Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Structure of SMAD4
The role of SMAD4 in TGF-β...
Crosstalk with other pathways
TGF-β/SMAD4 in cancer
TGF-β/SMAD4 in...
TGF-β/SMAD4 in DNA damage...
TGF-β/SMAD4 and microRNA
TGF-β/SMAD4 and cancer...
Conclusion and perspective
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(2):111-123. doi:10.7150/ijbs.23230 This issue Cite
Review
The role of TGF-β/SMAD4 signaling in cancer
Ming Zhao1,2, Lopa Mishra3 ![]() , Chu-Xia Deng1
, Chu-Xia Deng1 ![]()
1. Faculty of Health Sciences, University of Macau, Macau SAR, China;
2. Department of Oncology, The Affiliated Hospital of Southwest Medical University, Luzhou, China.
3. Center for Translational Research, Department of Surgery and GW Cancer Center, George Washington University, Washington DC, USA.
Received 2017-10-8; Accepted 2017-11-19; Published 2018-1-12
Abstract

Transforming growth factor β (TGF-β) signaling pathway plays important roles in many biological processes, including cell growth, differentiation, apoptosis, migration, as well as cancer initiation and progression. SMAD4, which serves as the central mediator of TGF-β signaling, is specifically inactivated in over half of pancreatic duct adenocarcinoma, and varying degrees in many other types of cancers. In the past two decades, multiple studies have revealed that SMAD4 loss on its own does not initiate tumor formation, but can promote tumor progression initiated by other genes, such as KRAS activation in pancreatic duct adenocarcinoma and APC inactivation in colorectal cancer. In other cases, such as skin cancer, loss of SMAD4 plays an important initiating role by disrupting DNA damage response and repair mechanisms and enhance genomic instability, suggesting its distinct roles in different types of tumors. This review lists SMAD4 mutations in various types of cancer and summarizes recent advances on SMAD4 with focuses on the function, signaling pathway, and the possibility of SMAD4 as a prognostic indicator.
Keywords: TGF-β, SMAD4, tumorigenesis, prognosis, mouse model
Introduction
The transforming growth factor-beta (TGF-β) superfamily contains over forty members, including TGF-βs, Nodal, Activin, and bone morphogenetic proteins (BMPs) [1, 2]. TGF-β/SMAD4 signaling pathway controls the signal transduction from cell membrane to nucleus, and is responsible for a wide range of cellular processes, including proliferation, differentiation, apoptosis, migration, as well as cancer initiation and progression [3]. TGF-β protein has dual functions on tumorigenesis where it exhibits a suppressive role by inducing cell cycle arrest and apoptosis at early stages of tumor formation. Later on, with tumor progression, tumor cells gradually become insensitive to it, and secreted TGF-β protein enhances tumor immunosuppression and facilitates tumor angiogenesis, invasion and metastasis [1]. As the core mediator of canonical TGF-β signaling pathway, SMAD4 plays a pivotal role in the switch of TGF-β function on tumorigenesis. Although the TGF-β/SMAD4 pathway is relatively simple, it can be regulated extensively by numerous classical pathways, such as MAPK, PI3K/AKT and WNT/β-catenin, to form a complex network [1, 4]. Here, we will review TGF-β/SMAD4 function, its related signaling network, and the impact of SMAD4 mutation on cancer.
Structure of SMAD4
SMAD4 gene consists of 12 exons and 10 introns. At first, only 11 exons were identified, and another exon was discovered years later upstream of exon 1, and was therefore named exon 0 [5, 6]. The protein encoded by this gene is composed of 552 amino acids, with a molecular weight of 60 KD. The primary structure of SMAD4 protein consists of three major parts, including the N-terminal MH1 domain, the C-terminal MH2 domain and the linker region between them (Figure 1A) [7]. The MH1 domain is responsible for DNA binding by recognizing the SMAD-binding DNA element (SBE) [8], while the MH2 domain is important for its transcriptional activity [9-11], and can interact directly with the MH1 domain of other SMAD proteins [12]. The SMAD activation domain (SAD), locating at the C-terminal of linker region, is necessary for SMAD4 transcriptional activity [9].
The structure of SMAD4 and its role as the common mediator for signaling of TGF-β superfamily. (A) Diagrammatic representation of the structure of SMAD4. Abbreviations: NLS, nuclear localization signal; NES, nuclear export signal; SAD, SMAD activation domain; SBE, SMAD binding DNA element. (B) Diagram showing SMAD4 as the common mediator for TGF-β and BMP signaling. TF: transcriptional factor.
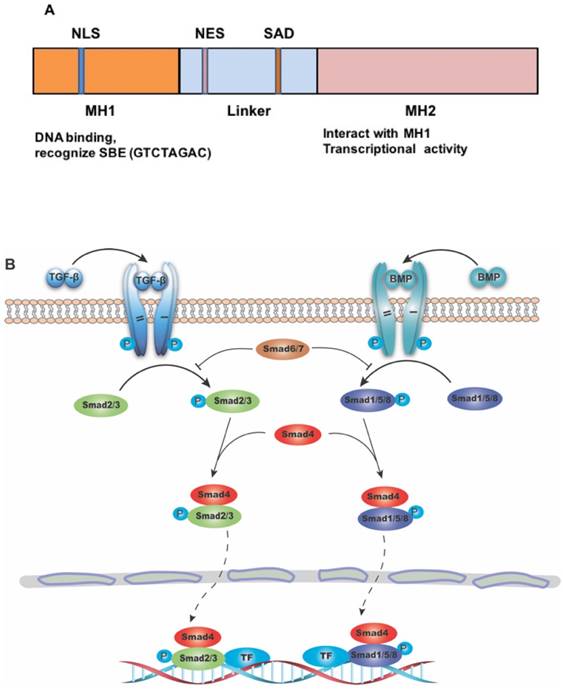
The role of SMAD4 in TGF-β signaling pathway
SMADs are a group of proteins transducing extracellular signals directly to the nucleus. There are eight different SMADs in mammalian cells, which can be divided to three categories: (1) the receptor-regulated SMAD (R-SMAD) including SMAD1, SMAD2, SMAD3, SMAD5, and SMAD8/9. While SMAD2/3 mainly mediate signaling from TGF-β subfamily members, SMAD1/5/8 transduct signaling from bone morphogenic protein (BMP) subfamily members; (2) the common-mediator SMAD (Co-SMAD) including only SMAD4, which is the central mediator of both TGF-β and BMP signaling pathway; and (3) the inhibitory SMAD (I-SMAD) including SMAD6 and SMAD7, which mainly function to inhibit receptor mediated R-SMAD phosphorylation, thus preventing the complex formation with Co-SMAD [13] (Figure 1B). Because many members in both TGF-β and BMP subfamilies play many important roles in various cancers, it is out of scope to describe specific functions of members in each of these subfamilies, therefore we will use TGF-β/SMAD4 to represent SMAD4 mediated signaling of the superfamily with an emphasis on signaling of TGF-β subfamily members.
Schematic diagram of the interaction between TGF-β/SMAD4 signaling pathway and MAPK (mitogen-activated protein kinase), PI3K/AKT (phosphatidylinositol-3 kinase/AKT) and WNT/β-catenin pathways. The canonical TGF-β/SMAD4 signal initiates from the TGF-β ligand activation and its binding to the type II and I receptors (T-βR II and T-βR I), which then phosphorylates SMAD2/3. The phosphorylated SMAD2/3 form a heterodimeric complex with SMAD4 and translocate to the nucleus and bind to SBE directly and regulate target genes transcription with the help of transcriptional factors. These target genes are mainly involved in growth arrest and apoptosis. (A) RAS/RAF/ERK1/2 axis regulates the TGF-β/SMAD4 pathway by (1) phosphorylating SMAD2 and SMAD3 to prevent its translocation into the nucleus; (2) mediating SMAD4 degradation; and (3) promoting AP-1 complex formation at the TGF-β1 promoter, thus boost the TGF-β1 transcription and secretion. As the substrate of JNK, c-Jun can directly bind to the transcriptional corepressor TG-interacting factor (TGIF) to inhibit SMAD2 dependent transcription. P38 can phosphorylate SMAD binding partners, such as activating transcription factor-2 (ATF-2), in nucleus and therefore facilitates TGF-β/SMAD4 induced genes transcription. (B) PI3K/AKT pathway suppresses the T-βR I mediated SMAD3 phosphorylation through its downstream molecule mTOR. AKT can directly phosphorylate FOXO and keep it in the cytoplasm to prevent its binding to the promoter of p27 and p21, thus blocks the TGF-β/SMAD4 mediated cytostatic signals. (C) β-catenin destruction complex consisting of AXIN, APC, GSK3β and CKIα causes SMAD7 ubiquitination and degradation. And in turn, SMAD7 can disassemble the complex by binding to AXIN, hence stablize β-catenin and promote its nucleus translocation.
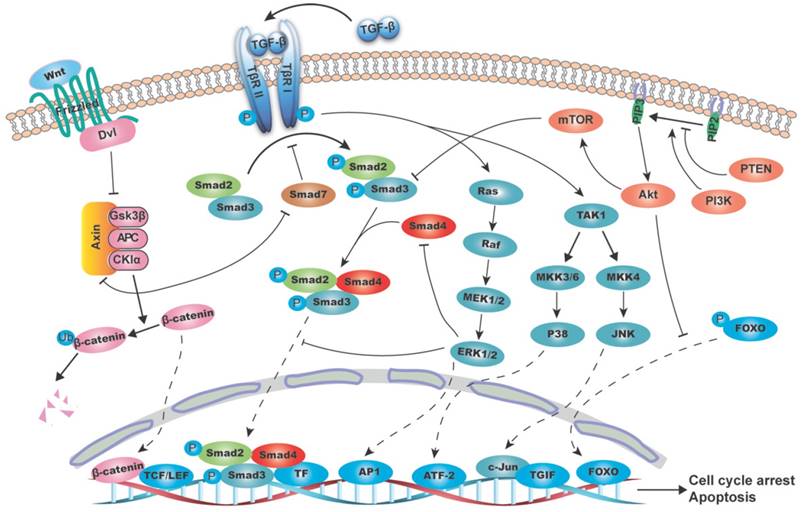
TGF-β subfamily signaling initiates from TGF-β ligand activation and its binding to the type II receptor (T-βR II). T-βR II then recruits and phosphorylates the type I receptor (T-βR I), which in turn phosphorylates its downstream target SMAD2/3. SMAD2/3 are phosphorylated at their conserved C-terminal SSXS motif (S is serine and X is any amino acid), leading to a conformational change in MH2 domain and its subsequent dissociation from the receptor [14]. Thereafter, the phosphorylated SMAD2/3 form a heterodimeric complex with SMAD4, and subsequently translocates to the nucleus [13, 15], where it binds to SBE directly and recurits co-transcriptional factors to transactivate or repress target genes [16].
The canonical TGF-β/SMAD4 signaling pathway plays a tumor suppressive role at early stages, mainly by inducing cell cycle arrest and apoptosis. SMAD4 functions at the G1/S checkpoint to make cells stay at the G1 phase, thus leading to cell cycle arrest [17]. Some CDK inhibitors, like p15, p21 and p27, are involved in this process [18]. For example, SMAD2/3/4 form a complex with transcriptional factor FoxO to promote transcription of p15 and p21. When ectopic overexpression of SMAD4 is induced in SMAD4-negative cells, it can bind to the promotor of p21 and enhance its transcription [19]. Inversely, TGF-β cannot induce the expression of p21 in pancreatic cancer cell lines, which lack SMAD4, with the result that cell growth is out of control [20]. Additionally, Lecanda et al. (2009) found that TGF-β/SMAD4 signal can enhance p27 expression, whereas inhibition of p27 by siRNA blocks the TGF-β/SMAD4 related cell growth arrest [21].
The nucleus localized SMAD2/3/4 complex induces apoptosis by promoting pro-apoptotic genes expression, such as TIEG (TGF-β-inducible early response gene), DAPK (the death-associated protein kinase), GADD45β and Bim. It was shown that expression of TIEG induced by TGF-β signaling causes apoptosis in pancreatic epithelial cells [22]. The rest three genes were reported to induce cytochrome C release from mitochondria and cause caspase activation [23-25].
Crosstalk with other pathways
In the past two decades, increasing number of classical pathways has been reported to interact with the canonical TGF-β/SMAD4 pathway. These crosslinks are along the whole TGF-β/SMAD4 signal transmision chain, from SMAD2/3 phosphorylation, SMAD complex formation, its translocation to nucleus and transcriptional activity. Here we will summarize the interaction between TGF-β/SMAD4 pathway with some well-established pathways including, but not limited to, MAPK (mitogen-activated protein kinase), PI3K/AKT (phosphatidylinositol-3 kinase/AKT) and WNT/β-catenin pathways.
MAPK pathway
MAPK is a superfamily that transduces extracellular signals to nucleus rapidly to regulate genes transcription, and affects a wide range of cellular processes including cell proliferation, differentiation and apoptosis. MAPK consists of three principal subfamilies: (1) the extracellular signal regulated kinase (ERK1/2); (2) c-Jun N-terminal kinase (JNK1/2/3) and (3) P38/MAPKs [26]. These subfamilies can phosphorylate a set of proteins to alter cell behaviors, and in verse, they can be activated by multiple extracellular molecules, such as TGF-β cytokines [27].
ERK pathway
ERK pathway is mainly responsible for cell growth and survival [28]. It can be activated by the RAS/RAF/MEK1/2 axis propagated signal that is initiated by membrane-associated receptor tyrosine kinases (RTKs) [29]. TGF-β peptide can mediate ERK activation rapidly in normal epithelial cells and in cancer cells [30]. It has been demonstrated that this regulation depends on the T-βR I recruited and activated ShcA/Grb2/Sos complex, which subsequently activate RAS/RAF/MEK1/2 signal [31, 32]. Inversely, ERK pathway inhibits the TGF-β/SMAD4 pathway by phosphorylating SMAD2 and SMAD3 at the serine or threonine residues in the linker region, thus preventing its translocation into the nucleus [33]. Subsequently, Matsuura et al. (2005) identified ERK phosphorylation sites in SMAD3 at Thr178, Ser203, and Ser207 in lung epithelial cells [34]. The TGF-β activated RAS/ERK cascade also promotes AP-1 complex formation at the TGF-β1 promoter, thus boosting the TGF-β1 transcription and secretion [35]. SMAD7, as the inhibitory SMAD, can be upregulated by ETS transcription factor ER81 through the HER2/Neu/ERK pathway in breast and ovarian cancer cell lines [36]. However, SMAD7 transcription is negatively regulated by ERK cascade in normal rat kidney (NRK) fibroblasts, suggesting the cell type dependence.
JNK pathway
JNK, as a stress-induced kinase, is responsive to various stress stimuli and is responsible for multiple cellular activities such as proliferation and apoptosis [37]. Principally, T-βR I can interact with TRAF6 and cause its autoubiquitylation, which subsequently activates TAK1 and JNK pathway [38]. Inversely, JNK is implicated to reduce the secretion of TGF-β1, as upon JNK inactivation, RAS gains ability to induce TGF-β1 autocrine [39]. JNK can also mediate phosphorylation of SMAD3 in the linker region and facilitate its activation by T-βR I, as well as nuclear translocation [40]. SMAD2 is another target of JNK, as it has been demonstrated that C-Jun, the substrate of JNK, can directly bind to the transcriptional corepressor TG-interacting factor (TGIF) to inhibit SMAD2 dependent transcription [41].
P38
Like JNK, p38 is also a stress-induced kinase that activated by various stress stimuli and accounts for diverse cellular processes. p38 can also be induced by TGF-β via T-βR I-TRAF6 interaction and TAK1 activation [38], and TGF-β induced P38 activation can strongly enhance SMAD4 sumoylation by PIAS family of E3 ligase, thus facilitating SMAD4 dependent transcription [42]. In human breast cancer cell line, SMAD2/3, phosphorylatd at Ser203/207 by Rho-dependent kinase (ROCK) and p38, synergize with TGF-β to induce cell growth arrest [43]. Apart from the crosstalk in cytoplasm, p38 also phosphorylates SMAD binding partners in nucleus and therefore affects TGF-β/SMAD4 induced genes transcription. For example, as the nuclear target of p38, activating transcription factor-2 (ATF-2) could be phosphorylated and bind to SMAD4 to activate TGF-β induced transcription [44].
PI3K/AKT pathway
PI3K/AKT pathway is an intracellular signaling pathway that is usually related to cell proliferation and apoptosis inhibition, and is important for the initiation and progression of many malignancies [45]. Upon activation of RTKs by a ligand, PI3K is activated and converts phosphatidylinositol-4,5-bisphosphate (PIP2) to phosphatidylinositol-3,4,5-triphosphate (PIP3), which then phosphorylates and activates AKT, and makes it localize on the plasma membrane [46]. The tumor suppressor PTEN can counteract PI3K and thereby alleviate AKT activation [47].
A multitude of studies have indicated that PI3K/AKT pathway is involved in TGF-β/SMAD4 pathway regulation. In mammary epithelial cell NmuMG, TGF-β1 administration induces AKT phosphorylation at Ser473 immediately, and transfection of dominant negative Rho inhibits this process [48]. Inactivating PI3K/AKT pathway by inhibitors affects TGF-β mediated EMT and cell migration [48]. This process is also affected by the TGF-β1 induced miR-487a overexpression, which directly binds to MAGI2 (Membrane-associated guanylate kinase inverted 2), and then stabilizes PTEN and suppresses PI3K/AKT pathway [49]. SMAD3 has been demonstrated to be the target of PI3K/AKT to affect TGF-β/SMAD pathway. Overexpression of AKT suppresses the T-βR II mediated SMAD3 phosphorylation, while the SMAD2 is not affected [50]. Treatment with rapamycin to activate the mammalian target of rapamycin (mTOR), which is the downstream of AKT, can suppress SMAD3 phosphorylation and activation [50]. Meanwhile, PI3K/AKT can also regulate SMAD3 function by phosphorylating some nuclear transcriptional factors, such as FOXO. AKT can directly phosphorylates FOXO and keep it in the cytoplasm [51]. Once dephosphorylated, FOXO enters the nucleus and binds to the promoter of p27 and p21 as a transcriptional factor, thus facilitating the TGF-β/SMAD4 mediated cytostatic signals [52].
WNT/β-catenin pathway
WNT is a type of secretory protein that initiates WNT/β-catenin signal from cell membrane to nucleus. WNT can bind to Frizzled family receptor (FZ) and pass the signal to Dishevelled (DVL) protein inside the cell and subsequently to the β-catenin destruction complex consisting of AXIN, APC, GSK3β and CKIα [53, 54]. The crosstalk between TGF-β/SMAD and WNT/β-catenin pathway mainly occurs in the nucleus, where SMADs form a complex with β-catenin and LEF protein to synergistically regulate a set of genes. For example, deletion of SMAD4 in the Müllerian duct led to reduced LEF1 signal, suggesting the alleviated WNT/β-catenin signaling pathway [55]. Meanwhile, these two pathways reciprocally regulate each other's ligand production and thus influence the pathway initiation [56, 57]. In the cytoplasm, SMAD7 is one of the points that link the two pathways. It is well known that SMAD7 functions to compete with SMAD2/3 for receptors, which leads to ubiquitination and degradation of receptors, and hence inhibiting SMAD2/3 phosphorylation and activation [3]. However, SMAD7 is found to disassemble the β-catenin destruction complex by binding to AXIN, thus stablizing β-catenin and promoting its nucleus translocation [56]. Meanwhile, the interaction also leads to SMAD7 ubiquitination and degradation by E3 ubiquitin ligase Arkadia [57]. Moreover, in the human pancreatic adenocarcinoma cells BxPC3, in which SMAD4 is homozigously deleted, chronic EGF stimulation induces β-catenin phosphorylation that can be antagonized by SMAD4 restoration, suggesting that SMAD4 homozigous deletion is critical to EGF induced WNT/β-catenin signaling pathway inhibition [58].
TGF-β/SMAD4 in cancer
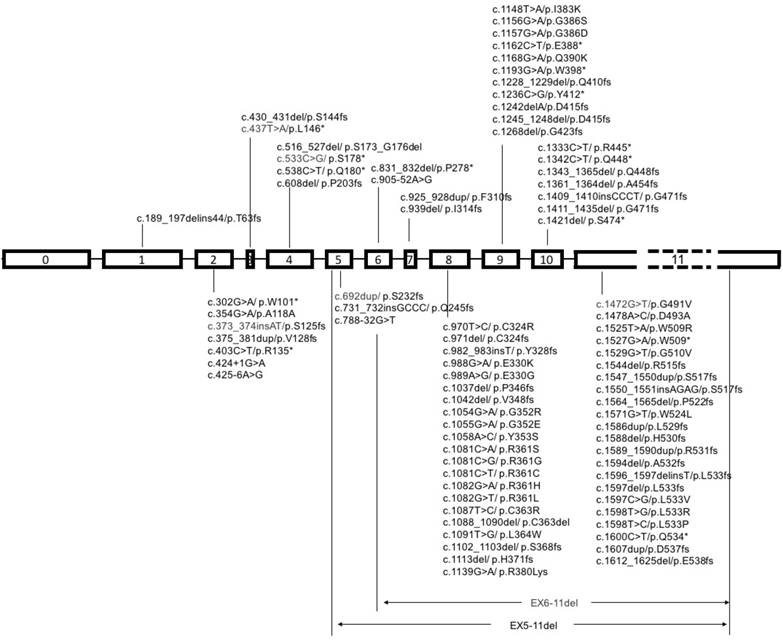
It is a common view that cancer is a notorious disease with an accumulation of critical genes alteration [59]. Heterozygous or homozygous deletion of SMAD4 was first discovered in pancreatic duct adenocarcinoma, and later detected in various types of cancers, such as colorectal cancer, cholangiocarcinoma, gastric cancer, prostate cancer and many others, though with lower frequencies to some extent (Table 1). Representative SMAD4 mutations in various cancers are summaried in Figure 3.
SMAD4 genetic alteration frequency in human cancers
| Cancer type | Total alteration frequency | Alteration type | Sample number | |||
|---|---|---|---|---|---|---|
| Complete deletion | Amplification | Mutation | Multiple alterations | |||
| Ampullary Carcinoma | 18.1% (29 cases) | 18.1% (29 cases) | 160 | |||
| Bladder Urothelial Carcinoma | 1.5% (4 cases) | 1.1% (1 cases) | 0.4% (1 cases) | 276 | ||
| Breast Cancer | 2.1% (98 cases) | 0.7% (33 cases) | 0.4% (17 cases) | 0.5% (22 cases) | 0.6% (26 cases) | 4728 |
| Cholangiocarcinoma | 4.2% (2 cases) | 4.2% (2 cases) | 48 | |||
| Colorectal Adenocarcinoma | 13.2% (167 cases) | 0.9% (11 cases) | 12.1% (152 cases) | 0.3% (4 cases) | 1261 | |
| Desmoplastic Melanoma | 10% (2 cases) | 10% (2 cases) | 20 | |||
| Esophageal Adenocarcinoma | 8.2% (12 cases) | 8.2% (12 cases) | 146 | |||
| Esophageal Carcinoma | 16.8% (31 cases) | 9.8% (18 cases) | 6% (11 cases) | 1.1% (2 cases) | 184 | |
| Esophagus-Stomach Cancers | 20.8% (55 cases) | 10.9% (29 cases) | 0.4% (1 cases) | 8.3% (22 cases) | 1.1% (3 cases) | 265 |
| Lymphoid Neoplasm Diffuse Large B-cell Lymphoma | 4.2% (2 cases) | 4.2% (2 cases) | 48 | |||
| Gallbladder Carcinoma | 3.1% (1 cases) | 3.1% (1 cases) | 32 | |||
| Glioblastoma | 0.7% (5 cases) | 0.1% (1 cases) | 0.6% (4 cases) | 708 | ||
| Advanced Germ Cell Tumors | 0.6% (1 cases) | 0.6% (1 cases) | 180 | |||
| Hepatocellular Carcinomas | 1.2% (10 cases) | 0.2% (2 cases) | 0.8% (7 cases) | 0.1% (1 cases) | 840 | |
| Head and Neck Squamous Cell Carcinoma | 6.2% (55 cases) | 3.8% (34 cases) | 2.4% (21 cases) | 889 | ||
| Recurrent and Metastatic Head & Neck Cancer | 1.5% (2 cases) | 1.5% (2 cases) | 132 | |||
| Kidney Cell Carcinoma | 0.8% (9 cases) | 0.3% (3 cases) | 0.1% (1 cases) | 0.5% (6 cases) | 1198 | |
| Kidney Chromophobe | 1.5% (1 cases) | 1.5% (1 cases) | 65 | |||
| Adrenocortical Carcinoma | 1.1% (1 cases) | 1.1% (1 cases) | 88 | |||
| Pan-Lung Cancer | 4.5% (52 cases) | 1.3% (15 cases) | 0.1% (1 cases) | 3.1% (35 cases) | 0.1% (1 cases) | 1144 |
| Lung Adenocarcinoma | 4.1% (33 cases) | 1% (8 cases) | 0.1% (1 cases) | 2.5% (20 cases) | 0.5% (4 cases) | 805 |
| Lung Squamous Cell Carcinoma | 2.6% (19 cases) | 0.7% (5 cases) | 0.3% (2 cases) | 1.8% (13 cases) | 733 | |
| Small Cell Lung Cancer | 3.4% (3 cases) | 3.4% (3 cases) | 139 | |||
| NSCLC young adult patients | 7.3% (3 cases) | 7.3% (3 cases) | 41 | |||
| Mesothelioma | 1.1% (1 cases) | 1.1% (1 cases) | 87 | |||
| Oral Squamous Cell Carcinoma | 5% (2 cases) | 5% (2 cases) | 40 | |||
| Ovarian Serous Cystadenocarcinoma | 1.3% (4 cases) | 0.3% (1 cases) | 0.9% (3 cases) | 316 | ||
| 4.8% (15 cases) | 3.9% (12 cases) | 1% (3 cases) | 311 | |||
| Pheochromocytoma and Paraganglioma | 0.6% (1 cases) | 0.6% (1 cases) | 162 | |||
| Pancreatic cancer | 27.8% (206 cases) | 6.2% (46 cases) | 20.4% (151 cases) | 1.2% (9 cases) | 740 | |
| Acinar Cell Carcinoma of the Pancreas | 26.1% (6 cases) | 26.1% (6 cases) | 23 | |||
| Pancreatic Neuroendocrine Tumors | 1% (1 cases) | 1% (1 cases) | 98 | |||
| Pediatric Ewing Sarcoma | 1% (1 cases) | 1% (1 cases) | 105 | |||
| Prostate Cancer | 1% (5 cases) | 0.2% (1 cases) | 0.8% (4 cases) | 501 | ||
| Prostate Adenocarcinoma | 2.1% (36 cases) | 0.9% (15 cases) | 0.5% (8 cases) | 0.8% (13 cases) | 1678 | |
| Prostate Adenocarcinoma, Metastatic | 4.9% (3 cases) | 1.6% (1 cases) | 1.6% (1 cases) | 1.6% (1 cases) | 61 | |
| Neuroendocrine Prostate Cancer | 10.3% (11 cases) | 10.3% (11 cases) | 107 | |||
| Skin Cutaneous Melanoma | 2.5% (3 cases) | 2.5% (3 cases) | 121 | |||
| Cutaneous squamous cell carcinoma | 10.3% (3 cases) | 10.3% (3 cases) | 29 | |||
| Sarcoma | 3.4% (11 cases) | 0.8% (2 cases) | 3.4% (9 cases) | 450 | ||
| Stomach Adenocarcinoma | 10.9% (99 cases) | 3.7% (34 cases) | 0.2% (2 cases) | 6% (55 cases) | 0.9% (8 cases) | 910 |
| Testicular Germ Cell Cancer | 1.3% (2 cases) | 0.7% (1 cases) | 0.7% (1 cases) | 149 | ||
| Uterine Carcinoma | 3.7% (28 cases) | 1.1% (8 cases) | 0.3% (2 cases) | 2.4% (18 cases) | 751 | |
Modified from http://www.cbioportal.org/index.do.
Schematic diagram of mutational pattern in exons of SMAD4. Note: SMAD4 gene contains 12 exons, 11 of which were identified at first, and another exon was discovered later at the upstream of exon 1, so it was called exon 0. The exon 11 is about fifty times longer than exon 10, so it is labeled with dash line.
Pancreatic cancer
Pancreatic cancer is a lethal disease with a 5-year survival rate of about 8%, and advance is slow for treatment [60]. So many studies have demonstrated that SMAD4 alteration is closely related with pancreatic cancer since its discovery in 1996. Loss of heterozygosity occurs in almost 60% human pancreatic cancers, and about 50% shows homozygous deletion or intragenic inactivating mutations [5, 61]. A further study revealed that SMAD4 mutation was associated with pancreatic pathologic stages. The inactivation of SMAD4 was 31% (9/29) in the high-grade stage neoplasms (Pan IN-3), while none was found in the rest 159 low-grade lesions (Pan IN-1 and 2) [62]. Genetically engineered mouse models have been used to investigate the mechanism (Table 3). Although knockout of SMAD4 by pancreatic-specific PDX1-Cre or P48-Cre did not initiate pancreatic cancer in mice [63, 64], they markedly facilitated tumor progression initiated by KRASG12D activation [65] or PTEN inactivation [66]. These reports favor the view that SMAD4 has a tumor suppressive function at the progressive stage.
Examples of SMAD4-deficient mouse models.
| Tissue type | Cre | SMAD4 status | Combined mutation | Phenotypes | Reference |
|---|---|---|---|---|---|
| Germline knockout | |||||
| Homozygous Deletion | _ | SMAD4-/- | _ | Embryonically lethal | [77] |
| Heterozygous Deletion | _ | SMAD4+/- | _ | Gastric and duodenal Polyps | [77, 78] |
| _ | SMAD4+/- | APC+/- | Intestinal Tumorigenesis | [79] | |
| Tissue-specific knockout | |||||
| Pancreatic progenitor cells | PDX1-Cre | SMAD4co/co | _ | No obvious phenotype in pancreas | [65] |
| PDX1-Cre | SMAD4co/co | KRASG12D | IPMN | [65, 121] | |
| PDX1-Cre | SMAD4co/co | PTENco/co | Accelerated tumor formation | [66] | |
| Pancreatic exocrine progenitor | P48-Cre | SMAD4co/co | KRASG12D | Enhanced IPMN development | [65] |
| P48-Cre | SMAD4+/co | KRASG12D | Mucinous cystic neoplasm | [122] | |
| Hepatocytes and bile duct epithelial cells | ALB-Cre | SMAD4co/co | _ | Increased iron accumulation in the liver | [123] |
| ALB-Cre | SMAD4co/co | PTENco/co | Intrahepatic cholangiocellular carcinoma | [67] | |
| Prostate epithelial cells | PB-Cre4 | SMAD4co/co | _ | No prostate neoplasia | [83] |
| PB-Cre4 | SMAD4co/co | PTENco/co | Accelerated progression and metastasis to lymph nodes and lung | [83] | |
| Epidermis and hair follicles | MMTV-Cre | SMAD4co/co | _ | Hair follicle defects and squamous cell carcinoma formation | [86, 87] |
| Mammary epithelial cells | MMTV-Cre | SMAD4co/co | _ | Breast squamous cell carcinoma | [124] |
| Esophageal and forestomach epithelia | K5-Cre | SMAD4co/co | _ | 19% of mice developed forestomach squamous cell carcinoma by 4 months | [88] |
| K5-Cre | SMAD4co/co | PTENco/co | All mice developed forestomach squamous cell carcinoma by 2 months | [88] | |
| Gastric LGR5+ stem cells | LGR5-CreERT2 | SMAD4co/co | PTENco/co | Invasive intestinal-type gastric cancer by 3 months | [89] |
Cholangiocarcinoma
Cholangiocarcinoma is a type of liver cancer originating from the bile duct. Although its prognosis is poor compared with that of hepatocellular carcinoma, few studies exist to address the molecular mechanism. SMAD4 has been regarded as a tumor suppressor in cholangiocarcinoma [67]. Loss of SMAD4 was noted in 19 of 42 (45.2%) cases with intrahepatic cholangiocarcinoma (ICC), and had a positive correlation with clinical stages (25% negative in stage I and II vs. 58% in stage III and IV) [68]. Additionally, loss of SMAD4 was inversely related with its prognosis [69]. Hepatolithiasis is commonly regarded as a risk factor for cholangiocarcinoma. One group examined SMAD4 deletion frequency in the normal intrahepatic bile duct (IHD), stone-containing IHD and ICC, with the result of 0% (0/24), 4.4% (2/46) and 33.3% (3/9) respectively, suggesting that loss of SMAD4 promotes transition from IHD to ICC [70]. However, tissue-specific deletion of SMAD4 along in mouse hepatocytes and bile duct epithelial cells did not cause discernable tumor formation. But homozygous deletion of both SMAD4 and PTEN in mouse liver induced intrahepatic cholangiocarcinoma [67]. Underlining the phenotype, PTEN and SMAD4 regulate each other through a negative feedback loop, therefore they can compensate each other's deletion. So far, little is known about the mechanism of SMAD4 in cholangiocarcinoma and more research studies examining this are required.
Colorectal cancer
The role of SMAD4 in colorectal cancer is similar to that in pancreatic cancer and cholangiocarcinoma. Accumulation of evidence has confirmed that the loss of SMAD4 in colorectal cancer occurs at a frequency of about 30% [71]. Royce et al examined SMAD4 expression by immunohistochemistry and RT-PCR in 109 patients with early-onset colorectal cancer, and detected 27 cases (accounting for 25%) had SMAD4 loss [72]. A study on colon cancer cell lines HCT116 and SW620 demonstrated TGF-β administration could activate ERK and P38 pathways and induce VEGF overexpression when SMAD4 was knocked down [73]. In addition, the SMAD4 loss was responsible for resistant to 5-fluoruracil mediated apoptosis in these two cell lines [73]. The rate of SMAD4 loss has a positive correlation with colorectal cancer progression, convincing that inactivation of SMAD4 is a late event in colorectal carcinogenesis [74, 75]. Although germline loss of SMAD4 in human [76] or heterozygous deletion in mice [77, 78] caused juvenile polyposis (JP), the risk of carcinoma is low and associated with a long latency, suggesting that SMAD4 loss alone may be insufficient for tumor initiation. It has been reported that heterozygous deletion of SMAD4 promotes progression of colorectal cancer initiated by germline mutation of APC [79]. SMAD4 status is also correlated with tumor microenvironment. SMAD4-deficient colorectal tumor cells secreted more CCL9 and CCL15, chemokines that recruit CCR1+ myeloid cells through CCL9-CCR1 and CCL15-CCR1 axis, leading to metastasis [80, 81]. Moreover, deletion of SMAD4 in human colorectal tumor cells reduced the number of S100A8-positive monocytes and attenuated the sensitivity of tumor cells to chemokine S100A8, suggesting the effect of SMAD4 on monocyte infiltrate [82].
Other cancers
SMAD4 loss occurs prevalently in above mentioned types of cancers and many other cancers with lower frequencies (Table 1). Similar to pancreatic and intrahepatic cholangiocarcinoma, prostate specific deletion of SMAD4 did not initiate cancer formation, but synergistically promotes tumorigenesis together with PTEN deletion [83]. SMAD4 mutations are rare in human prostate cancer, but SMAD4 promotor methylation is commonly detected, which may reduce SMAD4 expression [84]. Of note, SMAD4 downregulation was found at an early stage in human HNSCC, and deletion of SMAD4 alone in murine head and neck epithelia results in spontaneous HNSCC that mimics human HNSCC [85]. Keratinocyte-specific deletion of SMAD4 results in predominantly well-differentiated skin tumors, revealing that SMAD4 is a tumor initiator [86]. A very similar phenotype is obtained in the mouse mammary tumor virus (MMTV)-Cre mediated SMAD4 deletion [87]. In these types of cancers, SMAD4 loss occurs prior to tumor formation, and disrupts DNA repair mechanisms to enhance genomic instability and inflammation [85]. It was also reported that mice carrying K5-Cre mediated deletion of SMAD4 in keratinocytes developed forestomach squamous cell carcinoma (SCC) at 8 months, while deletion of PTEN only suffered dysplasia. However, concurrent deletion of SMAD4 and PTEN led to invasive SCC in 2 months [88]. Moreover, deletion of SMAD4 and PTEN by LGR5-Cre drived gastric LGR5+ stem cells transformation, thus causing invasive intestinal-type gastric cancer [89]. In the gastric cancer cell lines MGC-803 and BGC-823, SMAD4 serves as the direct target of miR-324-3p, which promotes gastric cancer progression. This miR-324-3p induced tumor growth can be rescued by restoration of SMAD4 [90]. These data prompt us to conclude that the role of SMAD4 on cancer initiation may be cancer type-dependent (Table 3).
TGF-β/SMAD4 in epithelial-mesenchymal transition
Epithelial-mesenchymal transition (EMT) is a well-coordinated process that epithelial cells lose their cell junctions and polarity, and are transformed to mesenchymal cells with the migratory and invasive abilities [91]. Molecularly, this process is accompanied with the cell junction switch from E-Cadherin to N-Cadherin [92, 93]. TGF-β signaling is known as an important molecule to induce EMT in a SMAD4-dependent manner through the induction of translocation of SMAD2/3/4 complex to the nucleus, leading to the expression of mesenchymal markers SNAIL, SLUG, TWIST and ZEB [94-96]. When knocking down SMAD4 in mammary gland epithelial cell NMuMG, the TGF-β induced EMT was potently blocked with the failure of E-cadherin reduction and N-cadherin induction, as well as morphologic transformation [97]. Similarly, SMAD4 knockdown in liver significantly reduced liver tumorigenesis through upregulating SNAIL, suggesting that TGF-β signal promotes EMT in a SMAD4-dependent manner [98]. Moreover, TGF-β induced EMT is also inhibited by the nuclear factor YY1, which can bind to SMAD complex and suppress its transcriptional activity [99, 100]. These reports reinforce the role of SMAD4 mediated TGF-β signal in the EMT process. A recent study revealed that TGF-β/SMAD4 signal also inhibits the transcription of KLF5 through SNAIL. With the low KLF5 level, SOX4 is switched from tumor promotor to suppressor. Therefore, TGF-β/SMAD4 actually induced lethal EMT in PDAC [101].
TGF-β/SMAD4 in DNA damage response and DNA damage repair
It has been shown that mice carrying specific disruption of SMAD4 in head and neck epithelia developed head and neck cancer that is associated with increased genomic instability and downregulated expression and function of genes encoding proteins in the Fanconi anemia/BRCA (FANC/BRCA) DNA repair pathway [85]. It was also shown mice carrying K14-Cre mediated disruption of SMAD4 (K14.SMAD4-/-) in keratinocytes suffered increased DNA damage and increased susceptibility to UV induced carcinogenesis [102]. The skin of the K14.SMAD4-/- mice had significantly reduced expression of excision repair cross-complementation group1 (ERCC1) gene that is correlated with reduced ERCC1-mediated DNA repair. Their further study demonstrated that SMAD4 deficiency impaired SNAIL binding to the ERCC1 regulatory elements, leading to the reduced ERCC1 expression. In breast cancer cells and primary breast tumor specimens, it was found that TGF-β downregulates the expression of several genes that are essential for DNA damage response (DDR), including ataxia telangiectasia-mutated (ATM), mutS homolog 2 (MSH2), and BRCA1 [103, 104]. Through downregulating these genes, TGF-β attenuates DDR in these breast cancer cells and enables them to escape the DNA damage checkpoint, allowing their proliferation in the presence of unrepaired DNA damage. Thus, TGF-β/SMAD4 signaling plays an important role in maintaining DDR and DNA damage repair through regulating transcription and activity of some key genes involved in these processes. This may be one of the main mechanisms through which TGF-β/SMAD4 signaling suppresses tumor formation and growth.
TGF-β/SMAD4 and microRNA
Recent studies revealed that another important function of TGF-β/SMAD4 signaling is in regulation of microRNA. It has been shown that TGF-β1 was able to induce miR-574-3p transcription in gastric cancer cell AGS to inhibit cell proliferation, which was dependent on the binding of SMAD4 to the promotor of miR-574-3p [105]. Moreover, TGF-β/SMAD4 signaling could induce a set of microRNAs in human hepatocellular carcinoma cell Huh-7. The transcription of miR-23a∼27a∼24 was upregulated quickly upon TGF-β treatment, and attenuated by SMAD4 deletion [106]. On the other hand, SMAD4 is also subject to regulation by microRNA. In colon cancer cells and tumor specimens, the upregulated miR-19b-3p directly targets SMAD4 to attenuate proliferation and sensitivity to oxaliplatin-based chemotherapy [107]. Besides miR-19b-3p, SMAD4 is also the target of some other microRNAs. For example, miR-27a, which is induced in human lymphatic endothelial cells (HLECs) by co-culturing with colon tumor cells, directly targetes SMAD4 and promotes lymphangiogenesis [108].
TGF-β/SMAD4 and cancer prognosis
Although the molecular mechanism of SMAD4 in carcinogenesis is still not clear, the alteration in types of cancers are studied exclusively, especially in pancreatic adenocarcinoma (Table 2). Wilentz et al (2000) studied 41 xenografted primary periampullary adenocarcinomas for the correlation between SMAD4 expression and prognosis [109]. Immunohistochemical staining of SMAD4 showed 22 cases were negative, accounting for 56% of the patients. The survival analysis revealed that the patients with positive SMAD4 signal lived longer than those with negative signal, with the median survival of 16.5 months versus 9 months. Recently, Singh's group focused on an Indian population with pancreatic duct adenocarcinoma for SMAD4 alteration and effect of its loss on patient survival [110]. The SMAD4 gene was altered by homozygous deletion in 5 patients and intragenic mutation in the MH2 domain in 3 patients, together accounting for 32% of the patients. The follow-up investigation provided a median survival of 5 months in the SMAD4 alteration group while 10 months in the SMAD4 wildtype group. Many other reports, as summarized in table 2, show the similar outcome in pancreatic duct adenocarcinoma.
Examples of the studies on SMAD4 and prognosis in pancreatic duct adenocarcinoma.
| Number of samples | Sex (female/male) | Age | Stage (I-II/III-IV) | Margin (negative/ positive) | N (N0/ N1) | Differentiation (well+moderate/poor) | SMAD4 (negative/positive) | Median survival (months, SMAD4 negative/positive) | P value | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 45 | 18/27 | 61 | NS | 25/20 | 24/21 | 33/12 | 35/10 | 6.4/13.6 | 0.0257 | [109] |
| 25 | 12/13 | 54.6 | 10/15 | 22/3 | 19/6 | 25/1 | 8/17 | 5/10 | 0.001 | [118] |
| 249 | 110/139 | 65.4 ± 10.5 | 59/190 | 168/80 | NS | NS | 138/111 | 14.7/19.2 | 0.03 | [110] |
| 34 | 12/22 | 55.2 ± 11.3 | 18/16 | NS | 20/14 | NS | 8/26 | 6.9/10.6 | 0.879 | [119] |
| 89 | 46/43 | 65.3 ±10.5 | NS | 58/31 | 18/71 | 55/34 | 40/49 | 11.5/14.2 | 0.006 | [120] |
SMAD4 expression level has a positive correlation with survival in colon cancer, and the loss of SMAD4 leads to poor prognosis [111]. Additionally, SMAD4 is also correlated with tumor metastasis [112]. A study involving 202 colorectal cancer patients revealed that cases with normal SMAD4 expression benefit from 5-fluorouracil based chemotherapy at a 3-fold higher improvement than those with SMAD4 loss [113]. These studies suggest that SMAD4 loss may predict poor outcome for 5-fluorouracil therapy in patients with colorectal cancer.
Recent studies indicated that SMAD4 could be a prognostic factor together with other genes. Through evaluating the expression level of SMAD4 and PTEN in patients bearing colorectal cdenocarcinoma, there was a significant difference in the overall survival, showing patients with loss of both SMAD4 and PTEN had worse outcome than cases with SMAD4 or PTEN loss alone [114]. Moreover, another study showed the patients with wildtype SMAD4 and KRAS had twice longer survival than those bearing SMAD4 loss and KRAS actication (83.8 vs. 36.7 months) [115].
Conclusion and perspective
Numerous studies during passed two decades provide strong evidence that SMAD4 is a tumor suppressor, whose mutations are found in at least 26 types of cancer, with higher frequencies in GI tract cancers, such as Esophagus, Stomach, pancrease and Colorectal cancers (Table 1). Additionally, loss of SMAD4 in tumors affects cancer progression and therapy, such as reduced response to adjuvant chemotherapy [116]. SMAD4 plays its function through acting as the central mediator for canonical TGF-β signaling pathway, and affects tumorigenesis through numerous mechanisms, such as inducing cell cycle arrest, apoptosis, EMT, and others. Thus, SMAD4 not only functions as a tumor suppressor, but also may serve as a prognostic indicator.
Even though the SMAD4 mediated TGF-β signaling pathway has been elucidated well, how SMAD4 alteration causes tumor formation and progression remains unclear due to the crosstalk with other pathways. For example, although SMAD4 is highly deleted in human pancreatic cancer, the mouse with SMAD4 deficiency in pancreas dose not develop a tumor, suggesting additional mutations may be required. Owing to the technology limitation, only a few genes, such as APC in colorectal cancer and PTEN in pancreatic cancer and cholangiocarcinoma, are currently identified. Fortunately, the recent well-developed CRISPR knockout library provides the opportunity to screen these SMAD4 synergistic genes at genome-scale level. Therefore, we can anticipate the discovery of more such genes. Moreover, the CRISPR knockout library can also be used to screen drug resistant or sensitive genes on the genetic background of SMAD4 deficiency, as the SMAD4 loss is reported to predict poor outcome for chemotherapy, such as 5-fluorouracil therapy in patients with colorectal cancer [113].
Although limited knowledge is availabale regarding the mechanism, some molecules associated to SMAD4, including UA62001 and UA62784, have been developed as candidates in cancer treatment [71, 117]. With the discovery of new components that could facilitate SMAD4 associated tumor progression and resistant genes in SMAD4 associated insensitivity to drugs, more and more SMAD4 associated molecules can be the potent target for cancer treatment.
Acknowledgements
We thank members of the Deng laboratory for critical reading and discussion. This work is supported by the Chair Professor Grant (CPG2017-00026-FHS), Startup Research Grant (SRG2015-00045-FHS), MYRG2016-00132-FHS and MYRG2016-00139 of University of Macau and FDCT grants (065/2015/A2 and 094/2015/A3) to Chu-Xia Deng.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Connolly EC, Freimuth J, Akhurst RJ. Complexities of TGF-beta targeted cancer therapy. Int J Biol Sci. 2012;8:964-78
2. Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616-30
3. Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685-700
4. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128-39
5. Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E. et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350-3
6. Mamot C, Mild G, Reuter J, Laffer U, Metzger U, Terracciano L. et al. Infrequent mutation of the tumour-suppressor gene Smad4 in early-stage colorectal cancer. Br J Cancer. 2003;88:420-3
7. Shi Y, Hata A, Lo RS, Massague J, Pavletich NP. A structural basis for mutational inactivation of the tumour suppressor Smad4. Nature. 1997;388:87-93
8. Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B. et al. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611-7
9. de Caestecker MP, Hemmati P, Larisch-Bloch S, Ajmera R, Roberts AB, Lechleider RJ. Characterization of functional domains within Smad4/DPC4. The Journal of biological chemistry. 1997;272:13690-6
10. de Caestecker MP, Yahata T, Wang D, Parks WT, Huang S, Hill CS. et al. The Smad4 activation domain (SAD) is a proline-rich, p300-dependent transcriptional activation domain. The Journal of biological chemistry. 2000;275:2115-22
11. Zhang Y, Musci T, Derynck R. The tumor suppressor Smad4/DPC 4 as a central mediator of Smad function. Curr Biol. 1997;7:270-6
12. Shi Y, Wang YF, Jayaraman L, Yang H, Massague J, Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-beta signaling. Cell. 1998;94:585-94
13. Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646-7
14. Souchelnytskyi S, Ronnstrand L, Heldin CH, ten Dijke P. Phosphorylation of Smad signaling proteins by receptor serine/threonine kinases. Methods Mol Biol. 2001;124:107-20
15. Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745-54
16. Massague J, Gomis RR. The logic of TGFbeta signaling. FEBS Lett. 2006;580:2811-20
17. Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295-309
18. Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nature Genetics. 2001;29:117-29
19. Hunt KK, Fleming JB, Abramian A, Zhang L, Evans DB, Chiao PJ. Overexpression of the tumor suppressor gene Smad4/DPC4 induces p21(waf1) expression and growth inhibition in human carcinoma cells. Cancer research. 1998;58:5656-61
20. Grau AM, Zhang L, Wang WX, Ruan SB, Evans DB, Abbruzzese JL. et al. Induction of p21(waf1) expression and growth inhibition by transforming growth factor beta involve the tumor suppressor gene DPC4 in human pancreatic adenocarcinoma cells. Cancer research. 1997;57:3929-34
21. Lecanda J, Ganapathy V, D'Aquino-Ardalan C, Evans B, Cadacio C, Ayala A. et al. TGF beta prevents proteasomal degradation of the cyclin-dependent kinase inhibitor p27(kip1) for cell cycle arrest. Cell Cycle. 2009;8:742-56
22. Tachibana I, Imoto M, Adjei PN, Gores GJ, Subramaniam M, Spelsberg TC. et al. Overexpression of the TGFbeta-regulated zinc finger encoding gene, TIEG, induces apoptosis in pancreatic epithelial cells. J Clin Invest. 1997;99:2365-74
23. Jang CW, Chen CH, Chen CC, Chen JY, Su YH, Chen RH. TGF-beta induces apoptosis through Smad-mediated expression of DAP-kinase. Nat Cell Biol. 2002;4:51-8
24. Yoo J, Ghiassi M, Jirmanova L, Balliet AG, Hoffman B, Fornace AJ Jr. et al. Transforming growth factor-beta-induced apoptosis is mediated by Smad-dependent expression of GADD45b through p38 activation. The Journal of biological chemistry. 2003;278:43001-7
25. Ohgushi M, Kuroki S, Fukamachi H, O'Reilly LA, Kuida K, Strasser A. et al. Transforming growth factor beta-dependent sequential activation of Smad, Bim, and caspase-9 mediates physiological apoptosis in gastric epithelial cells. Mol Cell Biol. 2005;25:10017-28
26. Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37-40
27. Javelaud D, Mauviel A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-beta: implications for carcinogenesis. Oncogene. 2005;24:5742-50
28. Lu Z, Xu S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life. 2006;58:621-31
29. Avruch J, Khokhlatchev A, Kyriakis JM, Luo Z, Tzivion G, Vavvas D. et al. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog Horm Res. 2001;56:127-55
30. Frey RS, Mulder KM. TGFbeta regulation of mitogen-activated protein kinases in human breast cancer cells. Cancer Lett. 1997;117:41-50
31. Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J. et al. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007;26:3957-67
32. Rojas A, Padidam M, Cress D, Grady WM. TGF-beta receptor levels regulate the specificity of signaling pathway activation and biological effects of TGF-beta. Biochim Biophys Acta. 2009;1793:1165-73
33. Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes & development. 1999;13:804-16
34. Matsuura I, Wang G, He D, Liu F. Identification and characterization of ERK MAP kinase phosphorylation sites in Smad3. Biochemistry. 2005;44:12546-53
35. Yue J, Mulder KM. Requirement of Ras/MAPK pathway activation by transforming growth factor beta for transforming growth factor beta 1 production in a Smad-dependent pathway. The Journal of biological chemistry. 2000;275:30765-73
36. Dowdy SC, Mariani A, Janknecht R. HER2/Neu- and TAK1-mediated up-regulation of the transforming growth factor beta inhibitor Smad7 via the ETS protein ER81. The Journal of biological chemistry. 2003;278:44377-84
37. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239-52
38. Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N. et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol. 2008;10:1199-207
39. Ventura JJ, Kennedy NJ, Flavell RA, Davis RJ. JNK regulates autocrine expression of TGF-beta1. Mol Cell. 2004;15:269-78
40. Matsuzaki K, Kitano C, Murata M, Sekimoto G, Yoshida K, Uemura Y. et al. Smad2 and Smad3 phosphorylated at both linker and COOH-terminal regions transmit malignant TGF-beta signal in later stages of human colorectal cancer. Cancer research. 2009;69:5321-30
41. Pessah M, Prunier C, Marais J, Ferrand N, Mazars A, Lallemand F. et al. c-Jun interacts with the corepressor TG-interacting factor (TGIF) to suppress Smad2 transcriptional activity. Proc Natl Acad Sci U S A. 2001;98:6198-203
42. Ohshima T, Shimotohno K. Transforming growth factor-beta-mediated signaling via the p38 MAP kinase pathway activates Smad-dependent transcription through SUMO-1 modification of Smad4. The Journal of biological chemistry. 2003;278:50833-42
43. Kamaraju AK, Roberts AB. Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-beta-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. The Journal of biological chemistry. 2005;280:1024-36
44. Hanafusa H, Ninomiya-Tsuji J, Masuyama N, Nishita M, Fujisawa J, Shibuya H. et al. Involvement of the p38 mitogen-activated protein kinase pathway in transforming growth factor-beta-induced gene expression. The Journal of biological chemistry. 1999;274:27161-7
45. Pene F, Claessens YE, Muller O, Viguie F, Mayeux P, Dreyfus F. et al. Role of the phosphatidylinositol 3-kinase/Akt and mTOR/P70S6-kinase pathways in the proliferation and apoptosis in multiple myeloma. Oncogene. 2002;21:6587-97
46. King D, Yeomanson D, Bryant HE. PI3King the lock: targeting the PI3K/Akt/mTOR pathway as a novel therapeutic strategy in neuroblastoma. J Pediatr Hematol Oncol. 2015;37:245-51
47. Brader S, Eccles SA. Phosphoinositide 3-kinase signalling pathways in tumor progression, invasion and angiogenesis. Tumori. 2004;90:2-8
48. Bakin AV, Tomlinson AK, Bhowmick NA, Moses HL, Arteaga CL. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. The Journal of biological chemistry. 2000;275:36803-10
49. Ma M, He M, Jiang Q, Yan Y, Guan S, Zhang J. et al. MiR-487a Promotes TGF-beta1-induced EMT, the Migration and Invasion of Breast Cancer Cells by Directly Targeting MAGI2. Int J Biol Sci. 2016;12:397-408
50. Song K, Wang H, Krebs TL, Danielpour D. Novel roles of Akt and mTOR in suppressing TGF-beta/ALK5-mediated Smad3 activation. EMBO J. 2006;25:58-69
51. Rafalski VA, Brunet A. Energy metabolism in adult neural stem cell fate. Prog Neurobiol. 2011;93:182-203
52. Atfi A, Abecassis L, Bourgeade MF. Bcr-Abl activates the AKT/Fox O3 signalling pathway to restrict transforming growth factor-beta-mediated cytostatic signals. EMBO Rep. 2005;6:985-91
53. Minde DP, Anvarian Z, Rudiger SG, Maurice MM. Messing up disorder: how do missense mutations in the tumor suppressor protein APC lead to cancer? Mol Cancer. 2011;10:101
54. Minde DP, Radli M, Forneris F, Maurice MM, Rudiger SG. Large extent of disorder in Adenomatous Polyposis Coli offers a strategy to guard Wnt signalling against point mutations. PLoS One. 2013;8(e):77257
55. Petit FG, Deng C, Jamin SP. Partial Mullerian Duct Retention in Smad4 Conditional Mutant Male Mice. Int J Biol Sci. 2016;12:667-76
56. Tang Y, Liu Z, Zhao L, Clemens TL, Cao X. Smad7 stabilizes beta-catenin binding to E-cadherin complex and promotes cell-cell adhesion. The Journal of biological chemistry. 2008;283:23956-63
57. Liu W, Rui H, Wang J, Lin S, He Y, Chen M. et al. Axin is a scaffold protein in TGF-beta signaling that promotes degradation of Smad7 by Arkadia. EMBO J. 2006;25:1646-58
58. Moz S, Basso D, Bozzato D, Galozzi P, Navaglia F, Negm OH. et al. SMAD4 loss enables EGF, TGFbeta1 and S100A8/A9 induced activation of critical pathways to invasion in human pancreatic adenocarcinoma cells. Oncotarget. 2016;7:69927-44
59. Croce CM. Oncogenes and cancer. N Engl J Med. 2008;358:502-11
60. Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F. et al. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115-32
61. Hahn SA, Seymour AB, Hoque AT, Schutte M, da Costa LT, Redston MS. et al. Allelotype of pancreatic adenocarcinoma using xenograft enrichment. Cancer research. 1995;55:4670-5
62. Wilentz RE, Iacobuzio-Donahue CA, Argani P, McCarthy DM, Parsons JL, Yeo CJ. et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer research. 2000;60:2002-6
63. Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447-57
64. Kawaguchi Y, Cooper B, Gannon M, Ray M, MacDonald RJ, Wright CV. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32:128-34
65. Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P. et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes & development. 2006;20:3130-46
66. Xu X, Ehdaie B, Ohara N, Yoshino T, Deng CX. Synergistic action of Smad4 and Pten in suppressing pancreatic ductal adenocarcinoma formation in mice. Oncogene. 2010;29:674-86
67. Xu X, Kobayashi S, Qiao W, Li C, Xiao C, Radaeva S. et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J Clin Invest. 2006;116:1843-52
68. Kang YK, Kim WH, Jang JJ. Expression of G1-S modulators (p53, p16, p27, cyclin D1, Rb) and Smad4/Dpc4 in intrahepatic cholangiocarcinoma. Human Pathology. 2002;33:877-83
69. Argani P, Shaukat A, Kaushal M, Wilentz RE, Su GH, Sohn TA. et al. Differing rates of loss of DPC4 expression and of p53 overexpression among carcinomas of the proximal and distal bile ducts. Cancer. 2001;91:1332-41
70. Lee KT, Chang WT, Wang SN, Chuang SC, Chai CY, Hu SW. Expression of DPC4/Smad4 gene in stone-containing intrahepatic bile duct. J Surg Oncol. 2006;94:338-43
71. Wang H, Stephens B, Von Hoff DD, Han H. Identification and characterization of a novel anticancer agent with selectivity against deleted in pancreatic cancer locus 4 (DPC4)-deficient pancreatic and colon cancer cells. Pancreas. 2009;38:551-7
72. Royce SG, Alsop K, Haydon A, Mead L, Smith LD, Tesoriero AA. et al. The role of SMAD4 in early-onset colorectal cancer. Colorectal Disease. 2010;12:213-9
73. Papageorgis P, Cheng KH, Ozturk S, Gong Y, Lambert AW, Abdolmaleky HM. et al. Smad4 Inactivation Promotes Malignancy and Drug Resistance of Colon Cancer. Cancer research. 2011;71:998-1008
74. Maitra A, Molberg K, Albores-Saavedra J, Lindberg G. Loss of Dpc4 expression in colonic adenocarcinomas correlates with the presence of metastatic disease. Am J Pathol. 2000;157:1105-11
75. Miyaki M, Iijima T, Konishi M, Sakai K, Ishii A, Yasuno M. et al. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene. 1999;18:3098-103
76. Le Goff C, Mahaut C, Abhyankar A, Le Goff W, Serre V, Afenjar A. et al. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat Genet. 2011;44:85-8
77. Takaku K, Miyoshi H, Matsunaga A, Oshima M, Sasaki N, Taketo MM. Gastric and duodenal polyps in Smad4 (Dpc4) knockout mice. Cancer research. 1999;59:6113-7
78. Xu X, Brodie SG, Yang X, Im YH, Parks WT, Chen L. et al. Haploid loss of the tumor suppressor Smad4/Dpc4 initiates gastric polyposis and cancer in mice. Oncogene. 2000;19:1868-74
79. Takaku K, Oshima M, Miyoshi H, Matsui M, Seldin MF, Taketo MM. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998;92:645-56
80. Kitamura T, Kometani K, Hashida H, Matsunaga A, Miyoshi H, Hosogi H. et al. SMAD4-deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nat Genet. 2007;39:467-75
81. Inamoto S, Itatani Y, Yamamoto T, Minamiguchi S, Hirai H, Iwamoto M. et al. Loss of SMAD4 Promotes Colorectal Cancer Progression by Accumulation of Myeloid-Derived Suppressor Cells through the CCL15-CCR1 Chemokine Axis. Clin Cancer Res. 2016;22:492-501
82. Ang CW, Nedjadi T, Sheikh AA, Tweedle EM, Tonack S, Honap S. et al. Smad4 loss is associated with fewer S100A8-positive monocytes in colorectal tumors and attenuated response to S100A8 in colorectal and pancreatic cancer cells. Carcinogenesis. 2010;31:1541-51
83. Ding Z, Wu CJ, Chu GC, Xiao Y, Ho D, Zhang J. et al. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 2011;470:269-73
84. Aitchison AA, Veerakumarasivam A, Vias M, Kumar R, Hamdy FC, Neal DE. et al. Promoter methylation correlates with reduced Smad4 expression in advanced prostate cancer. Prostate. 2008;68:661-74
85. Bornstein S, White R, Malkoski S, Oka M, Han G, Cleaver T. et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J Clin Invest. 2009;119:3408-19
86. Yang L, Mao C, Teng Y, Li W, Zhang J, Cheng X. et al. Targeted disruption of Smad4 in mouse epidermis results in failure of hair follicle cycling and formation of skin tumors. Cancer research. 2005;65:8671-8
87. Qiao W, Li AG, Owens P, Xu X, Wang XJ, Deng CX. Hair follicle defects and squamous cell carcinoma formation in Smad4 conditional knockout mouse skin. Oncogene. 2006;25:207-17
88. Teng Y, Sun AN, Pan XC, Yang G, Yang LL, Wang MR. et al. Synergistic function of Smad4 and PTEN in suppressing forestomach squamous cell carcinoma in the mouse. Cancer research. 2006;66:6972-81
89. Li XB, Yang G, Zhu L, Tang YL, Zhang C, Ju Z. et al. Gastric Lgr5(+) stem cells are the cellular origin of invasive intestinal-type gastric cancer in mice. Cell Res. 2016;26:838-49
90. Sun GL, Li Z, Wang WZ, Chen Z, Zhang L, Li Q. et al. miR-324-3p promotes gastric cancer development by activating Smad4-mediated Wnt/beta-catenin signaling pathway. J Gastroenterol. 2017
91. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131-42
92. Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429-37
93. Gravdal K, Halvorsen OJ, Haukaas SA, Akslen LA. A switch from E-cadherin to N-cadherin expression indicates epithelial to mesenchymal transition and is of strong and independent importance for the progress of prostate cancer. Clin Cancer Res. 2007;13:7003-11
94. Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF. et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer research. 2008;68:7846-54
95. Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J. et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nature cell biology. 2009;11:943-50
96. Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH, Moustakas A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol. 2006;174:175-83
97. Deckers M, van Dinther M, Buijs J, Que I, Lowik C, van der Pluijm G. et al. The tumor suppressor Smad4 is required for transforming growth factor beta-induced epithelial to mesenchymal transition and bone metastasis of breast cancer cells. Cancer research. 2006;66:2202-9
98. Moon H, Ju HL, Chung SI, Cho KJ, Eun JW, Nam SW. et al. Transforming Growth Factor-beta Promotes Liver Tumorigenesis in Mice via Up-regulation of Snail. Gastroenterology. 2017;153:1378-91 e6
99. Kurisaki K, Kurisaki A, Valcourt U, Terentiev AA, Pardali K, Ten Dijke P. et al. Nuclear factor YY1 inhibits transforming growth factor beta- and bone morphogenetic protein-induced cell differentiation. Mol Cell Biol. 2003;23:4494-510
100. Takeda M, Mizuide M, Oka M, Watabe T, Inoue H, Suzuki H. et al. Interaction with Smad4 is indispensable for suppression of BMP signaling by c-Ski. Mol Biol Cell. 2004;15:963-72
101. David CJ, Huang YH, Chen M, Su J, Zou Y, Bardeesy N. et al. TGF-beta Tumor Suppression through a Lethal EMT. Cell. 2016;164:1015-30
102. Mitra D, Fernandez P, Bian L, Song N, Li F, Han G. et al. Smad4 loss in mouse keratinocytes leads to increased susceptibility to UV carcinogenesis with reduced Ercc1-mediated DNA repair. J Invest Dermatol. 2013;133:2609-16
103. Wang SE, Xiang B, Guix M, Olivares MG, Parker J, Chung CH. et al. Transforming growth factor beta engages TACE and ErbB3 to activate phosphatidylinositol-3 kinase/Akt in ErbB2-overexpressing breast cancer and desensitizes cells to trastuzumab. Mol Cell Biol. 2008;28:5605-20
104. Liu L, Zhou W, Cheng CT, Ren X, Somlo G, Fong MY. et al. TGFbeta induces "BRCAness" and sensitivity to PARP inhibition in breast cancer by regulating DNA-repair genes. Mol Cancer Res. 2014;12:1597-609
105. Zhang R, Wang M, Sui P, Ding L, Yang Q. Upregulation of microRNA-574-3p in a human gastric cancer cell line AGS by TGF-beta1. Gene. 2017;605:63-9
106. Huang S, He X, Ding J, Liang L, Zhao Y, Zhang Z. et al. Upregulation of miR-23a approximately 27a approximately 24 decreases transforming growth factor-beta-induced tumor-suppressive activities in human hepatocellular carcinoma cells. Int J Cancer. 2008;123:972-8
107. Jiang T, Ye L, Han Z, Liu Y, Yang Y, Peng Z. et al. miR-19b-3p promotes colon cancer proliferation and oxaliplatin-based chemoresistance by targeting SMAD4: validation by bioinformatics and experimental analyses. J Exp Clin Cancer Res. 2017;36:131
108. Xu Q, Tong JL, Zhang CP, Xiao Q, Lin XL, Xiao XY. miR-27a induced by colon cancer cells in HLECs promotes lymphangiogenesis by targeting SMAD4. PLoS One. 2017;12(e):186718
109. Wilentz RE, Su GH, Dai JL, Sparks AB, Argani P, Sohn TA. et al. Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol. 2000;156:37-43
110. Singh P, Srinivasan R, Wig JD. SMAD4 genetic alterations predict a worse prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas. 2012;41:541-6
111. Isaksson-Mettavainio M, Palmqvist R, Dahlin AM, Van Guelpen B, Rutegard J, Oberg A. et al. High SMAD4 levels appear in microsatellite instability and hypermethylated colon cancers, and indicate a better prognosis. International Journal of Cancer. 2012;131:779-88
112. Li XM, Liu BQ, Xiao JB, Yuan Y, Ma J, Zhang YF. Roles of VEGF-C and Smad4 in the Lymphangiogenesis, Lymphatic Metastasis, and Prognosis in Colon Cancer. Journal of Gastrointestinal Surgery. 2011;15:2001-10
113. Boulay JL, Mild G, Lowy A, Reuter J, Lagrange M, Terracciano L. et al. SMAD4 is a predictive marker for 5-fluorouracil-based chemotherapy in patients with colorectal cancer. British Journal of Cancer. 2002;87:630-4
114. Chung Y, Wi YC, Kim Y, Bang SS, Yang JH, Jang K. et al. The Smad4/PTEN Expression Pattern Predicts Clinical Outcomes in Colorectal Adenocarcinoma. J Pathol Transl Med. 2017
115. Masetti M, Acquaviva G, Visani M, Tallini G, Fornelli A, Ragazzi M. et al. Long-term survivors of pancreatic adenocarcinoma show low rates of genetic alterations in KRAS, TP53 and SMAD4. Cancer Biomark. 2017
116. Toga T, Nio Y, Hashimoto K, Higami T, Maruyama R. The dissociated expression of protein and messenger RNA of DPC4 in human invasive ductal carcinoma of the pancreas and their implication for patient outcome. Anticancer Res. 2004;24:1173-8
117. Wang H, Han H, Von Hoff DD. Identification of an agent selectively targeting DPC4 (deleted in pancreatic cancer locus 4)-deficient pancreatic cancer cells. Cancer research. 2006;66:9722-30
118. Biankin AV, Morey AL, Lee CS, Kench JG, Biankin SA, Hook HC. et al. DPC4/Smad4 expression and outcome in pancreatic ductal adenocarcinoma. J Clin Oncol. 2002;20:4531-42
119. Tascilar M, Skinner HG, Rosty C, Sohn T, Wilentz RE, Offerhaus GJ. et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2001;7:4115-21
120. Hua Z, Zhang YC, Hu XM, Jia ZG. Loss of DPC4 expression and its correlation with clinicopathological parameters in pancreatic carcinoma. World J Gastroenterol. 2003;9:2764-7
121. Kojima K, Vickers SM, Adsay NV, Jhala NC, Kim HG, Schoeb TR. et al. Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia. Cancer research. 2007;67:8121-30
122. Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM. et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer cell. 2007;11:229-43
123. Wang RH, Li C, Xu X, Zheng Y, Xiao C, Zerfas P. et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2:399-409
124. Li W, Qiao W, Chen L, Xu X, Yang X, Li D. et al. Squamous cell carcinoma and mammary abscess formation through squamous metaplasia in Smad4/Dpc4 conditional knockout mice. Development. 2003;130:6143-53
Author contact
![]() Corresponding authors: Lopa Mishra, MD; Director, Center for Translational Medicine; Professor, Surgery and GW Cancer Center; George Washington University and VAMC, Washington DC. lopamishra2com, lmishraedu and Chu-Xia Deng, PhD. Faculty of Health Sciences, University of Macau, Macau SAR, China. cxdengmo
Corresponding authors: Lopa Mishra, MD; Director, Center for Translational Medicine; Professor, Surgery and GW Cancer Center; George Washington University and VAMC, Washington DC. lopamishra2com, lmishraedu and Chu-Xia Deng, PhD. Faculty of Health Sciences, University of Macau, Macau SAR, China. cxdengmo