Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(6):599-607. doi:10.7150/ijbs.23419 This issue Cite
Research Paper
Evaluation of the bioactivity of a mastoparan peptide from wasp venom and of its analogues designed through targeted engineering
Xiaoling Chen1*, Luyao Zhang1*, Yue Wu1, Lei Wang1, Chengbang Ma1 ![]() , Xinping Xi1
, Xinping Xi1 ![]() , Olaf R.P. Bininda-Emonds2, Chris Shaw1, Tianbao Chen1, Mei Zhou1
, Olaf R.P. Bininda-Emonds2, Chris Shaw1, Tianbao Chen1, Mei Zhou1 ![]()
1. Natural Drug Discovery Group, School of Pharmacy, Queen's University Belfast, Belfast, Northern Ireland, UK
2. AG Systematik und Evolutionsbiologie, IBU—Faculty V, Carl von Ossietzky University Oldenburg, Oldenburg, Germany
* These authors contributed equally to this work
Received 2017-10-20; Accepted 2018-2-21; Published 2018-4-25
Abstract

Mastoparan is a typical cationic and amphipathic tetradecapeptide found in wasp venom and exhibits potent biological activities. Yet, compared with other insect-derived peptides, such as melittin from the bee venom, this family have been underrated. Herein, we evaluated the biological activities of mastoparan-C (MP-C), which was identified from the venom of the European Hornet (Vespa crabro), and rationally designed two analogues (a skeleton-based cyclization by two cysteine residues and an N-terminal extension via tat-linked) for enhancing the stability of the biological activity and membrane permeability, respectively. Three peptides possessed broadly efficacious inhibiting capacities towards common pathogens, resistant strains, as well as microbial biofilm. Although, cyclized MP-C showed longer half-life time than the parent peptide, the lower potency of antimicrobial activity and higher degree of haemolysis were observed. The tat-linked MP-C exhibited more potent anticancer activity than the parent peptide, but it also loses the specificity. The study revealed that MP-C is good candidate for developing antimicrobial agents and the targeted-design could improve the stability and transmembrane delivery, but more investigation would be needed to adjust the side effects brought from the design.
Keywords: mastoparan-C, peptide design, antimicrobial, anticancer
Introduction
Wasp venom contains numerous bioactive substances that are of importance to the animal for hunting and defending against intruders, but have also attracted attention because of their potential physiological, pharmacological, and therapeutic applications 1-4. Of the many small venom-derived peptides that are known, mastoparan and mastoparan-like peptides are the best characterised candidates 4, 5. Although there is much sequence heterogeneity among mastoparans, the family nevertheless displays several common structural characteristics among its members. For example, the majority of mastoparans are 14 amino acids in length, most of which are the hydrophobic residues leucine, isoleucine, valine, and alanine 4, 6, 7. In addition, the peptides tend to be polycationic because of the numerous lysine residues and also possess an amidated C-terminus 8. Finally, whereas mastoparans typically possess a random-coil structure in an aqueous solution, they partially convert to an α-helical conformation in an amphiphilic environment 9.
Pharmacologically, mastoparans also represent a promising group of small peptides, with those mastoparans that have been characterised variously displaying broad-spectrum action against microorganisms, inhibitory effects against tumour proliferation, and stimulating serotonin release from platelets and mast cell degranulation 10-12. Effective active concentrations are also often low, particularly for antimicrobial activity where suitable inhibitory concentrations are on the order of micromolars or even lower 13-15. Importantly, recent studies have also shown that mastoparans are effective against some clinically resistant microbial strains. For instance, Lin and colleagues demonstrated that multidrug-resistant Acinetobacter baumanni, one of major contributors to nosocomial infections, is compromised by mastoparan-AF in low dosages 15.
However, the promise shown by mastoparans as potential drug candidates is offset currently by their haemolytic activity and other reported toxic effects 6, 16. In addition, like with other natural antimicrobial peptides, additional non-negligible issues, including restricted half-lives and stability in addition to potential, general toxicity must be addressed prior to therapeutic application in clinical settings 17, 18. A useful strategy in this regard is targeted engineering, which proximately aims to optimize therapeutic activity, in part by reducing toxicity toward normal cells, and ultimately enables us to further explore the structure-function interrelationship of small peptides with the goal of improving targeted design changes. Commonly implemented changes in this context include amino-acid replacement to reduce toxicity (e.g., by replacing highly hydrophobic amino acids with isoleucine) or to alter the α-helical content to enhance cell-selectivity or anti-biofilm property 19, 20as well as altering the length of the primary structure. For instance, KS-30, a truncated sequence that was derived from human cathelicidin LL-37 by removing seven residues at the N-terminus of the latter, was shown to exert more potent antimicrobial activity 21. Alternatively, short foreign peptide sequences can also be added to antimicrobial peptides as part of a directed therapeutic drug design, particularly CPP/PTD application (frequently artificial sequence (polyarginine and polylysine)) to improve drug delivery, but also the virus fragments VP22 and Tat or the insect peptide penetratin 22, 23. Finally, skeleton-based end-capping of either the N- or C-terminus (e.g., via cyclization) has also been exploited to limit conformational flexibility and to avert enzymatic degradation.
In this paper, we broadly investigate the antimicrobial peptide mastoparan-C (MP-C) from the European Hornet (Vespa crabro) venom with respect to its therapeutic potential against common pathogens, antibiotic-resistant strains, and microbial biofilms. In addition to our examinations of the natural peptide, we also derived and tested two analogues of it that were designed for either improved stability in serum (by introducing a disulphate bridge between N- and C- terminus) or enhanced intracellular delivery to possibly improve anticancer activity (by introducing a TAT sequence). For both analogues, we assessed the degree to which the specific goals of the designed changes were met and compared their biological activities to the natural, parent peptide. Altogether, this study yields important information regarding the potential of MP-C as a drug candidate as well as how it is affected by possible design changes to increase its therapeutic potential.
Experimental approach
Design and synthesis of MP-C and the two analogues
We obtained the primary sequence for MP-C (LNLKALLAVAKKIL-NH2) from the literature 24, 25. From this natural, parent sequence, we derived two artificial analogues designed either to enhance its stability or its cell-based targeting activity. In the former case, given that a cyclic structure makes polypeptides more resistant to degradation, we introduced one cysteine at each of the N- and C-termini causing MP-C to form a loop structure (cMP-C; CLNLKALLAVAKKILC-NH2). In the latter case, we added a short cell-penetrating peptide (RKKRRQRRR) to the N-terminal of the parent sequence (tMP-C; RKKRRQRRRLNLKALLAVAKKIL-NH2). This sequence is derived from the basic domain (residues 48-57) of the HIV-1 TAT protein and is indicated to be the smallest fragment of the protein that maintains the ability to be assimilated by eukaryotic cell membranes 26-29 without any biological response 30, 31.
Sufficient quantities to evaluate the bioactivities of all three peptides were obtained using the automatic PS4 peptide synthesizer (Protein Technologies, USA) along with Rink amide resin and standard Fmoc-chemistry. Cleavage of the primary products from the resin and subsequent deprotection used a mixture of trifluoroacetic acid (TFA), ethanedithiol (EDT), triisopropylsilane (TIPS) and water (94:2:2:2 (v/v)). Prior to lyophilisation, cMP-C was dissolved in 0.2% of hydrogen peroxide in 0.05/19.95/80.00 (v/v/v) TFA/ water/ acetonitrile for 0.5h to get the loop structure via sulfhydryl oxidization. Finally, each synthetic peptide was purified using RP-HPLC (Phenomenex C-5 column, 0.46 cm × 25 cm) and its purity was confirmed using MALDI-TOF mass spectrometry (supplementary in Figure S1) (Perseptive Biosystems, MA, USA).
Circular Dichroism (CD) analysis of synthetic peptides
The secondary structure of each peptide was estimated using a CD spectrometer (Jasco J851, USA). Specifically, the parameters were One hundred µM of each peptide, which was dissolved in 10 mM ammonium acetate (NH4AC) buffer or 50 % trifluoroehanol (TFE) (v/v in 10 mM NH4AC) respectively, was loaded in a cuvette (1-mm path length). For the analysis, three passes (“accumulation”) within the range of 190-260 nm were made at 20 °C at a scanning speed of 200 nm/min, a bandwidth of 1 nm, and a data pitch of 0.5 nm. The percentage of the α-helix structure was predicted via K2D method in DichroWeb website (http://dichroweb.cryst.bbk.ac.uk/html/home.shtml).
Stability in serum/reaction kinetics
Fifty μM of each peptide was incubated in RPMI-1640 culture medium (Invitrogen, Paisley, UK) supplemented with 10% (v/v) of fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO, USA) at 37 ºC for 24 h. At four-hour time intervals, 300 μL of the reaction solution was added to 600 μL of 96% ethanol to precipitate the serum proteins. The cloudy reaction sample was then cooled at 4 ºC for 15 min before being spun at 18,000× g for 2 min to remove the precipitated serum proteins in the pellet. Subsequently, each reaction supernatant was analysed using RP-HPLC and MALDI-TOF to provide both quantitative and qualitative results of the degradation process. The percentage of each peptide that remained was calculated by comparing the corresponding peak area at a given time point with that of the original stock solution without serum.
Antimicrobial assays
The minimal inhibitory concentrations (MICs) of all three synthesized peptides were determined against S. aureus (NCTC 10788), E. coli (NCTC 10418) and C. abicans (NCTC 1467) as well as against the resistant microorganisms methicillin-resistant S. aureus (MRSA; ATCC 12493), P. aeruginosa (ATCC 27853) and E. faecalis (NCTC 12697), each of which had been cultured in Mueller-Hinton Broth (MHB). Cultures of each microorganism (105 colony forming units (CFU)/mL) were inoculated with peptide solutions in a concentration range of 1 to 512 μM (in two-fold dilutions) in a 96-well plate (100 μL per well) and incubated at 37 °C in a humidified atmosphere for 16-24 h. Thereafter, the absorbance values of each well was determined at 550 nm using a Synergy HT plate reader (Biotech, USA) and the MIC was defined as the lowest concentration of the respective peptide that resulted in no apparent growth of the microorganism. In addition, 20 uL of a mixture from each well was inoculated on Mueller Hinton agar (MHA) plates. The corresponding peptide concentration where no bacterial communities grew was defined as the minimum bactericidal concentration (MBC).
Anti-biofilm assay
For measuring the minimum biofilm inhibitory concentration (MBIC), we used the two common biofilm-forming bacteria P. aeruginosa and S. aureus, which were cultured in Luria Broth (LB) and Tryptic Soy Broth (TSB), respectively 32-34. Peptide solutions from 1 - 512 μM (again in two-fold dilution) were incubated with 106 CFU/mL of each bacterial culture in the corresponding medium and again placed in the wells of 96-well plate (100 μL per well). After 18 h of incubation (at 37 °C and 200 rpm), each well was rinsed twice with deionized water and stained with 125 μL of 0.1% Crystal Violet. Excess stain was removed by further rinsing with deionized water. After air drying overnight, 150 μL of 30% glacial acetic acid was added to each well and the absorbance at 595 nm was measured using the Synergy HT plate reader.
The assays to determine the minimum biofilm eradication concentration (MBEC) followed a similar procedure except that 100 μL of each bacterial culture was first seeded in a 96-well plate and incubated for 18 h (at 37 °C and 200 rpm) to form the biofilm. After the planktonic phase was removed, peptide solutions in the standard concentrations were added to the biofilms that had formed in each well. After 24 h incubation, the biofilms were stained and measured as described above for MBIC.
Both MBIC and MBEC were defined in an analogous fashion to MIC and MBC, respectively.
Membrane permeability assay
Membrane permeability was evaluated using a SYTOX Green Nucleic Acid Stain (Life Technologies, UK) assay. Cells from a culture of S. aureus that was in the logarithmic growth phase were centrifuged, washed and re-suspended in 5% TSB in 0.85% NaCl solution. Thereafter, 50 µL of the bacterial suspension (1 x 107 CFU/mL) was incubated for 2 h at 37 ˚C with 40 µL of peptide solution in final concentrations of 1-, 2- and 4-fold of the respective MICs in a black 96 well plate (Sterilin, UK) that was shielded from any light. Equivalent bacteria cells treated with 70% isopropanol or 5% TSB only served as positive and negative controls, respectively. After two hours, SYTOX green nucleic acid stain was added to each well at the final concentration of 5 µM and allowed to incubate for 5 min after which the fluorescent intensity (excitation at 485 nm and emission at 528 nm) was recorded using an ELISA plate reader (Biolise BioTek EL808).
On the other hand, to obtain the fluorescence kinetics of membrane permeabilisation, 50 µL of bacterial suspension was added to 40 µL of peptide solution in final concentrations of 4-fold of the respective MICs in a black 96 well plate. And the 5 µM SYTOX green nucleic acid stain was mixed with the reaction immediately. Thereafter, changes in membrane permeability were quantified via time-course analyses over a period of 40 min with data collection occurring at one-minute intervals and the examination method of fluorescent intensity see above.
MTT anti-cancer assay
Each of the five cancer cell lines — non-small cell lung cancer H157, melanocyte MDA-MB-435S, human prostate carcinoma PC-3, human glioblastoma astrocytoma U251MG, human breast cancer MCF-7 — as well as the cell line for normal human microvessel endothelial cells HMEC-1 were seeded into a 96-well plate at densities of 5000 cells/well. After incubation for 24 h at 37 ºC with 5% CO2, the cells were starved for 6 h by replacing the medium with serum-free medium. Thereafter, synthesized peptides (in ten-fold concentrations from 10-4 to 10-9 M in serum-free medium) were incubated with the cells for 24 h after which 10 μL of MTT solution (5mg/ml) was added to each well under dark conditions. Following a further 4-6 h of incubation, 100 μl of DMSO superseded medium was added to each well to dissolve the formazan crystals. The OD value of each well was read by the Synergy HT plate reader at 550 nm.
Haemolysis assay
Finally, to determine the toxicity of the peptides to normal mammalian cells, a 2% solution of horse erythrocytes was re-suspended in PBS solution and incubated with each peptide at the standard concentrations (i.e., from 1 to 512 μM) for 2 h at 37 ºC. Equivalent cells treated with PBS or 1% Triton X-100 served as the positive and negative controls, respectively. Following centrifugation at 900× g for 5 min, 100 μL of the supernatant from each tube was transferred to a 96-well plate, which was then read at 550 nm using the Synergy HT plate reader.
Results
Physicochemical properties and secondary structures of MP-C and its analogues
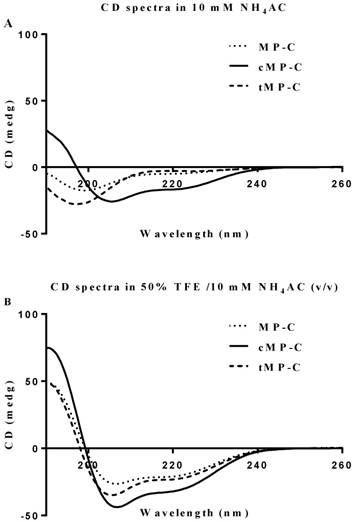
Both MP-C and cMP-C possessed the same net positive charge of +4, which increased to +12 in the case of tMP-C (Table 1). Similarly, MP-C and cMP-C had a similar degree of hydrophobicity, which was dramatically reduced in tMP-C. All three peptides adopted α-helical conformations in membrane-mimic solution, presenting obviously negative peaks at 222 nm and 208 nm, with cMP-C presenting the largest proportion of an α-helical domain (76% of its secondary structure). Despite being designed to be a cyclic peptide, 28% of the secondary structure of cMP-C unexpectedly retained an α-helical domain in an aqueous environment, whereas MP-C and tMP-C mainly adopted a random coil structure (Figure 1).
Physicochemical properties of MP-C and its two analogues.
| Peptide | Residues | Hydrophobicity | Net charge | % α-helix | |
|---|---|---|---|---|---|
| 10 mM NH4AC | 50% TFE (v/v) | ||||
| MP-C | 14 | 0.634 | +4 | 7 | 33 |
| cMP-C | 16 | 0.748 | +4 | 28 | 76 |
| tMP-C | 23 | 0.027 | +12 | 9 | 28 |
CD spectra for MP-C (dotted line), cMP-C (solid line) and tMP-C (dashed line) in 100 µM concentrations in (A) 10mM NH4AC buffer and (B) 50% TFE/10mM NH4AC solution at 20 ºC.
Serum stability of peptides
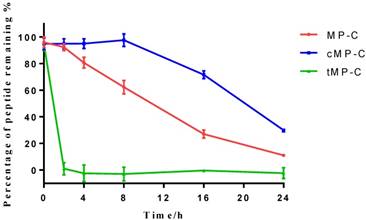
Peptide degradation analyses showed that MP-C began to degrade at a more-or-less linear rate (3.54 % h-1) upon being mixed with serum, but that cMP-C was not degraded significantly within the first 8 h. Thereafter, however, cMP-C also began to degrade at about the similar linear rate (4.23 % h-1) such that both peptides were degraded significantly (to less than 40% remaining) after 24 h (Figure 2). On the other hand, tMP-C demonstrated poor serum stability, which possibly co-precipitated with serum protein by intense electrostatic adsorption.
The stability of MP-C (red), cMP-C (blue) and tMP-C (green) in 10% FBS in RMPI-1640 culture medium at 37 ºC. Data points represent the average of three independent experiments with error bars presenting the s.d.
Antimicrobial activity
Both parent peptide and its analogues displayed at least moderate bioactivity again all the microorganisms that we examined (Table 2). This was especially the case for both MP-C and tMP-C, where MIC and MBC values were at most 16 µM and often much lower. By contrast, the comparative activity of cMP-C was at least 4× lower. Importantly, MP-C and tMP-C maintained similar levels of potency against the three antibiotic resistant microorganisms, whereas the MICs of cMP-C were 4× higher in two of the three cases compared to the standard test organisms.
Antimicrobial activity of the parent mastoparan peptide and its two analogues against various microorganisms.
| Microorganisms | MIC/MBC (µM) | ||
|---|---|---|---|
| MP-C | cMP-C | tMP-C | |
| S. aureus | 2/2 | 32/64 | 4/4 |
| E. coli | 4/8 | 32/128 | 2/2 |
| C. albicans | 4/4 | 32/128 | 2/2 |
| MRSA | 4/4 | 128/128 | 4/8 |
| P. aeruginosa | 8/16 | 32/>512 | 4/4 |
| E. faecalis | 8/8 | 128/128 | 8/16 |
Anti-biofilm activity
Similarly, both MP-C and tMP-C were able to broadly suppress biofilm formation by S. aureus and P. aeruginosa (maximum MBIC of 32 µM), but were noticeably less effective at eliminating an already existing biofilm (Table 3). Again, cMP-C showed less potency than the other two peptides and was only able to inhibit the formation of biofilm by S. aureus at the concentrations that we examined.
Membrane permeabilisation of S. aureus
Two distinct behaviours were observed among the peptides, with cMP-C again differentiating itself from the parent peptide and tMP-C (Figure 3). cMP-C caused almost immediate and severe membrane permeabilisation, even at the lowest concentration of it that we tested (1-fold of its MIC versus S. aureus). Significant permeabilisation also occurred at higher concentrations of cMP-C, albeit noticeably reduced in comparison the base concentration. Analogously, relative florescent intensity, which was maximal after only a few minutes, also decreased slightly over time in the case of cMP-C (Figure 3B) (** P<0.01, comparison between the maximum point in fluorescence intensity at the 19th min and end point at the 40th min).
Anti-biofilm activity of three peptides against the biofilm of S. aureus and P. aeruginosa.
| Microorganisms | MBIC/MBEC (µM) | ||
|---|---|---|---|
| MP-C | cMP-C | tMP-C | |
| S. aureus | 4/64 | 32/>512 | 32/256 |
| P. aeruginosa | 32/128 | >512/>512 | 8/128 |
Membrane permeabilisation of the Gram-positive bacteria as indicated by an increase in the fluorescence of SYTOX Green. (A) Permeabilisation after 2 h of incubation with MP-C (solid), cMP-C (hatched) and tMP-C (shaded) at 1-, 2- and 4-fold concentrations of their MICs against S. aureus. The statistical significance shows in Table S1. (B) Relative fluorescent intensity over time of 4-fold MIC of MP-C (red), cMP-C (blue) and tMP-C (green) at 4-fold concentrations of the MICs against S. aureus.
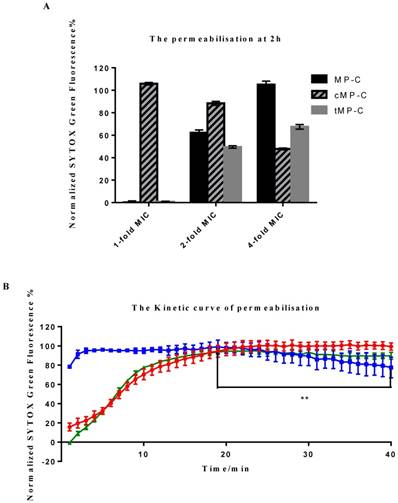
By contrast, membrane permeabilisation was negligible for both MP-C and tMP-C at the basal concentration but increased greatly at concentrations of 2- and especially 4-fold their MICs against S. aureus, with the increase being larger for MP-C. The time-series analysis for these two peptides were also similar, with the degree of permeabilisation initially increasingly slowly over time such that full permeabilisation occurred only after 20 minutes.
Anti-tumor and haemolytic activity
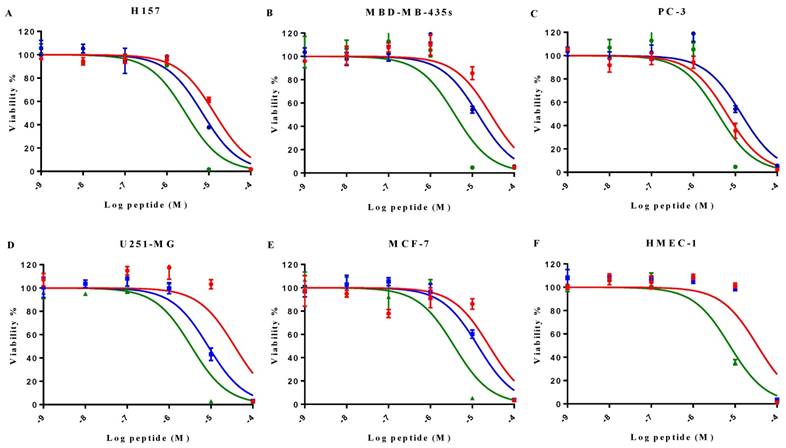
MP-C and analogues showed broad-spectrum anticancer activities as evidenced by their ability to inhibit the proliferation of five diverse human cancer cell lines (Figure 4). IC50s for MP-C ranged from 6.26 to 36.65 µM and were even lower for the two analogues (Table 4), 2x in the case of cMP-C (with the exception of PC-3) and from 5-11x for tMP-C. For ther latter peptide, all IC50s for the cancer cell lines were less than 4 µM.
Induced cytotoxicity of MP-C and analogues on a panel of human cancer cells and normal cells (human HMEC-1 and horse erythrocytes). IC50 and HC50 were calculated from the normalized curves in Figures 4 and 5 using GraphPad Prism 6 (GraphPad Software, USA).
| Cell lines | IC50 (μM) | ||
|---|---|---|---|
| MP-C | cMP-C | tMP-C | |
| H157 | 13.57 | 7.02 | 2.79 |
| MBD-MB-435S | 27.70 | 13.87 | 3.86 |
| PC-3 | 6.29 | 13.87 | 3.86 |
| U251-MG | 36.65 | 8.56 | 3.36 |
| MCF-7 | 25.27 | 13.66 | 3.70 |
| HMEC-1 | 57.15 | 39.53 | 9.18 |
| Haemolysis (HC50) of horse erythrocytes | 40.11 | 9.19 | 77.94 |
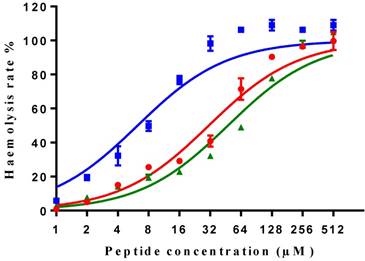
All three peptides showed relatively weaker activity against the normal cell line HMEC-1, although tMP-C still displayed significant cytotoxicity with an IC50 < 10 µM and a 60% inhibitory effect at a concentration of 10 µM. By contrast, MP-C and tMP-C exhibited moderate hemolytic effects on horse erythrocytes, with the potency of tMP-C being greatly reduced compared to that against the cancer cells lines (Figure 5; Table 4). cMP-C showed high activity against the red blood cells at low concentrations (IC50 < 10 µM) and concentrations that were often lower than those for the cancer cell lines.
Discussion
Clearly, the venom produced by venomous creatures including snakes, jellyfish, cone snails, spiders, scorpions as well as some insects like wasps have evolved because of their direct benefits to these organisms in terms of self-defence and/or prey acquisition. However, because the bioactive substances from the venom often also possess strong antimicrobial and anti-cancer properties (e.g., cupiennin 1a from spider 35), they also serve as promising drug candidates, justifying their identification, isolation, and testing in this context. Indeed, some bioactive peptides have even reached the clinical trial stage, such as MVIIA (commercial name ziconotide 36) and magainin (commercial name Pexiganan 37) for the treatment of intractable pain and diabetic foot ulcers, respectively. The bioactive peptides from wasp venom, like mastoparan, should represent a promising source for new drug leads discovery and development, having been shown to be potent substances with obvious antimicrobial and anti-cancer properties 19, 20, 38. Yet, mastoporan have not been researched to the same degree in this context as have other bioactive peptides derived from other insects, and the bee-venom protein mellitin in particular, possibly because of potential haemolytic activity and other toxic effects 39.
Indeed, exactly this scenario was mirrored in our results, which showed that the natural, parent peptide, MP-C, shows strong antimicrobial and anticancer properties, with effective concentrations in the low micromolar range. MP-C as a typical cationic antimicrobial venom Importantly, it displayed about the same level of efficacy against both stock and resistant bacteria as well as only a slightly reduced efficacy against biofilms. However, effective concentrations against both the normal human cell line HMEC-1 and horse erythrocytes were generally also in the low micromolar range, which is problematic for the use of MP-C a clinical context. Similar results were obtained by Yoon et al. (antimicrobial activity against E.coli ATCC 11775 (MIC>1000 µM), S.aureus ATCC 12600 (MIC=500 µM), C.albicans ATCC 10231 (MIC=100 µM) and causing no hemolysis to human erythrocytes within 200 µM 25), although effective concentrations were up to two orders of magnitude higher than what we observed. We have no ready explanation for this latter discrepancy.
To potentially ameliorate this situation, we turned to two targeted modifications of MP-C in an attempt to increase their potency or, more likely given its already strong bioacitivties, to reduce its toxicity to the host organism. In this regard, targeted modification represents an important extension of natural drug discovery given that medical bioactivities are often useful side-effects of many natural compounds that could be improved further. However, because the exact mechanism of action of most bioactive peptides remains unknown, our modifications are often restricted to following general guidelines (e.g., beneficial properties that include an increased amphipathic nature or cationic charge), which was also the case here insofar as our modifications were made to either increase the stability of the peptide in serum solution (cMP-C) or to increase its intracellular delivery (tMP-C).
The effect on cell proliferation of MP-C (red), cMP-C (blue) and tMP-C (green) on the cancer cell lines (A) H157, (B) MDA-MB-435S, melanocyte, (C) PC-3, (D) U251MG, and (E) MCF-7 as well as (F) the normal human microvessel endothelial cell HMEC-1. Data points represent the average of three independent experiments with error bars presenting the s.d. The statistical significance shows in Table S2.
Hemolytic activity of MP-C (red), cMP-C (blue) and tMP-C (green) against horse red blood cells. Data points represent the average of three independent experiments and error bars represent the s.d. The statistical significance shows in Table S3.
The results of our modifications were mixed. Previous work has indicated that a cysteine knot resulting from paired two cysteine amino acids in N- and C- termini in a peptide can play an important role in maintaining or inducing the bioactive structure of the peptide 40, largely by preventing its degradation by diverse proteases 41, 42. However, our modification of the parent peptide to contain such a head-to-tail cyclized structure (as cMP-C) reduced its antimicrobial activity notably, despite the latter always displaying the greatest α-helical nature of the three peptides (even in an aqueous environment), a conformation that is supposed to facilitate interaction with the cell membrane 43. After confirmation of the formation of disulphide bridge in cMP-C by MALDI-TOF, we assume that the disulphide bridge might be remarkable for stabilising the helical structure of cMP-C instead of turning the backbone for a β-sheet conformation. Similarly, non-helical coil wasp venom peptides, examples like OdVP4, EpVP3, EpVP3S, EpVP4a or b, and EpVP5, have neither antimicrobial and hemolytic, nor cell lytic activities, suggesting helical conformation is crucial property for membrane interaction 44, 45. The latter, however, probably did occur with non-bacterial (i.e., mammalian) cells, where cMP-C showed higher (toxic) activity than did the parent peptide as well as much higher membrane permeabilisation capabilities. (The decreasing ability of cMP-C to permeabilise cell membranes at increasingly higher concentrations might derive from fluorescence quenching arising from the increasing formation of peptide-DNA aggregates given that long regions of an α-helical conformation could bind to DNA stably 46.) By contrast, we hypothesize in the case of bacterial cells that the cyclical nature of cMP-C causes it to become trapped in the bacterial cell wall or biofilm (unlike the linear MP-C and MP-C) following its aggregation on the cell surface via electrostatic interaction, requiring higher concentrations to reach the cell membrane. Further experiments with other (labelled) bioactive peptides are needed to determine if this is indeed the problem with a cyclical structure and if this problem occurs frequently enough such that the increased stability offered by the cyclical structure is not justified in the face of reduced antimicrobial activities. Further investigation, including enzymatic treatments or incubation with mimicking-membrane 47, 48, is also needed to determine whether cMP-C does indeed exhibit increased stability and longevity in vivo.
Our addition of the TAT peptide to the N-terminal of the parent MP-C resulted in little change, on average, to the already good antimicrobial activities of MP-C, but dramatically improved its anticancer properties by a factor of at least five-fold. It has previously been reported that the anticancer activity of mastoporan occurs generally via lysis of the outer cell membrane 49. Instead, our results showed a large relative decrease in membrane permeabilisation by tMP-C despite the fact that the increased cationicity of its N-terminus should enhance its electrostatic attraction to the cell membrane, thereby reinforcing its cytolytic effects 50. Balancing this, however, is that most antimicrobial peptides contain a relative hydrophobic segment in their N-terminus, presumably to interact with the cell membrane 51, 52. Thus, given that the TAT is known to be a short cell-penetrating peptide, we suspect an additional, intracellular mechanism to explain the increased anticancer properties of tMP-C. This in line with reports that mastoparan can interact with the phospholipid phase of the mitochondrial membrane, which has been implicated as the leading cause for inducing apoptosis in B16F10-Nex2 melanoma cells 38, 53, and can selectively activate phospholipase or inhibit ATPase activity to cause failure in cell proliferation and metabolism 54, 55. The addition of the TAT peptide to MP-C would presumably increase access to such intracellular targets to induce apoptotic cell death 56, 57 while leaving the outer cell membrane more intact in comparison to the native peptide. However, the poor stability in blood serum and the sometimes severe cytolytic effects of tMP-C (e.g., versus the normal human cell line HMEC-1) speak against its clinical application without further modification.
In summary, our study has reinforced the general utility of mastoporan as an alternative to conventional antibiotics given its strong antimicrobial action that was also evident against resistant bacterial strains and those forming biofilms. A necessary step in this process, however, is targeted modifications of the native peptide to either minimize its potential toxic side effects and/or to improve its potency. In the latter instance, our addition of the short, cell-penetrating peptide TAT to the N-terminus of the native peptide increased its activity against cancer cells dramatically without any adverse effect on the generally good antimicrobial activity. Unfortunately, our modifications could not ameliorate the occasionally strong cytolytic effects against either normal human cells or hose erythrocytes and more work here is obviously needed. Another aspect that must be considered in the potential clinical application of any mastoporan derivative derive from reports that MP-C can release histamine from mammalian mast cells 24, possibly leading to an immune system response. Nevertheless, it is our view that mastoporan remains a viable clinical candidate as an alternative to conventional antibiotics as well as ideal template for designing therapeutic drugs through targeted modification.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Habermann E. Bee and wasp venoms. Science. 1972Jul28;177(4046):314-22
2. Mortari MR, Cunha AOS, De Oliveira L, Vieira EB, Gelfuso EA, Coutinho-Netto J, Ferreira dos Santos W. Anticonvulsant and behavioural effects of the denatured venom of the social wasp polybia occidentalis (polistinae, vespidae). Basic & Clinical Pharmacology & Toxicology. 2005;97(5):289-95
3. Piek T. Venoms of the hymenoptera: Biochemical, pharmacological and behavioural aspects. Elsevier. 2013
4. Moreno M, Giralt E. Three valuable peptides from bee and wasp venoms for therapeutic and biotechnological use: Melittin, apamin and mastoparan. Toxins. 2015;7(4):1126-50
5. Palma MS. Insect venom peptides. The Handbook of Biologically Active Peptides. 2006:409-16
6. Hirai Y, YASUHARA T, YOSHIDA H, NAKAJIMA T, FUJINO M, KITADA C. A new mast cell degranulating peptide" mastoparan" in the venom of vespula lewisii. Chemical and Pharmaceutical Bulletin. 1979;27(8):1942-4
7. Ma R, Mahadevappa R, Kwok HF. Venom-based peptide therapy: Insights into anti-cancer mechanism. Oncotarget. 2017;8(59):100908-30
8. da Silva AV, De Souza BM, dos Santos Cabrera, Marcia P, Dias NB, Gomes PC, Neto JR, Stabeli RG, Palma MS. The effects of the C-terminal amidation of mastoparans on their biological actions and interactions with membrane-mimetic systems. Biochimica Et Biophysica Acta (BBA)-Biomembranes. 2014;1838(10):2357-68
9. de Souza BM, dos Santos Cabrera, Marcia Perez, Neto JR, Palma MS. Investigating the effect of different positioning of lysine residues along the peptide chain of mastoparans for their secondary structures and biological activities. Amino Acids. 2011;40(1):77-90
10. Higashijima T, Uzu S, Nakajima T, Ross EM. Mastoparan, a peptide toxin from wasp venom, mimics receptors by activating GTP-binding regulatory proteins (G proteins). J Biol Chem. 1988May15;263(14):6491-4
11. dos Santos Cabrera, Marcia P, Alvares DS, Leite NB, Monson de Souza B, Palma MS, Riske KA, Ruggiero Neto J. New insight into the mechanism of action of wasp mastoparan peptides: Lytic activity and clustering observed with giant vesicles. Langmuir. 2011;27(17):10805-13
12. Kawakami H, Goto SG, Murata K, Matsuda H, Shigeri Y, Imura T, Inagaki H, Shinada T. Isolation of biologically active peptides from the venom of japanese carpenter bee, xylocopa appendiculata. Journal of Venomous Animals and Toxins Including Tropical Diseases. 2017;23(1):29
13. Konno K, Hisada M, Naoki H, Itagaki Y, Fontana R, Rangel M, Oliveira JS, dos Santos Cabrera, Marcia Perez, Neto JR, Hide I. Eumenitin, a novel antimicrobial peptide from the venom of the solitary eumenine wasp eumenes rubronotatus. Peptides. 2006;27(11):2624-31
14. Čeřovský V, Slaninová J, Fučík V, Hulačová H, Borovičková L, Ježek R, Bednárová L. New potent antimicrobial peptides from the venom of polistinae wasps and their analogs. Peptides. 2008;29(6):992-1003
15. Lin C, Lee M, Tzen JT, Lee H, Chang S, Tu W, Lin C. Efficacy of mastoparan-AF alone and in combination with clinically used antibiotics on nosocomial multidrug-resistant acinetobacter baumannii. Saudi Journal of Biological Sciences. 2017;24(5):1023-9
16. Mendes MA, de Souza BM, Palma MS. Structural and biological characterization of three novel mastoparan peptides from the venom of the neotropical social wasp protopolybia exigua (saussure). Toxicon. 2005;45(1):101-6
17. Diamond G, Beckloff N, Weinberg A, Kisich KO. The roles of antimicrobial peptides in innate host defense. Curr Pharm Des. 2009;15(21):2377-92
18. Peters BM, Shirtliff ME, Jabra-Rizk MA. Antimicrobial peptides: Primeval molecules or future drugs? PLoS Pathogens. 2010;6(10e):1001067
19. Henriksen JR, Etzerodt T, Gjetting T, Andresen TL. Side chain hydrophobicity modulates therapeutic activity and membrane selectivity of antimicrobial peptide mastoparan-X. PloS One. 2014;9(3e):91007
20. Zhang S, Song J, Gong F, Li S, Chang H, Xie H, Gao H, Tan Y, Ji S. Design of an α-helical antimicrobial peptide with improved cell-selective and potent anti-biofilm activity. Scientific Reports. 2016;6:27394
21. Feng X, Sambanthamoorthy K, Palys T, Paranavitana C. The human antimicrobial peptide LL-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant acinetobacter baumannii. Peptides. 2013;49:131-7
22. Bechara C, Sagan S. Cell-penetrating peptides: 20 years later, where do we stand? FEBS Lett. 2013;587(12):1693-702
23. Copolovici DM, Langel K, Eriste E, Langel U. Cell-penetrating peptides: Design, synthesis, and applications. ACS Nano. 2014;8(3):1972-94
24. Argiolas A, Pisano JJ. Isolation and characterization of two new peptides, mastoparan C and crabrolin, from the venom of the european hornet, vespa crabro. J Biol Chem. 1984Aug25;259(16):10106-11
25. Yoon KA, Kim K, Nguyen P, Seo JB, Park YH, Kim K, Seo H, Koh YH, Lee SH. Comparative bioactivities of mastoparans from social hornets vespa crabro and vespa analis. Journal of Asia-Pacific Entomology 2015 December. 2015;18(4):825-9
26. Vogel BE, Lee SJ, Hildebrand A, Craig W, Pierschbacher MD, Wong-Staal F, Ruoslahti E. A novel integrin specificity exemplified by binding of the alpha v beta 5 integrin to the basic domain of the HIV tat protein and vitronectin. J Cell Biol. 1993Apr;121(2):461-8
27. Vives E, Brodin P, Lebleu B. A truncated HIV-1 tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272(25):16010-7
28. Chang HC, Samaniego F, Nair BC, Buonaguro L, Ensoli B. HIV-1 tat protein exits from cells via a leaderless secretory pathway and binds to extracellular matrix-associated heparan sulfate proteoglycans through its basic region. Aids. 1997;11(12):1421-31
29. Jung HJ, Park Y, Hahm K, Lee DG. Biological activity of tat (47-58) peptide on human pathogenic fungi. Biochem Biophys Res Commun. 2006;345(1):222-8
30. Verdurmen WP, Brock R. Biological responses towards cationic peptides and drug carriers. Trends Pharmacol Sci. 2011;32(2):116-24
31. Risso A, Braidot E, Sordano MC, Vianello A, Macri F, Skerlavaj B, Zanetti M, Gennaro R, Bernardi P. BMAP-28, an antibiotic peptide of innate immunity, induces cell death through opening of the mitochondrial permeability transition pore. Mol Cell Biol. 2002Mar;22(6):1926-35
32. Bjarnsholt T. The role of bacterial biofilms in chronic infections. Apmis. 2013;121(s136):1-58
33. Gjødsbøl K, Christensen JJ, Karlsmark T, Jørgensen B, Klein BM, Krogfelt KA. Multiple bacterial species reside in chronic wounds: A longitudinal study. International Wound Journal. 2006;3(3):225-31
34. Harrison JJ, Stremick CA, Turner RJ, Allan ND, Olson ME, Ceri H. Microtiter susceptibility testing of microbes growing on peg lids: A miniaturized biofilm model for high-throughput screening. Nature Protocols. 2010;5(7):1236
35. Kuhn-Nentwig L, Willems J, Seebeck T, Shalaby T, Kaiser M, Nentwig W. Cupiennin 1a exhibits a remarkably broad, non-stereospecific cytolytic activity on bacteria, protozoan parasites, insects, and human cancer cells. Amino Acids. 2011;40(1):69-76
36. Pope JE, Deer TR. Ziconotide: A clinical update and pharmacologic review. Expert Opin Pharmacother. 2013;14(7):957-66
37. Flamm RK, Rhomberg PR, Simpson KM, Farrell DJ, Sader HS, Jones RN. In vitro spectrum of pexiganan activity when tested against pathogens from diabetic foot infections and with selected resistance mechanisms. Antimicrob Agents Chemother. 2015Mar;59(3):1751-4
38. Yamamoto T, Ito M, Kageyama K, Kuwahara K, Yamashita K, Takiguchi Y, Kitamura S, Terada H, Shinohara Y. Mastoparan peptide causes mitochondrial permeability transition not by interacting with specific membrane proteins but by interacting with the phospholipid phase. The FEBS Journal. 2014;281(17):3933-44
39. Gajski G, Garaj-Vrhovac V. Melittin: A lytic peptide with anticancer properties. Environ Toxicol Pharmacol. 2013;36(2):697-705
40. Hill TA, Shepherd NE, Diness F, Fairlie DP. Constraining cyclic peptides to mimic protein structure motifs. Angewandte Chemie International Edition. 2014;53(48):13020-41
41. Weber AE, Halgren TA, Doyle JJ, Lynch RJ, Siegl PK, Parsons WH, Greenlee WJ, Patchett AA. Design and synthesis of P2-P1'-linked macrocyclic human renin inhibitors. J Med Chem. 1991;34(9):2692-701
42. Maryanoff BE, Qiu X, Padmanabhan KP, Tulinsky A, Almond HR Jr, Andrade-Gordon P, Greco MN, Kauffman JA, Nicolaou KC, Liu A. Molecular basis for the inhibition of human alpha-thrombin by the macrocyclic peptide cyclotheonamide A. Proc Natl Acad Sci USA. 1993Sep1;90(17):8048-52
43. Shai Y. Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by α-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochimica Et Biophysica Acta (BBA)-Biomembranes. 1999;1462(1):55-70
44. Baek JH, Lee SH. Isolation and molecular cloning of venom peptides from orancistrocerus drewseni (hymenoptera: Eumenidae). Toxicon. 2010;55(4):711-8
45. Lee SH, Baek JH, Yoon KA. Differential properties of venom peptides and proteins in solitary vs. social hunting wasps. Toxins. 2016;8(2):32
46. Niidome T, Ohmori N, Ichinose A, Wada A, Mihara H, Hirayama T, Aoyagi H. Binding of cationic α-helical peptides to plasmid DNA and their gene transfer abilities into cells. J Biol Chem. 1997;272(24):15307-12
47. Svangård E, Burman R, Gunasekera S, Lövborg H, Gullbo J, Göransson U. Mechanism of action of cytotoxic cyclotides: Cycloviolacin O2 disrupts lipid membranes. J Nat Prod. 2007;70(4):643-7
48. Manabe T, Kawasaki K. D-form KLKLLLLLKLK-NH2 peptide exerts higher antimicrobial properties than its L-form counterpart via an association with bacterial cell wall components. Sci Rep. 2017;7:43384
49. Hilchie AL, Sharon AJ, Haney EF, Hoskin DW, Bally MB, Franco OL, Corcoran JA, Hancock RE. Mastoparan is a membranolytic anti-cancer peptide that works synergistically with gemcitabine in a mouse model of mammary carcinoma. Biochimica Et Biophysica Acta (BBA)-Biomembranes. 2016;1858(12):3195-204
50. Lee Y, Johnson G, Pellois J. Modeling of the endosomolytic activity of HA2-TAT peptides with red blood cells and ghosts. Biochemistry (NY). 2010;49(36):7854-66
51. Wallner M, Meera P, Toro L. Determinant for beta-subunit regulation in high-conductance voltage-activated and ca(2+)-sensitive K+ channels: An additional transmembrane region at the N terminus. Proc Natl Acad Sci USA. 1996;93(25):14922-7
52. Yeaman MR, Yount NY. Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev. 2003;55(1):27-55
53. de Azevedo RA, Figueiredo CR, Ferreira AK, Matsuo AL, Massaoka MH, Girola N, Auada AV, Farias CF, Pasqualoto KF, Rodrigues CP. Mastoparan induces apoptosis in B16F10-Nex2 melanoma cells via the intrinsic mitochondrial pathway and displays antitumor activity in vivo. Peptides. 2015;68:113-9
54. Longland CL, Mezna M, Michelangeli F. The mechanism of inhibition of the Ca2+-ATPase by mastoparan. mastoparan abolishes cooperative ca2+ binding. J Biol Chem. 1999;274(21):14799-805
55. Schmitt J, Noble A, Otsuka M, Berry P, Maitland NJ, Rumsby MG. Phorbol ester stimulates ethanolamine release from the metastatic basal prostate cancer cell line PC3 but not from prostate epithelial cell lines LNCaP and P4E6. Br J Cancer. 2014Oct14;111(8):1646-56
56. Altieri DC, Kang BH. Mitochondria-Targeted Anti-Tumor Agents. 2013.
57. Hao X, Yan Q, Zhao J, Wang W, Huang Y, Chen Y. TAT modification of alpha-helical anticancer peptides to improve specificity and efficacy. PloS One. 2015;10(9e):0138911
Author contact
![]() Corresponding authors: c.maac.uk (C. Ma); x.xiac.uk (X. Xi)
Corresponding authors: c.maac.uk (C. Ma); x.xiac.uk (X. Xi)