annotation
Target genes of miRNAs of particular interest were predicted using miRnada [27] and RNAhybrid [28]. Functional annotations of miRNA target genes were performed by the following database searches: Nr (NCBI non-redundant protein sequences), Nt (NCBI non-redundant nucleotide sequences), KO (KEGG Ortholog database), GO (Gene Ontology).
GO enrichment analysis and KEGG Pathway Enrichment Analysis
Gene Ontology (GO) enrichment analysis of the target genes of DIE-miRNAs was applied using the GOseq R packages, which can adjust for gene length biases [29]. KEGG pathways were determined using the KOBAS software [30].
Verification of miRNA expression by qPCR
A total of 24 differentially expressed miRNAs, including 15 up-regulated and 9 down-regulated genes were validated by qPCR. The verification was designed and conducted in compliance with the MIQE (minimum information for publication of quantitative real-time PCR experiments) guidelines [31].
Total miRNA was extracted using the miRcute miRNA isolation kit (Tiangen Biotech Co., Ltd. Beijing, China) according to the manufacturer's instructions. No mammalian homologue cel-miR-39 (5′-UCACCGGGUGUAAAUCAGCUUG) was used to normalize the different RNA samples [13, 32, 33]. The procedures of qPCR for miRNAs were performed as previously described [13]. Briefly, a total of 5 μL of miRNA (625 ng) per sample was used to perform First-Strand cDNA Synthesis using the miRcute miRNA First-Strand cDNA Synthesis Kit (Tiangen Biotech Co., Ltd. Beijing, China) according to the manufacturer's instructions. Then, 1 μL of first-strand cDNA was used to perform the miRNA qPCR in an ABI 7500 Real-Time PCR System (Applied Biosystems, USA) using the miRcute miRNA qPCR Detection Kit (SYBR Green) (Tiangen Biotech Co., Ltd. Beijing, China) according to the manufacturer's instructions. Cel-miR-39 was used as a reference gene to normalize the expression of the miRNAs. The primers for this assay are listed in Table S1. At the end of the protocol, a dissociation melt curve of the products was determined. The amplification efficiency of the qPCR reactions for each gene (or primer pair) in this study were ranged from 99.6% to 104.3% with R2 > 0.99 (data not shown). No-template control as well as no-reverse transcriptase control were included in all plates to verify the absence of contamination. All melt curves showed a single peak, indicating a single amplicon in each qPCR reaction. All amplifications were run in triplicate, and any questionable curves were excluded.
The expression levels of miRNAs were normalized to the mean of spiked-in miRNA cel-miR-39 and were calculated using the 2-ΔΔCt method [34]. All data were expressed as mean ± SD. Statistical significance in the data was determined by the Student's t-test, with P values of < 0.05 considered to be statistically significant.
Results
Establishment of an in vitro bovine mastitis model
At 7 dpi with either S. aureus or E. coli, the infected cows exhibited clinical signs consistent with mastitis (redness, swelling, warmth and pain in the teats, fever, and flocculent precipitate in milk, and milk somatic cell counts >2,000,000 cells/mL). These clinical signs indicated the mastitis model was successfully established. For confirmation of the clinical observations, H&E staining of mammary tissue biopsies was used to verify the development of mastitis. The results showed that pathological changes were evident in the S. aureus and E. coli infected compared to control group (Figure 1). Taken together, these observations indicate that mastitis was successfully induced.
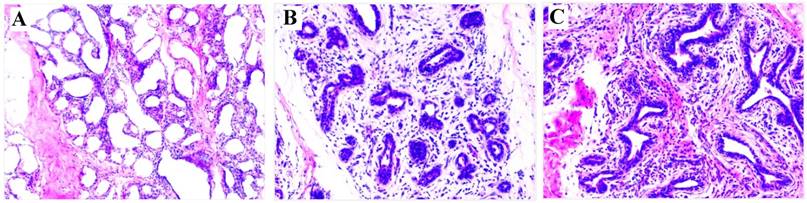
HE staining of mammary gland tissue (200×). (A) Control; (B) E. coli; (C) S. aureus. Comparing with (A), (B) and (C) have a few pathological changes, including disruption of the lobular structure of the mammary gland, separation of the mammary epithelial cells, reduced size of acinar cavities, damaged mammary gland acini, as well as inflammatory infiltrates including neutrophils and macrophages.
miRNA sequencing
To study expression levels of miRNA in the mammary gland of dairy cattle during mastitis, three cDNA libraries of small RNAs were constructed from pooled total RNA for each group. Through sequencing of cDNA libraries using the Illumina Hiseq 2500 platform, 24,672,171, 18,365,120 and 19,677,779 raw reads were obtained from the control, S. aureus, and E. coli infected samples, respectively. After removal of low-quality reads, adaptor sequences, and reads with sequences <18 nt or >30 nt, an average of greater than 11.89 M of clean data per sample were obtained (Table 1). This included 20,608,579, 15,600,273 and 16,681,613 clean reads (Q30 > 94.37) from control, S. aureus and E. coli infected samples, respectively. Of these, 16,158,450 (78.41%), 12,552,468 (80.46%) and 13,650,442 (81.83%) clean reads of small RNAs from control and S. aureus and E. coli infected samples, respectively, were unannotated, including miRNA (Table 1). Furthermore, the clean reads of all samples were compared with the bovine reference genome UMD3.1 (http://www.ensembl.org/Bos_taurus/Info/Index). The results showed that 13,048,593 (63%) clean reads of the control sample, 10,420,961 (67%) clean reads of S. aureus and 11,309,412 (68%) clean reads of the E. coli infected sample were able to be mapped to the bovine genome (Table 1).
Overview of miRNA sequencing data
| Samples | Raw reads | Q30 (%) | Clean reads | Annotated readsa | Unannotated readsb | Mapped Small RNA Reads |
|---|---|---|---|---|---|---|
| Control | 24,672,171 | 94.37 | 20,608,579 (100.00%) | 4,450,129 (21.59%) | 16,158,450 (78.41%) | 13,048,593 (63%) |
| S. aureus | 18,365,120 | 94.57 | 15,600,273 (100.00%) | 3,047,805 (19.65%) | 12,552,468 (80.46%) | 10,420,961 (67%) |
| E. coli | 19,677,779 | 94.46 | 16,681,613 (100.00%) | 3,031,171 (18.17%) | 13,650,442 (81.83%) | 11,309,412 (68%) |
a Annotated reads include rRNA, tRNA, snRNA, snoRNA, ncRNA, repbase.
b miRNAs are contained in unknown unannotated reads.
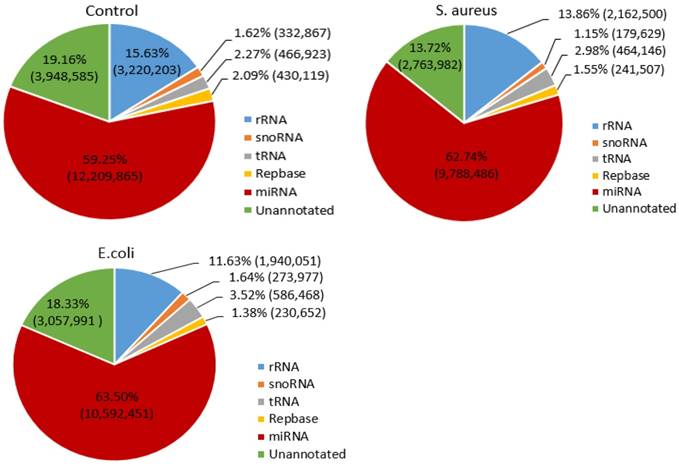
To determine the distribution of small RNAs, such as rRNA, tRNA, snRNA, snoRNA, ncRNA and miRNA, the Bowtie software was used to align the clean reads with the Silva, GtRNAdb, Rfam and Repbase databases. The results indicated that the miRNA reads in the control, S. aureus and E. coli infected groups were 12,209,865 (59.25%), 9,788,486 (62.74%) and 10,592,451 (63.50%), respectively (Figure 2).
Distribution of small RNAs among different categories in the three treatment groups. The numbers in parentheses indicate the read numbers for each type of small RNA.
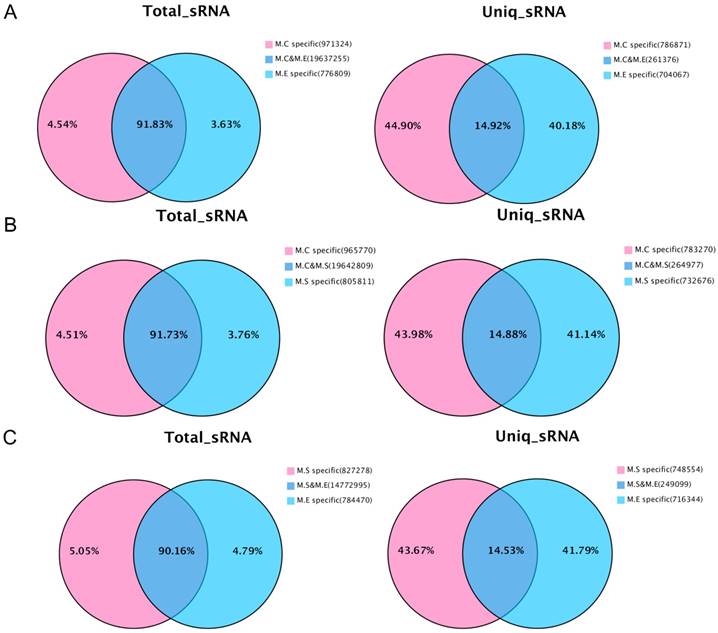
In addition, the analysis was performed on the common sequence and specific sequences of small RNAs by pairwise comparisons of the three sets of sequencing data. The results are presented in Figure 3. With respect to the total number of sRNA sequence reads, most sequences (90.16%~91.83%) were common among the groups, while a small proportion of the sequences (3.63%~5.05%) were specific. For the total number of sequence types, the greatest proportion (40.18%~44.90%) is specific, whereas a small proportion of the sequences (14.53%~14.92%) were common. The above results suggest that the copy number of the specific sequence types between samples is much larger than that of the common sequence types.
Venn diagram representation of the statistics of common and specific sequences of small RNAs between samples. (A) Control mammary tissue (M.C) vs E. coli infected mammary tissue (M.E); (B) Control mammary tissue (M.C) vs S. aureus infected mammary tissue (M.S); (C) S. aureus infected mammary tissue (M.S) vs E. coli infected mammary tissue (M.E). Uniq_sRNA means type of sRNA sequences, total_sRNA means quantity of sRNA sequences.
Identification of miRNAs
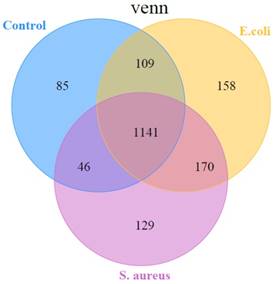
In the present study, the miRDeep2 software package [24] was used to detect known miRNAs, as well as to identify novel miRNAs. The Randfold tools software was used to predict the secondary structure of novel miRNAs. The results showed that there were, in total, 1838 miRNAs detected in the three groups, including 580 known-miRNAs and 1258 predicted novel miRNAs (Table S2). Among them, 1141 miRNAs were common among the 3 groups, while 85 miRNAs, 129 miRNAs, and 158 miRNAs were specific in control, S. aureus infected, and E. coli infected groups, respectively (Figure 4).
A representation of the number of all miRNAs overlapping in different comparisons
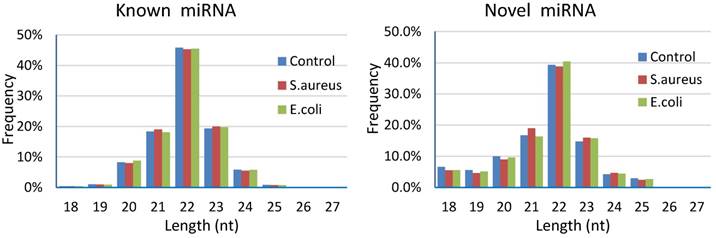
Results of length distribution of the mapped miRNAs indicated that the majority of the mapped known miRNAs and novel miRNAs were 20 to 24 nucleotides in length (comprising 97.58%~97.77% and 84.94%~86.62% of their total number, respectively). The length of the majority of known or novel miRNAs was 22 nucleotides, followed by 23, 21, 20 or 24 nucleotides (Figure 5).
Length distribution of the mapped miRNAs in mammary glands of cows
Analysis of miRNA expression
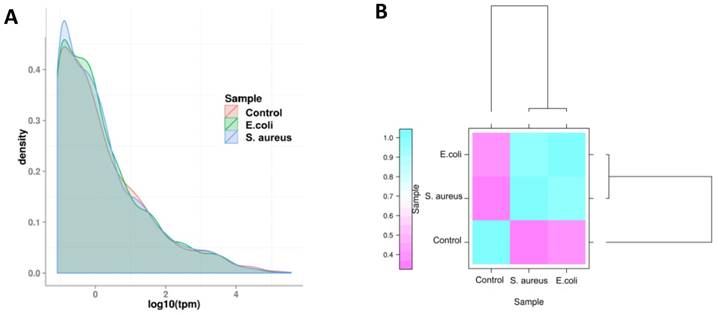
The expression level of miRNAs were calculated based on the frequency of clean tags, and were subsequently normalized to TPM [25]. The overall expression pattern of miRNAs in the samples is presented in Figure 6. It can be seen that the miRNA expression within the three samples are highly consistent (Figure 6 A). In comparison to the control group, the overall expression levels in the infected groups were similar, indicating that both E. coli and S. aureus infection had similar effects on the expression of miRNAs in dairy cows (Figure 6 B). Furthermore, the most abundant miRNAs (at least one group of TPM > 1,000) are presented in Table S3. The top 10 most highly expressed miRNAs among all of the samples were bta-miR-148a, bta-miR-143, bta-miR-21-5p, bta-miR-26a, bta-miR-30a-5p, bta-miR-99a-5p, bta-let-7f, bta-let-7g, bta-miR-10b and bta-miR-126-3p. In addition, the novel miRNA conservative_X_30752 (the sequence is presented in Table S4) was the most highly expressed miRNA in mammary gland tissues of all samples.
The overall distribution of miRNA expression in each group. (A) TPM density distribution. The curves of different colours represent the different experimental samples. The abscissa of the points on the curve represents the logarithm of TPM of miRNA in the corresponding sample, and the ordinate of the point represents the probability density. (B) Comparison of correlation between samples, which measured with Pearson correlation coefficient as calculated by the TPM of miRNA.
DIE-miRNAs in mammary glands of infected animals
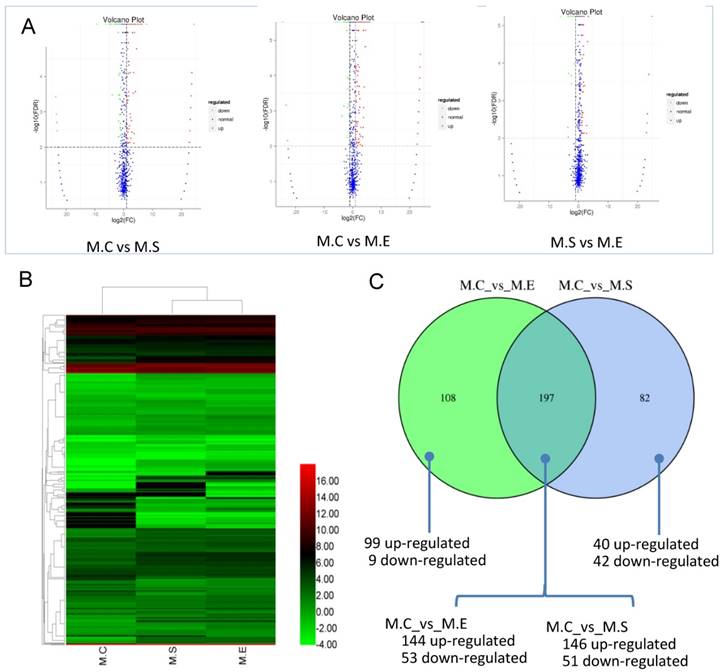
The expression levels of miRNAs were compared between the S. aureus or E. coli infected groups in relation to the control group in order to identify DIE-miRNAs. Based on the criteria of a two-fold or greater change, FDR<0.01, and TPM>1, a total of 279 DIE-miRNAs were identified, with 186 being up-regulated and 93 down-regulated, between the S. aureus-infected and control groups (Figure 7, Table S5). Among them, 173 were known miRNAs, whereas 106 were predicted novel miRNAs. It is worth noting that bta-miR-7863 was not detected in the control sample, however, in the S. aureus infected group, expression increased more than 24-fold. This suggests that bta-miR-7863 could be a sensitive biomarker of S. aureus mastitis. Many known miRNAs (including bta-miR-184, bta-miR-223, bta-miR-2344, bta-miR-147, bta-miR-665, bta-miR-370, bta-miR-9-5p, bta-miR-214, bta-miR-154c, bta-miR-27a-5p, bta-miR-493, bta-miR-494, bta-miR-155, bta-miR-221, bta-miR-146a, bta-miR-199a, bta-miR-378, etc.) as well as three novel miRNAs (conservative_14_6048, conservative_21_15256, conservative_X_30874) (Table S4) were stably and highly expressed in bovine mammary glands. It was observed that these miRNAs were further significantly up-regulated in S. aureus infected animals. Moreover, in comparison to the control, many of the known miRNAs, including bta-miR-375, bta-miR-885, bta-miR-6522, bta-miR-6516, bta-miR-2285t, bta-miR-29, bta-miR-30, bta-miR-200, etc., were significantly down-regulated. For example, bta-miR-375, bta-miR-885, and bta-miR-6522, were the most down-regulated miRNAs (3.4~6.2 fold change). The expression of novel miRNA conservative_26_19257 was significantly decreased (9.2-fold change) in the S. aureus infected group (Table S5).
Differential expression of miRNAs (DIE-miRNAs) among samples. (A) The plot showing the expression levels of DIE-miRNAs. The differential expression of each point in the volcanic map represents a miRNA, the abscissa represents a logarithmic value of a difference in expression between the two samples; the ordinate represents -log(FDR). The greater the absolute value of the abscissa, the greater the difference in the expression between the two samples. The greater the ordinate value, the more significant the difference, and the more accurately the miRNAs were screened. The blue plots in the figure represents miRNAs that are not differentially expressed, the red dots represent up-regulated miRNAs, and the green points represent down-regulated miRNAs. (B) Clustering analysis of expression pattern of DIE-miRNAs among the three groups. (C) Comparison of the number of DIE-miRNAs between S. aureus and E. coli infected samples compared with controls, respectively. The heatmap is based on the log2TPM of each DIE-miRNA. M.C, control sample (Healthy mammary gland); M.E, E. coli infected mammary gland; M.S, S. aureus infected mammary gland.
In response to experimental E. coli infection, a total of 305 DIE-miRNAs were detected, with 243 being up-regulated and 62 down-regulated, in comparison to the control group (Figure 7, Table S6). Among the up-regulated miRNAs, 2 known miRNAs (miR-202, miR-2357) were only detected in E. coli infected samples, making them promising biomarkers for E. coli induced mastitis in cattle. Moreover, 6 miRNAs (miR-7863, miR-1247-3p, miR-541, miR-2320-5p, miR-431, and miR-2448-5p) were detected in both E. coli and S. aureus infected samples, suggesting these miRNAs have similar roles in the regulation of mastitis induced by the 2 types of bacterium.
Comparative analysis of DIE-miRNAs between S. aureus and E. coli infected mammary glands
It is well established that both S. aureus and E. coli cause mammary inflammation. However, the former is Gram-positive bacteria, the latter is Gram-negative bacteria. In order to investigate the differences in the molecular mechanism of mastitis caused, we compared the DIE-miRNAs of M.C (control, healthy mammary gland) vs M.S (S. aureus infected mammary gland) and M.C vs M.E (E. coli infected mammary gland). The data indicated that 197 DIE-miRNAs are identical, 108 DIE-miRNAs are specific to E. coli, and 82 DIE-miRNAs are specific to S. aureus. Surprisingly, it was observed that bta-miR-144 and bta-miR-451 were significantly up-regulated in S. aureus infected mammary glands, but were significantly down-regulated in E. coli infected mammary glands (Table S5, S6, and Figure 7C), suggesting that they play different roles in the regulatory mechanisms of mastitis caused by S. aureus and E. coli.
When the DIE-miRNAs of the S. aureus and E. coli infected groups were compared, it was observed that 70 up-regulated and 21 down-regulated DIE-miRNAs were uniquely identified in the E. coli mastitis samples compared with those of S. aureus mastitis (Table S7). These results suggest that different miRNAs exert different molecular functions with respect to inflammatory responses in mammary glands caused by different pathogens, namely E. coli and S. aureus.
Verification of differentially expressed miRNAs by qPCR
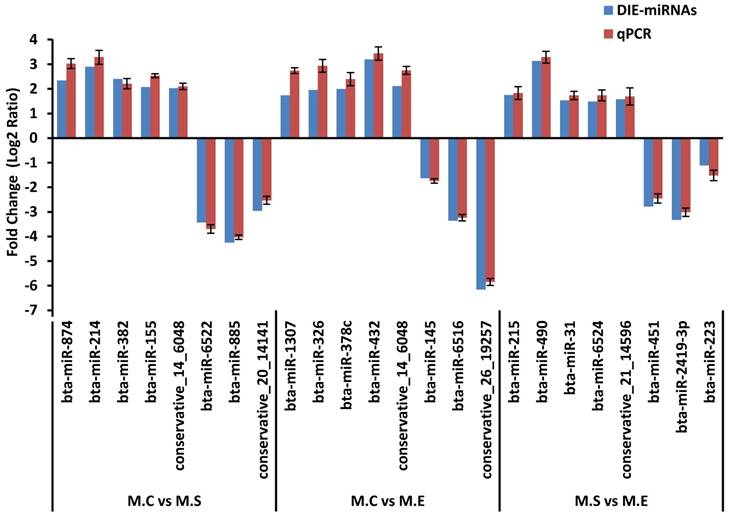
To verify the results of the deep sequencing and comparative analyses, we randomly selected 24 miRNAs for qPCR confirmation: 5 up-regulated and 3 down-regulated from the three comparisons of M.C vs M.S, M.C vs M.E and M.E vs M.S. As is presented in Figure 8, the qPCR detection of the DIE-miRNAs were consistent with those identified through RNAseq, confirming the reliability of our DIE-miRNAs data.
Expression pattern validation of miRNAs selected from the DIE analysis of RNAseq by qPCR. The relative expression level (Fold change=Log2 Ratio) of each miRNA was normalized to the spiked-in cel-miR-39.
Functional annotation and KEGG pathway enrichment analysis of DIE-miRNA target genes
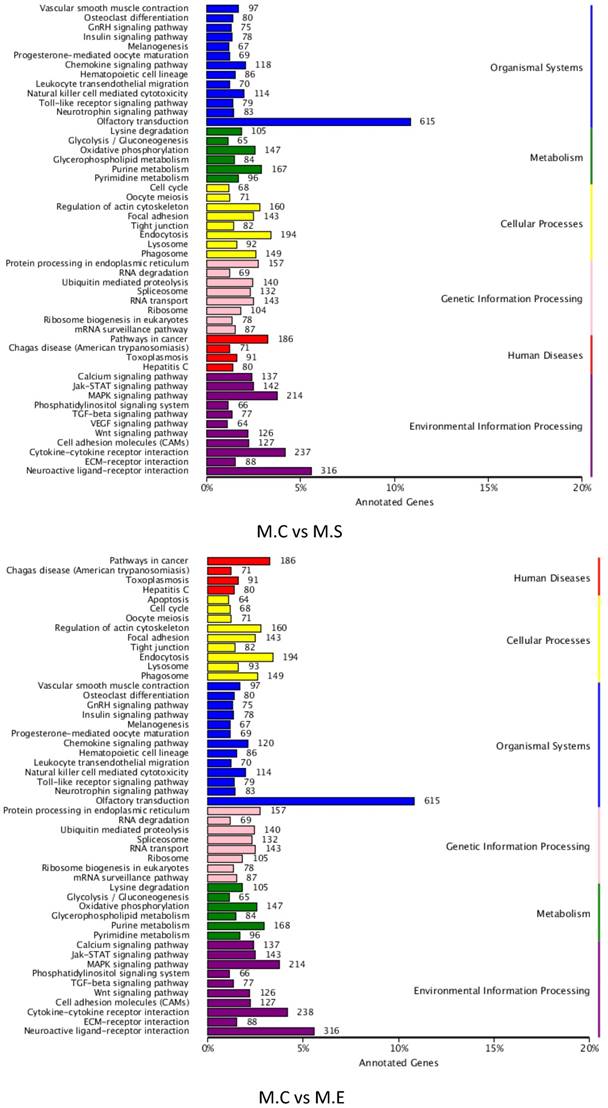
To better understand the roles of the identified DIE-miRNAs in bovine mammary glands, the target genes of DIE-miRNAs were predicted. The results revealed a total of 18,385 target genes of the 305 DIE-miRNAs of M.C vs M.E, and 18,399 target genes of the 279 DIE-miRNAs of M.C vs M.S. The predicted target genes were divided into 6 groups, including genetic information processing, organismal systems, disease, metabolism, cellular processes, and environmental information processing. The top 6 clusters observed were as follows: olfactory transduction, neuroactive ligand-receptor interaction, cytokine-cytokine receptor interaction, MAPK signalling pathway, endocytosis and pathways in cancer (Figure 9). It is interesting and supportive of our findings that many immune signalling pathways are included, such as TLR signalling pathway, cell adhesion molecules, MAPK signalling pathway, TGF-β signalling pathway, leukocyte trans endothelial migration, cytokine-cytokine receptor interaction, and chemokine signalling pathways. Thus, it can be inferred that DIE-miRNAs likely participate in the immune response through regulation of their target genes.
KEGG pathway annotation of DIE-miRNA target genes
Discussion
RNAseq technologies have enabled the discovery of novel miRNAs, the detection of those expressed at low levels in a high-throughput system, as well as the detection of differentially expressed miRNAs [35, 36]. In the case of bovine mastitis, at the time of publication, a limited number of studies have applied these approaches to study the expression patterns and roles of miRNAs. Lawless et al. [36] used the Illumina HiSeq 2000 platform to detect 21 significant differentially expressed miRNAs in response to Streptococcus uberis infected bMECs. Jin et al. [35] detected 113 novel miRNAs, and identified 10 and 10 differentially expressed miRNAs in E. coli and S. aureus challenged bMECs, respectively. However, the study of bMECs alone cannot fully reveal the molecular mechanisms of mammary gland infection due to the relative lack of complexity compared to in vivo models of mastitis. Immune cells in tissues are also involved in the regulation of the mammary gland inflammatory response process. With respect to bovine mammary tissues, only one study to date has utilized solexa sequencing to detect the differential expression of miRNAs in response to high dose (5 mL suspension of 107 CFU/mL S. aureus) and short-term (24 hpi) S. aureus infection [37]. However, mastitis is a gradual process of repeated infections with low dose pathogens in the dairy industry. Therefore, we sought to investigate the function of miRNA in an in vivo model of mastitis caused by either S. aureus or E. coli, that most closely resembles natural disease progression. The mastitis models employed here were induced in dairy cows with a single low dose infection of E. coli or S. aureus, which slowly develops into mastitis. Mammary glands presenting with mastitis were sequenced using RNAseq technology to estimate the expression of miRNAs. In this study, 580 known miRNAs and 1258 predicted miRNAs were detected in the infected mammary glands of dairy cattle. Of these, 74 were very highly expressed (TPM > 1,000) (Table S3), suggesting their relative importance in the development of inflammation in the bovine mammary gland.
Although DIE-miRNAs in mammary tissue challenged with a high dose S. aureus have been previously reported [37], our results differed slightly with respect to the quantities and types of DIE-miRNAs, suggesting that dose if the bacterial inoculum, or the duration of infection can affect the expression of miRNAs in mammary glands. Therefore, the current study contributes additional useful information with regard to the molecular regulation of mastitis in dairy cows. The expression patterns of miRNAs in control and S. aureus infected groups included a total of 279 DIE-miRNAs includes 186 up-regulated and 93 down-regulated ones (Figure 7, Table S5). Furthermore, a total of 305 DIE-miRNAs, including 243 up-regulated and 62 down-regulated, were identified in E. coli infected samples compared with controls (Figure 7, Table S6). Of these DIE-miRNAs, it appears as though bta-miR-7863 might be a specific biomarker of the two types of mastitis examined here due to the expression between the S. aureus and E. coli infected groups increased by 24-fold over the control group. The potential target genes predicted by the bioinformatics analysis showed that bta-miR-7863 might regulate multiple immune-related genes, including members of the interleukin family, IRAK1, TLR7, LBP, ect., which are key molecules of the animal immune system (Table S8). Moreover, miRNAs with high expression levels in the control and S. aureus infected groups, including bta-miR-223, bta-miR-146a, bta-miR-184, bta-miR-155, bta-miR-214, bta-miR-147 and bta-miR-378, were also differentially expressed in both groups. The expression levels of many of the miRNAs characterized in this study were consistent with findings reported in other previous studies. For example, several studies reported that the expressions of bovine miR-223 in mammary gland with mastitis was up-regulated in comparison to that of healthy cows [18, 37, 38], and have also been observed to regulate the mammary innate immune response by directly targeting HMGB1 [39]. Furthermore, our previous study identified that the expression of bovine miR-146a was significantly increased in bovine mammary tissues from cases of mastitis caused by S. aureus, E. coli or mixed bacterial infection [11], and supports the idea that bovine miR-146a regulates the secretion of inflammatory cytokines such as TNF-α, IL-6, and IL-8 in bMECs [13]. It is also noteworthy that bta-miR-375 is one of most down-regulated DIE-miRNAs (6.20 and 4.67-fold change in S. aureus and E. coli infected mammary tissue, respectively), suggesting that it participates in the regulation of immune responses and inflammation. However, very little information has been reported on the function of bta-miR-375 in bovine immunity, and comprehensive studies are required to elucidate its role in the regulation of bovine mammary inflammation.
In this study, we first compared the DIE-miRNAs of M.C vs M.S and M.C vs M.E. The observed results showed that most DIE-miRNAs are identical between the two datasets, and a small number of DIE-miRNAs is unique to each group. This indicates that these DIE-miRNAs function among a common or specific regulatory pathway in the immune system in response to bovine mastitis following E. coli and S. aureus infection. Moreover, we observed that expression changes of bta-miR-144 and bta-miR-451 in the two groups were opposite of each other (up-regulated in S. aureus infected mammary gland, while down-regulated in E. coli infected mammary gland), suggesting that they play different roles in the mechanisms regulating the two types of mastitis examined here. Sontakke et al. [40] reported that bta-miR-144 and bta-miR-451 were up-regulated in large healthy follicles relative to small follicles. However, there are no reports at the time of publication regarding their expression and regulatory functions in mastitis. In order to explore their roles, potential target genes were predicted using bioinformatics analyses. The results indicated that bta-miR-451 might regulate ATF2, CDKN2D and MEF2D (Table S8). Interestingly, it had been reported that human miR-451 (has-miR-451) directly target the 3′UTR of the ATF2 gene [41], in addition to regulating inflammatory cytokine secretion in rheumatoid arthritis [42]. Meanwhile, the MEF2D gene was demonstrated to be involved in regulating inflammatory responses [43, 44]. As far as bta-miR-144 is concerned, total of 31 potential target genes involved in immunity, such as EZH2, NKRF, and so on (Table S8), are predicted. Several studies have reported that the role of EZH2 is the regulation of the differentiation and function of T cells [45], as well as in negatively regulating immune responses [46]. Moreover, NKRF inhibits the activity of NF-κB, and subsequently regulates the NF-κB signaling pathway, which is the key signaling pathway of the innate immune response [47, 48]. According the results of bioinformatics analysis, we speculate that bta-miR-144 and bta-miR-451 might participate in the regulation of immune responses. Thus, comprehensive studies of the immunological functions of bta-miR-144 and bta-miR-451 are needed to reveal the significant differences in the immune regulatory mechanisms responsive to S. aureus and E. coli infections.
To investigate the regulatory function of DIE-miRNAs in dairy cattle, we predicted potential targets of these miRNAs. Among the putative targets, bta-miR-146a and bta-miR-214 were identified to control genes responsible for regulating inflammation in bovine mammary epithelial cells [11-13]. In the present study, the putative target genes of DIE-miRNAs were annotated as being involved in various classes of immune signalling pathways, TLR signalling pathway, TGF-β signalling pathway, cytokine-cytokine receptor interaction, MAPK signalling pathway, cell adhesion molecules, leukocyte trans endothelial migration, and chemokine signalling pathways. It has been previously reported that these signalling pathways described above are likely associated with the development of mastitis [3, 49-51]. The functions of the putative targets and the corresponding miRNAs' role in the regulation of mammary inflammation indicate that the DIE-miRNAs may play multiple roles in immunity related gene regulation networks.
In summary, we detected the miRNAs expressed in bovine mammary glands infected with S. aureus or E. coli, and identified 580 known miRNAs and 1258 novel miRNAs. Furthermore, 279 DIE-miRNAs were identified in S. aureus infected mammary glands, and 305 DIE-miRNAs in E. coli infected mammary gland. These miRNAs might be involved in the regulation of various immune signaling pathways, including TLR signaling pathways, MAPK signaling pathway, cell adhesion molecules, TGF-β signaling pathway, cytokine-cytokine receptor interactions, leukocyte trans endothelial migration, and chemokine signaling pathways, according the KEGG pathway enrichment analysis of target genes of the detected DIE-miRNAs. Furthermore, many DIE-miRNAs, such as bta-miR-144, bta-miR-451 and bta-miR-7863, might be biomarkers of the two types of mastitis examined here. This study provides an experimental basis for revealing underlying causes and regulatory mechanisms leading to the development of mastitis. We also uncovered a potential role for miRNAs as biomarkers in the diagnosis of mastitis, as well as for the development of control measures.
Abbreviations
miRNAs: MicroRNAs; E. coli: Escherichia coli; S. aureus: Staphylococcus aureus; CFU: Colony-Forming Units; DIE-miRNAs: differentially expressed miRNAs; dpi: days post-infection; hpi: hours post-infection; KEGG: Kyoto Encyclopedia of Genes and Genomes; GO: Gene Ontology; TLR: Toll-like receptor; MAPK: bMECs: bovine mammary epithelial cells; qPCR: quantitative real-time PCR; TPM: the number of transcripts per million clean tags; M.C: control, healthy mammary gland; M.S: S. aureus infected mammary gland; M.E: E. coli infected mammary gland; S. uberis: Streptococcus uberis; IRAK1: interleukin-1 receptor-associated kinase 1; TLR7: toll-like receptor 7; LBP: lipopolysaccharide binding protein; ATF2: activating transcription factor 2; CDKN2D: cyclin-dependent kinase inhibitor 2D; MEF2D: myocyte enhancer factor 2D; EZH2: enhancer of zeste homolog 2; NKRF: NF-κB repressing factor.
Supplementary Material
Supplementary tables.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31501935), National Science-technology Support Plan Projects (2012BAD12B07), China Postdoctoral Science Foundation funded project (2014M562466) and the Construct Program of the Key Discipline in Hunan province.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hogeveen H, Huijps K, Lam T. Economic aspects of mastitis: New developments. New Zeal Vet J. 2011;59(1):16-23
2. Seegers H, Fourichon C, Beaudeau FO. Production effects related to mastitis and mastitis economics in dairy cattle herds. Vet Res. 2003;34(5):475-491
3. Gilbert FB, Cunha P, Jensen K. et al. Differential response of bovine mammary epithelial cells to Staphylococcus aureus or Escherichia coli agonists of the innate immune system. Vet Res. 2013;44(1):40
4. Sutra L, Poutrel B. Virulence factors involved in the pathogenesis of bovine intramammary infections due to Staphylococcus aureus. J Med Microbiol. 1994;40(2):79-89
5. Fu Y, Zhou E, Liu Z. et al. Staphylococcus aureus and Escherichia coli elicit different innate immune responses from bovine mammary epithelial cells. Vet Immunol Immunop. 2013;155(4):245-252
6. Ibeagha-Awemu EM, Ibeagha AE, Messier S. et al. Proteomics, genomics, and pathway analyses of Escherichia coli and Staphylococcus aureus infected milk whey reveal molecular pathways and networks involved in mastitis. J Proteome Res. 2010;9(9):4604-4619
7. Günther J, Esch K, Poschadel N. et al. Comparative kinetics of Escherichia coli- and Staphylococcus aureus-specific activation of key immune pathways in mammary epithelial cells demonstrates that S. aureus elicits a delayed response dominated by interleukin-6 (IL-6) but not by IL-1A or tumor necrosis factor alpha. Infect Immun. 2011;79(2):695-707
8. Lahouassa H, Moussay E, Rainard P. et al. Differential cytokine and chemokine responses of bovine mammary epithelial cells to Staphylococcus aureus and Escherichia coli. Cytokine. 2007;38(1):12-21
9. Lawless N, Vegh P, Oâ Farrelly C. et al. The role of microRNAs in bovine infection and immunity. Front Immunol. 2014;5:611
10. O'Connell RM, Rao DS, Chaudhuri AA. et al. Physiological and pathological roles for microRNAs in the immune system. Nat Rev Immunol. 2010;10(2):111-122
11. Wang XP, Luoreng ZM, Zan LS. et al. Expression patterns of miR-146a and miR-146b in mastitis infected dairy cattle. Mol Cell Probe. 2016;30(5):342-344
12. Song N, Wang X, Gui L. et al. MicroRNA-214 regulates immunity-related genes in bovine mammary epithelial cells by targeting NFATc3 and TRAF3. Mol Cell Probe. 2017;35:27-33
13. Wang XP, Luoreng ZM, Zan LS. et al. Bovine miR-146a regulates inflammatory cytokines of bovine mammary epithelial cells via targeting the TRAF6 gene. J Dairy Sci. 2017;100(9):7648-7658
14. Guan L, Hu X, Liu L. et al. Bta-miR-23a involves in adipogenesis of progenitor cells derived from fetal bovine skeletal muscle. Sci Rep. 2017;7:43716
15. Taganov KD, Boldin MP, Chang KJ. et al. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci U S A. 2006;103(33):12481-12486
16. Dai Y, Jia P, Fang Y. et al. miR-146a is essential for lipopolysaccharide (LPS)-induced cross-tolerance against kidney ischemia/reperfusion injury in mice. Sci Rep. 2016;6:27091
17. Zhou R, O'Hara SP, Chen X. MicroRNA regulation of innate immune responses in epithelial cells. Cell Mol Immunol. 2011;8(5):371-379
18. Naeem A, Zhong K, Moisá SJ. et al. Bioinformatics analysis of microRNA and putative target genes in bovine mammary tissue infected with Streptococcus uberis1. J Dairy Sci. 2012;95(11):6397-6408
19. Li Z, Zhang H, Song N. et al. Molecular cloning, characterization and expression of miR-15a-3p and miR-15b-3p in dairy cattle. Mol Cell Probe. 2014;28(5-6):255-258
20. Guo L, Zhang Y, Zhang L. et al. MicroRNAs, TGF-β signaling, and the inflammatory microenvironment in cancer. Tumor Biol. 2016;37(1):115-125
21. Dachs GU, Chaplin DJ. Microenvironmental control of gene expression: implications for tumor angiogenesis, progression, and metastasis. Semin Radiat Oncol. 1998;8(3):208-216
22. Zhao L, Lu H, Meng Q. et al. Profilings of microRNAs in the liver of Common Carp (Cyprinus carpio) infected with flavobacterium columnare. Int J Mol Sci. 2016;17(4):566
23. Wang Y, Wang Q, Gao L. et al. Parsing the regulatory network between small RNAs and target genes in Ethylene pathway in tomato. Front Plant Sci. 2017;8:527
24. Friedländer MR, Mackowiak SD, Li N. et al. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37-52
25. Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 2012;131(4):281-285
26. Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440-9445
27. Betel D, Wilson M, Gabow A. et al. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2007;36(Database issue):D149-D153
28. Rehmsmeier M, Steffen P, Hochsmann M. et al. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10(10):1507-1517
29. Young MD, Wakefield MJ, Smyth GK. et al. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14
30. Mao X, Cai T, Olyarchuk JG. et al. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics. 2005;21(19):3787-3793
31. Bustin SA, Benes V, Garson JA. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611-622
32. Mitchell PS, Parkin RK, Kroh EM. et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105(30):10513-10518
33. Ioannidis J, Donadeu FX. Circulating miRNA signatures of early pregnancy in cattle. BMC Genomics. 2016;17(1):184
34. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402-408
35. Jin W, Ibeagha-Awemu EM, Liang G. et al. Transcriptome microRNA profiling of bovine mammary epithelial cells challenged with Escherichia coli or Staphylococcus aureus bacteria reveals pathogen directed microRNA expression profiles. BMC Genomics. 2014;15(1):181
36. Lawless N, Foroushani AB, McCabe MS. et al. Next generation sequencing reveals the expression of a unique miRNA profile in response to a gram-positive bacterial infection. Plos One. 2013;8(3):e57543
37. Li R, Zhang C, Liao X. et al. Transcriptome microRNA profiling of bovine mammary glands infected with Staphylococcus aureus. Int J Mol Sci. 2015;16(3):4997-5013
38. Chen L, Liu X, Li Z. et al. Expression differences of miRNAs and genes on NF-κB pathway between the healthy and the mastitis Chinese Holstein cows. Gene. 2014;545(1):117-125
39. Li L, Huang J, Zhang X. et al. One SNP in the 3′-UTR of HMGB1 gene affects the binding of target bta-miR-223 and is involved in mastitis in dairy cattle. Immunogenetics. 2012;64(11):817-824
40. Sontakke SD, Mohammed BT, McNeilly AS. et al. Characterization of microRNAs differentially expressed during bovine follicle development. Reproduction. 2014;148(3):271-283
41. Sun X, Lou L, Zhong K. et al. MicroRNA-451 regulates chemoresistance in renal cell carcinoma by targeting ATF-2 gene. Exp Biol Med (Maywood). 2017;242(12):1299-1305
42. Wang ZC, Lu H, Zhou Q. et al. MiR-451 inhibits synovial fibroblasts proliferation and inflammatory cytokines secretion in rheumatoid arthritis through mediating p38MAPK signaling pathway. Int J Clin Exp Pathol. 2015;8(11):14562-14567
43. Zhu HX, Shi L, Zhang Y. et al. Myocyte enhancer factor 2D provides a cross-talk between chronic inflammation and lung cancer. J Transl Med. 2017;15(1):65
44. Yang S, Gao L, Lu F. et al. Transcription factor myocyte enhancer factor 2D regulates interleukin-10 production in microglia to protect neuronal cells from inflammation-induced death. J Neuroinflammation. 2015;12:33
45. Karantanos T, Chistofides A, Barhdan K. et al. Regulation of T cell differentiation and function by EZH2. Front Immunol. 2016;7:172
46. Chen S, Sheng C, Liu D. et al. Enhancer of zeste homolog 2 is a negative regulator of mitochondria-mediated innate immune responses. J Immunol. 2013;191(5):2614-2623
47. Huang L, Liu Y, Wang L. et al. Down-regulation of miR-301a suppresses pro-inflammatory cytokines in Toll-like receptor-triggered macrophages. Immunology. 2013;140(3):314-322
48. Qiu W, He JH, Zuo H. et al. Identification, characterization, and function analysis of the NF-kappaB repressing factor (NKRF) gene from Litopenaeus vannamei. Dev Comp Immunol. 2017;76:83-92
49. Lai J, Liu Y, Liu C. et al. Indirubin inhibits LPS-induced inflammation via TLR4 abrogation mediated by the NF-kB and MAPK signaling pathways. Inflammation. 2017;40(1):1-12
50. Wu J, Li L, Sun Y. et al. Altered molecular expression of the TLR4/NF-kappaB signaling pathway in mammary tissue of Chinese Holstein cattle with mastitis. Plos One. 2015;10(2):e118458
51. Fang L, Hou Y, An J. et al. Genome-wide transcriptional and post-transcriptional regulation of innate immune and defense responses of bovine mammary gland to Staphylococcus aureus. Front Cell Infect Microbiol. 2016;6:193
Author contact
![]() Corresponding author: Prof. Lin-Sen Zan, E-mail: zanlinsencom, Tel: +86-29-87091923.
Corresponding author: Prof. Lin-Sen Zan, E-mail: zanlinsencom, Tel: +86-29-87091923.