Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(6):667-681. doi:10.7150/ijbs.23815 This issue Cite
Research Paper
VRTN is Required for the Development of Thoracic Vertebrae in Mammals
Yanyu Duan*, Hui Zhang*, Zhen Zhang, Jun Gao, Jie Yang, Zhongping Wu, Yin Fan, Yuyun Xing, Lin Li, Shijun Xiao, Yong Hou, Jun Ren ![]() , Lusheng Huang
, Lusheng Huang ![]()
State Key Laboratory of Pig Genetic Improvement and Production Technology, Jiangxi Agricultural University, Nanchang, 330045, China.
* These authors contributed equally to this work.
Received 2017-11-12; Accepted 2018-3-13; Published 2018-4-30
Abstract

Vertnin (VRTN) variants are associated with thoracic vertebral number (TVN) in pigs. However, the biological function of VRTN remains poorly understood. Here we first conducted a range of experiments to demonstrate that VRTN is a responsible gene for TVN and two causative variants in the regulatory region of VRTN additively regulate TVN. Then, we show that VRTN is a novel DNA-binding transcription factor as it localizes exclusively in the nucleus, binds to DNA on a genome-wide scale and regulates the transcription of a set of genes that harbor VRTN binding motifs. Next, we illustrate that VRTN is essential for the development of thoracic vertebrae. Vrtn-null embryos display somitogenesis defect with the failure of axial rotation and fewer somites at the thoracic somite stage. Half of Vrtn heterozygous mice show abnormal spinal development with fewer thoracic vertebrae and ribs than their wild-type littermates. Lastly, we reveal that VRTN could modulate somite segmentation via the Notch signaling pathway. The findings advance our understanding of the mechanisms underlying the development of thoracic vertebrate in mammals, and VRTN causative variants provide a robust tool to improve pork production by selecting the alleles increasing the number of thoracic vertebrae and ribs.
Keywords: VRTN, transcription factor, thoracic vertebrae, somitogenesis
Introduction
A segmented body structure is one of the most notable features of vertebrates. Each vertebra is derived from a vertebral segment, called a somite, which in turn is formed from the presomitic mesoderm (PSM) during early embryogenesis [1]. The number of somites - and, hence, vertebrae - is highly conserved among mammalian species and is usually fixed within each species [2, 3]. However, the pig (Sus scrofa) has variable number of thoracolumbar vertebrae, ranging from 19 in wild boars and most indigenous breeds to between 21 and 23 in European commercial breeds [4, 5]. This phenotypic variation is presumably caused by intensive selection for a greater body size, to increase meat production in commercial animals [6]. This variation in vertebral number within a single species is an ideal system in which to investigate the mechanisms controlling the formation of somites and vertebrae in mammals. Understanding these mechanisms also might have implications for understanding abnormal vertebral development in humans, and mammalian evolution.
In previous studies [7-9], researchers had showed that a quantitative trait locus (QTL) on pig chromosome 7 (SSC7) has a major effect on thoracic vertebral number (TVN) (approximately 0.5 vertebra per allele). Vertnin (VRTN) has been suggested as a strong candidate gene for this locus [7, 9]. We have also previously identified two candidate quantitative trait nucleotides (QTNs) that could account for the QTL effect in the VRTN gene [7], and showed that the candidate QTNs may have introgressed from Chinese pigs into European pigs [10]. However, the causality of VRTN in relation to TVN has not been established, and the molecular features of VRTN and its role in controlling the formation of thoracic somites have remained elusive.
In this study, we provide further evidence that VRTN is the gene responsible for the QTL affecting TVN on SSC7 and confirm the causality of the two VRTN candidate QTNs in relation to the QTL effect. Furthermore, we show that VRTN is a transcription factor that relates to the Notch signaling molecules at the thoracic somite stage. Together, our findings reveal that VRTN is essential for embryogenesis and is absolutely required for the development of thoracic vertebrae, advancing our understanding of the mechanisms controlling segment number in mammals. The VRTN QTNs are of considerable economic significance in the pig industry as breeding companies would like to increase the frequencies of the mutant alleles because of their desirable effects on TVN and pork production.
Materials and methods
Ethics approval statement
All experiments involving animals were carried out in accordance with the approved guidelines by the Ministry of Agriculture in China. Approval was obtained from the ethics committee of Jiangxi Agricultural University prior to the study.
GWAS mapping
A genome-wide association study (GWAS) was conducted on 609 European hybrid derived from a three-way cross pigs: Duroc × (Landrace × Large White) (thereafter referred to as DLY). The pedigree, management and phenotype recording information about these pigs has been described in our previous publication [10]. Briefly, these pigs were purchased from a commercial company (Guohong, Nanchang) and were raised under standard and consistent feeding conditions. At the age of 180 ± 3 days, all animals were slaughtered in the same abattoir (Guohong, Nanchang) to determine TVN in each animal.
Genomic DNA was extracted from ear tissue using a routine phenol/chloroform method. The genomic DNA of each pig was genotyped for 61,565 SNPs on porcine SNP60K Beadchips V2 (Illumina, USA) as previously described [7]. Nine additional SNPs around the VRTN gene were genotyped for all DLY pigs via Sanger sequencing using the primers listed in Table S1. The quality control criteria for the SNP data for the GWAS mapping were assessed using GenABEL, an R library for whole genome association analyses [11]. Animals with SNP call rates of greater than 95% and familial Mendelian error rates of less than 0.1, and SNPs with call rates of greater than 95%, minor allele frequencies of greater than 0.1 and significant levels of deviation from Hardy-Weinberg equilibrium that were greater than 10-6 were included for further association analysis.
The association analysis adjusted for population stratification by modeling similarities between individuals on the basis of genome-wide SNP data. In this analysis, the allelic effect of each SNP on phenotypic traits was tested using a general linear mixed model as previously described [10]. The formula of the model is as follows: y = μ + Xb + sc + Za + e, where y is the vector of phenotypes; μ is the overall mean; b is the vector of fixed effects, including sex and batch effects; c is the effect of each SNP; a is the vector of random additive genetic effects, with a ~ N (0, Gσα2), where G is the genomic relationship matrix calculated from the Illumina Porcine 60K SNP Beadchips and σα2 is the polygenetic additive variance; e is the vector of residual errors, with e ~ N (0, Iσe2), where I is the identity matrix and σe2 is the residual variance; X and Z are incidence matrices for b and a, respectively; and s is the vector representing the SNP genotype for each individual. Bonferroni-corrected P-values were adopted for the genome-wide and chromosome-wide significant thresholds, which were set as 0.05/N and 1/N, respectively, where N is the number of informative SNPs in the data set. The 95% confidence interval was determined by a LOD score drop-off of 2 from the value of the most significant SNP. Linkage disequilibrium was analyzed using Haploview software [12]. The phenotypic variance explained by the VRTN variants was estimated using the following formula.
Var% = (Vreduce- Vfull)/Vreduce✕100
Where Vfull and Vreduce are the residual variances of ordinary linear models with and without including the genotypes of VRTN variants as predictor variables, respectively.
Cell culture
Pig kidney cells (PK-15, Xiangf Bio) and human embryonic kidney cells 293T (HEK293T, SIBS) cells were cultured in high-glucose DMEM (Gibco) supplemented with 10% fetal bovine serum (Hyclone) and an antibiotic solution.
Dual luciferase reporter assay
The VRTN promoter segment spanning -610 to +1558 bp relative to the transcription start site (TSS) was amplified using pig genomic DNA and the primers listed in Table S1. PCR products were purified and ligated into pGEM-T vectors (Promega). Site-directed mutagenesis was performed to generate recombinant constructs with the qQ and Qq combinations for the two QTN sites via overlap extension PCR using the pGEM-T-VRTN clone as a template. Four QTN allelic forms (QQ, qq, Qq and qQ) of 2,173 bp fragments were subcloned into promoterless luciferase pGL4.20 reporter vectors (Promega) to generate VRTN-Luc plasmids for the subsequent luciferase reporter assay.
To detect the activity of luciferase reporter gene controlled by the VRTN promoter, VRTN-Luc and pRL-tk constructs were co-transfected into HEK293T cells (2×105 cells/well) grown to 90% confluence using Lipofectamine 2000 (Invitrogen) in 24-well plates. Twenty-four hours after transfection, the activity of firefly luciferase was measured using the Dual-Luciferase Reporter Assay System (Promega) on an Infinite 200 PRO multimode reader (Tecan). Relative luciferase activity was determined based on the firefly/Renilla luciferase activity ratio. Each reporter plasmid was transfected in triplicate, and three independent reporter assays were performed.
To verify that VRTN is involved in the Notch signaling pathway, a NOTCH2-responsive reporter, HES1-Luc, derived from human and pig genomes was constructed as described by Solecki et al. [13]. The NOTCH2-responsive HES1-Luc and pRL-tk constructs were co-transfected into PK-15 cells (1×105 cells/well) containing empty or VRTN-overexpression vectors [13]. The reporter activity of HES1-Luc was evaluated as described above.
Digital droplet PCR
Total RNA was isolated from mouse C57BL/6J embryos at 5.5-12.5 dpc using TRIzol reagent (Invitrogen) according to manufacturer's instructions. One μg of total RNA was used for cDNA synthesis with random hexamers (TaKaRa). Digital droplet PCR (ddPCR) was used to assess Vrtn expression levels because Vrtn mRNA can be faint and hard to measure using routine quantitative real-time PCR (qPCR) techniques. The ddPCR reaction contained 1× ddPCR Supermix for probes (Bio-Rad), 50 nM Vrtn Taqman probe (ABI), 4 μl cDNA and double-distilled H2O. The experiment was conducted as previously described [14]. Briefly, the ddPCR reaction mix was partitioned into 20,000 nanoliter-sized droplets using a QX200 droplet generator (Bio-Rad). Then, the emulsions were transferred to a 96-well PCR plate and sealed with foil. The ddPCR was performed in a T100 Thermal Cycler (Bio-Rad) with a ramp rate of 2.5 °C/s between all steps. The final PCR product was detected in a QX200 droplet reader (Bio-Rad), and the results were analyzed using QuantaSoft software Version 1.4 (Bio-Rad). Reactions were run in three independent experiments. The primer and probe sequences used in this study are given in Table S1.
Production of VRTN QTN-LacZ transgenic mice
A LacZ transgenic mouse assay was performed as reported in the VISTA Enhancer Browser database [15]. Briefly, two copies of the Q allelic form of 291 bp at the VRTN QTN site (g.20311_20312ins291) were subcloned into upstream regions of a LacZ reporter gene using the Gateway Recombination System (Invitrogen). The VRTN QTN-LacZ plasmids were linearized and then injected into pronuclear C57BL/6 embryos. At 9.5 dpc, embryos were harvested and dissected in cold PBS, followed by LacZ staining. Yolk sacs were dissected from embryos and genotyped via PCR using primers listed in Table S1.
Real-time quantitative PCR
Total RNA was isolated from pig embryos harvested at 17.5 dpc using TRIzol (Invitrogen) and reverse transcribed with random hexamers (TaKaRa). cDNA (5 µl) was pre-amplified using the Taqman PreAmp Master Mix (ABI), primers (50 nM each) and VRTN or NOTCH2 probes. The PCR parameters were as follows: 95 °C for 10 min, followed by 14 cycles of 95 °C for 15 s and 60 °C for 2 min. The qPCRs were conducted in a total volume of 10 µl, containing 5 µl Taqman Master Mix (ABI), 2.5 µl cDNA, 0.5 µl Taqman probe and the primers and 2 µl distilled water. The cycling conditions were as follows: 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. All PCRs were performed on a 7900HT RT-qPCR system (ABI). BACT or 18S was used to compare the relative levels of mRNA. Primer and probe sequences are given in Table S1.
Immunofluorescence staining
To collect VRTN mutant (QQ) and wild-type (qq) pig embryos at the thoracic somite stage, we slaughtered two heterozygous Qq Large White sows at 17.5 days after mating with two Large White Qq boars. Embryos were harvested from the uterus with PBS containing 1% BSA and stored in liquid nitrogen for RNA extraction. The extra-embryonic tissues of each embryo were collected for DNA extraction and genotyping of the VRTN QTNs via Sanger sequencing using the primers listed in Table S1. Pig embryos were sectioned on a freezing microtome (Leica). Frozen sections of these embryos were incubated with anti-VRTN rabbit antibody (Sigma) at dilutions of 1:200 at 4 °C overnight. For negative controls, the antibody was replaced with normal non-immune serum. After extensive washing, these samples were incubated with anti-rabbit IgG-FITC antibody (Sigma) and DAPI. Fluorescence images were viewed and assessed with a TCS SP8 confocal microscope (Leica).
Subcellular localization
To determine the subcellular localization of pig VRTN, the full-length coding sequence of pig VRTN was artificially synthesized (Genescript) and then subcloned into pEGFP-C1 vectors (Addgene). The pEGFP-C1-VRTN vectors were transiently transfected into PK-15 cells using Lipofectamine 2000 transfection reagent (Invitrogen) following the manufacturer's instructions. After 24 hours, cells were fixed and stained with DAPI according to a standard protocol [16]. Fluorescence images were viewed and assessed with a TCS SP5 confocal microscope (Leica).
Fluorescence recovery after photobleaching (FRAP)
PK-15 cells were transfected with pEGFP-C1 or pEGFP-C1-VRTN vectors as described above. At 24 hours post-transfection, cells were re-seeded onto glass-bottomed dishes and then cultured in a live-cell imaging chamber for a FRAP assay. The assay was conducted using a TCS SP5 confocal microscope equipped with a 63/1.4 NA oil immersion objective (Leica). The 488-nm laser lines from an argon ion laser bleached the fluorescein of the complex in a defined rectangular strip of the nucleus. Fluorescence images were captured before and after the FRAP experiment to determine the photobleaching and recovery levels. The mean fluorescence intensity values of the strips were analyzed at each time point with SP5 software (Leica). Background intensities at each time point were subtracted from each image before analysis. To allow for the direct comparisons of individual FRAP curves, the raw data were normalized to the mean pre-bleach intensity of each cellular measurement.
Expression chip analysis
The full-length coding sequence of pig VRTN was subcloned into pNTAP-2×flag (kindly provided by Kai Lei at Fudan University, China) expression vectors. HEK293T cells were transfected with pNTAP-2×flag-VRTN or empty vectors. Total RNA was isolated from cells 24 hours after transfection using TRIzol reagent (Invitrogen). A total of 2 µg RNA was labeled with reagents from Affymetrix according to the manufacturer's instructions. Hybridization cocktails containing 5-5.5 µg of fragmented, end-labeled single-stranded cDNA were prepared and hybridized to GeneChip Human Exon 1.0 ST arrays (Affymetrix). The arrays contained approximately 5.4 million 5-µm features (probes) grouped into 1.4 million probe sets, interrogating over 1 million exon clusters. Processed arrays were scanned using the GeneChip Scanner 3000 7G system (Affymetrix). Affymetrix Expression Console Software (version 1.0) was used to perform the quality assessments. A random variance model (RVM) t-test was applied to discriminate DEGs between the treatment and control groups. The RVM t-test can effectively raise the degrees of freedom in the case of a small sample size [17]. After a false-discovery rate (FDR) analysis, we selected DEGs only in cases with a P-value <0.05.
ChIP sequencing
HEK293T cells were transfected with pNTAP-2×flag-VRTN vectors as described above. ChIP samples were prepared from VRTN-overexpressing HEK293T cells using the SimpleChIP Enzymatic Chromatin IP kit (CST) according to the manufacturer's instructions. The cells were harvested and chemically cross-linked with 1% formaldehyde for 10 minutes at 16-20°C. The cell nuclei were released via sonication using an Ultrasonic Processor (SONXI). The collected nuclei were digested with micrococcal nuclease (CST) for 20 minutes at 37 °C. Sheared chromatin was incubated with Anti-FLAG M2 Affinity Gel (Sigma) for 4 hours. Immunoprecipitated DNA was treated with RNase A (CST) and purified with the SimpleChIP kit (CST). ChIP sequencing was performed at Beijing Institute BGI (Wuhan, China). Briefly, a sequencing library was constructed from enriched DNA fragments using the TruSeq ChIP Library Prep kit (Illumina). Final DNA libraries were validated and sequenced at 50 bases per sequence read using a HiSeq 2000 sequencer (Illumina) at a depth of approximately 20 million sequences.
ChIP-seq reads were mapped to the human genome (GRCh38.p5; http://asia.ensembl.org/Homo_sapiens/Info/Index) using BEDtools [18]. Only the alignments with fewer than two mismatches were considered for enriched-region (peak) identification (peak calling) using the Model-based Analysis of ChIP-seq (MACS) algorithm [19]. MEME 4.6.1 software was used to identify the VRTN binding motif [20]. PeakAnalyzer software was used for peak annotation and the analysis of the nearest downstream gene [21]. 'Peak proximity scores' were calculated for the DEGs identified in the expression chip analysis using an algorithm as previously described [22]. If a DEG had a peak proximity score >10, it was considered a direct target of VRTN.
TALEN-mediated Vrtn knockout mice
TALEN repeats were designed to bind the exon 1 of the Vrtn gene as depicted in Figure S1 and then linked into pcDNA6.0 vectors (Invitrogen) to obtain in vitro transcribed mRNA. The transcripts were generated using the T7 Message Max kit and a polyA tailing kit (Epicentre) following the manufacturer's instructions. Washed and dried RNA was dissolved in water, and 4 ng TALEN mRNA was microinjected into the male pronuclei of C57BL/6 mouse zygotes. Injected eggs were then transferred to pseudopregnant females at 0.5 dpc. A total of 316 injected embryos were transferred into 14 pseudo-pregnant mice, which gave birth to 37 offspring. DNA extracted from tail samples of newborn mice was amplified using the primers listed in Table S1, and the PCR products were sequenced to identify heterozygous or knockout descendants. Only three mice carried the induced loss-of-function mutations at the TALEN targeting site in one allele (Figure S1), two of which were used to generate Vrtn -null mouse strains.
Morphological analysis of Vrtn-null and heterozygous mice
Embryos at 9.5 dpc were harvested to observe the thoracic somites of Vrtn -null mice. Whole embryos were fixed with 10% formalin and observed using light microscopy (Nikon). The yolk sac of each embryo was collected for DNA extraction and genotyping of the TALEN targeting site via Sanger sequencing using the primers listed in Table S1. Skeletal analyses were performed for five-month-age wild-type and heterozygous mice using the alcian blue/alizarin red staining method as previously described [23].
RNA sequencing
Total RNA was isolated from three wild-type (qq) and three mutant (QQ) pig embryos at 17.5 dpc, which was then used to construct cDNA libraries using the TruSeq RNA Sample Preparation kit (Illumina). Single-end reads (50SE) were generated on a HiSeq 2000 sequencer (Illumina) at a depth of approximately 20 million sequences. Adapter contaminated reads, reads of more than half of bases with Sanger quality smaller than 10, and reads with more than 5% of 'N' and less than 30 bp were removed. Next, trimmed, clean reads were mapped to the pig reference genome (Sscrofa 10.2; http://asia.ensembl.org/Sus_scrofa/Info/Annotation/) using the SOAP2 software and allowing two mismatches [24]. Gene expression levels were further calculated using the reads per kilobase of transcript per million mapped reads (RPKM) method to identify DEGs between the wild-type and mutant RNA samples as previously described [25]. A FDR of no more than 0.001 (FDR ≤ 0.001) and a difference ratio of the RPKM between the wild-type and mutant samples of no less than 1 (|log2 ratio| ≥ 1) were adopted as the significance thresholds to identify DEGs. A GO functional enrichment analysis was performed using Blast2GO [26]. KEGG pathway analyses of the DEGs were performed using Cytoscape software (http://www.cytoscape.org/).
Results
VRTN is the causative gene determining TVN on SSC7
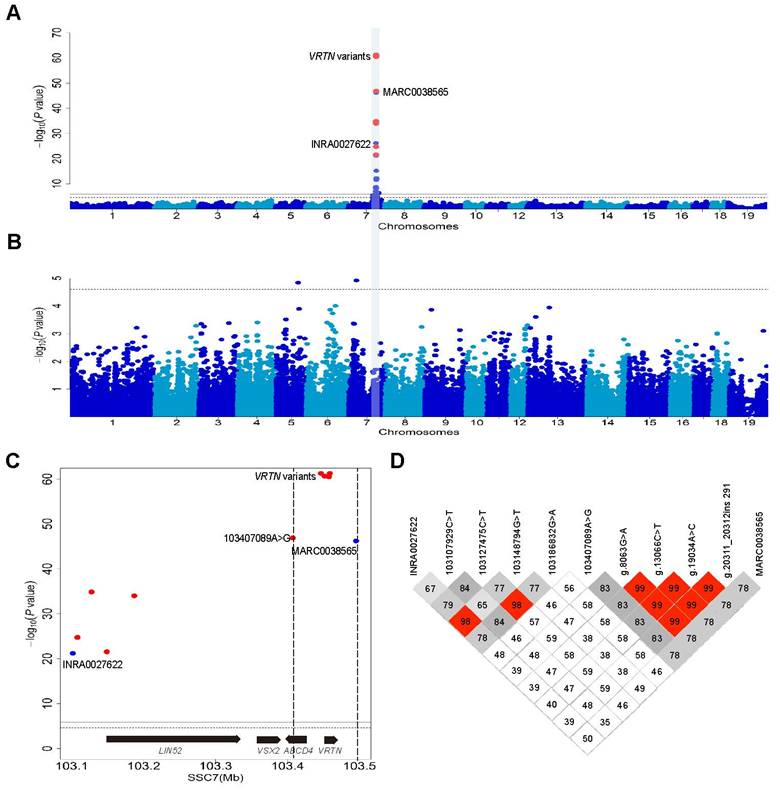
We recorded TVN in 609 European DLY hybrid pigs that were genotyped for 39,367 informative SNPs on an Illumina Porcine 60K chip. We conducted a genome-wide association study (GWAS) and identified a genome-wide significant QTL for TVN that was associated with the top SNP (MARC0038565) at 103.5 Mb on SSC7 (Figure S2). One allele at this locus increased TVN by 0.5. The 95% confidence interval for the major QTL was defined within a 400 kb segment containing the VRTN gene (Figure S2). This result is consistent with previous studies [7, 9, 27-30]. To refine the location of the QTL, we increased the SNP density from 2 to 11 in the 400 kb region and conducted an association analysis in the DLY population. Four variants in the VRTN gene, including the two previously identified candidate QTNs, showed the strongest association signal (P = 5.79×10-62, Figure 1A) and explained 68.7% of phenotypic variance in this DLY population. When the effect of the VRTN variants was removed from the analysis, no genome-wise significant association signal was observed for TVN across the genome (Figure 1B). The logarithm of the odds ratio (LOD) values (association strength) of the VRTN variants were much (>14) greater than those of their flanking SNPs within an interval of 88 kb that contains only one intact gene, VRTN (Figure 1C). These four VRTN variants defined a single haplotype (r2 > 0.98, Figure 1D). These observations strongly support that VRTN is the gene responsible for the QTL on SSC7 that affects TVN.
Genome-wide association study (GWAS) mapping supports VRTN as one of the genes responsible for the number of thoracic vertebrae. (A) Manhattan plots of a GWAS for the number of thoracic vertebrae in 609 European hybrid pigs. Negative log10 P-values of the qualified SNPs were plotted against their genomic positions. Different colors indicate distinct chromosomes. The red dots represent the nine variants including four VRTN variants within a region of 400 kb that were not included in Illumina Porcine 60K Beadchips, and the top SNP (MARC0038565) identified in the GWAS using the Beadchip is indicated. (B) When the results were conditional on the effect of the VRTN candidate QTN, no other SNPs on chromosome 7 showed an association signal. The solid and dashed lines indicate the 5% genome-wide and chromosome-wide Bonferroni-corrected thresholds, respectively. (C) Regional association plot for the number of thoracic vertebrae within the 95% confidence interval of the QTL on chromosome 7. The vertical dashed lines delineate the boundary region of the top GWAS SNPs, which are four VRTN variants. The two flanking markers (MARC0038565 and 103407089A>G) within an interval of 88 kb have a much lower association strength (LOD less than 14) values than the VRTN variants. The genes encoded on this part of the chromosome are indicated. (D) A haplotype view of all polymorphisms shown in panel C. The top four significant SNPs including g.8063G>A, g.13066C>T, g.19034A>C and g.20311_20312ins291 in the VRTN gene define a single haplotype block (r2 > 0.98).
Two causative mutations additively affect the expression of VRTN
Using a series of genetic analyses, we have previously identified two candidate QTNs in the pig VRTN gene that are associated with an increase in TVN [7]. The two variants are in nearly complete linkage disequilibrium in diverse populations [7] and were the most significantly associated markers in our previous studies [7, 10] and this current GWAS. One of the two variants is a SNP (g.19034A>C) in the promoter region, whereas the other (g.20311_20312ins291) is an indel in intron 1 of VRTN. The SNP is 105 bp upstream and the indel 1,169 bp downstream of the transcription start site, and both reside in conserved functional elements [7]. This led us to speculate that the two candidate QTNs affect the expression of VRTN.
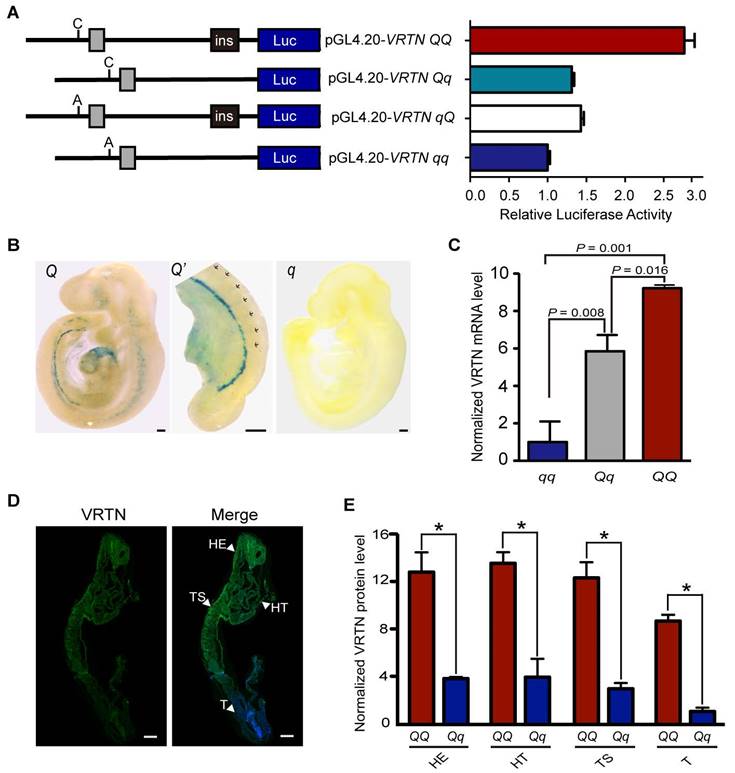
To test this hypothesis, we performed a dual luciferase reporter assay in HEK293T cells. We first cloned both allelic forms (the double mutant, C/ins, denoted QQ; and wild-type, A/-, denoted qq) of a 2,173 bp and 1,882 bp fragment containing the two candidate QTNs into the pGL4.20 luciferase vector. Then, we generated two recombinant constructs (Qq and qQ) to determine the relative contribution of the two candidate QTNs (Figure 2A). We transfected the four constructs into HEK293T cells and measured the luciferase reporter activity after 24 hours. Compared with wild type, the two individual mutant alleles were associated with a similar increase (approximately 1.5-fold) in luciferase activity, and when combined, they were associated with a three-fold increase in luciferase activity (Figure 2A). This indicates that the two candidate QTNs are functional variants, and that they additively affect the expression of VRTN.
Two candidate causative variants additively affect the expression of VRTN. (A) Luciferase activity driven by four pig promoter sequences in human HEK293T cells. The schematic diagram (left panel) illustrates the sequence of each construct, and the right panel shows the relative luciferase activity of each construct. Ins indicates the ins allele at the g.20311_20312ins291 mutation site. C or A represents the C or A allele at the g.19034A>C mutation site. The relative luciferase activity (firefly luciferase light units/Renilla luciferase light units) is represented as the mean ± standard error (S.E) of triplicate experiments. (B) The VRTN candidate causative mutations direct the expression of LacZ reporter in transgenic mouse embryos at E9.5. Two copies of 291 bp fragment corresponding to the Q allelic form at the candidate QTN site (g.20311_20312ins291) were cloned into LacZ reporter plasmids (HSP68-lacZ), which were then used to generate Q mice. Lateral views of representative X-gal-stained transgenic (Q) and control (q) embryos at 9.5 dpc are shown with sections of stained tissues. One enlarged areas (Q') correspond to the region indicated by dashed boxes in the left panel. Three independent transgenic lines displayed virtually identical expression patterns (Fig. S6). (C) mRNA expression levels of VRTN in mutant (QQ, n = 3), heterozygous (Qq, n = 18), and wild-type (qq, n = 7) pig embryos at E17.5. Values were determined via RT-qPCR and are expressed as the mean ± S.E. of the triplicate experiments after normalization to 18S mRNA levels. The expression level of the qq samples was set as 1. Significance of difference between each paired group was determined by student's t-test. A P value of smaller than 0.05 was considered statistically significant. (D) VRTN expression in whole pig embryos at E17.5. Whole mount immunohistochemistry was performed with an anti-VRTN antibody. VRTN expression is shown in green and nuclei in blue. The full names of the abbreviations used by the arrowheads are as follows: HE, head; HT, heart; T, tail; TS, thoracic somites. Embryos were all imaged via confocal microscopy at the same magnification. Scale bar: 100 µm. (E) Quantitative analysis of relative VRTN immunostaining intensities in the head (HE), thoracic somite (TS), heart (HT) and tail (T, presomitic mesoderm) regions of the mutant (QQ) and heterozygous (Qq) pig embryos at E17.5. The data represent the fold increase (mean ± S.E. of triplicate experiments) relative to the staining intensity in the tails of Qq embryos.*, P < 0.05 as determined by student's t-test.
We also cloned two fragments - one 194 bp and the other 666 bp, each containing one of the other notable VRTN SNPs identified in the GWAS (g.8063G>A or g.13066C>T) - into the upstream region of the wild-type VRTN promoter in the reporter plasmid. No obvious difference in luciferase activity was observed between the two SNPs and wild type (Figure S3), indicating that these SNPs are not functional variants.
VRTN is conserved in mammals (Figure S4, Figure S5). It is much more time-consuming and costly to conduct experiments using pig embryos than mouse embryos at multiple time points. To evaluate the potential role of VRTN in the embryonic development of thoracic vertebrae, we first quantified mRNA transcripts of Vrtn in mice (C57BL/6) over 8 time points from day 5.5 to day 12.5 post coitum, i.e. from blastocyst implantation to late somite formation, via droplet digital PCR. We found that Vrtn was highly expressed from 7.5 dpc to 9.5 dpc (Theiler stage 15, 21-29 somites) (Figure S6). This phase exactly corresponds to the thoracic-somite stage (11-24 somites) in the mouse, indicating that Vrtn functions at the critical developmental stage of the formation of thoracic somites. Next, we examined whether the two QTNs up-regulated the expression of Vrtn at this embryonic stage (9.5 dpc). For this, we generated VRTN QTN-LacZ transgenic mice as previously described [15]. To mimic the additive effect of the two candidate QTNs, we cloned two copies of a 291-bp fragment corresponding to the mutant allele at the QTN site (g.20311_20312ins291) into an upstream region of a LacZ reporter gene. The reporter constructs were linearized and injected into fertilized mouse oocytes. At 9.5 dpc, the embryos were stained to visualize LacZ reporter gene activity. We noted that LacZ expression was clearly present in the thoracic somatic portion, the hindbrain/cervical somatic region, the caudal intermediate mesoderm and the developing head and heart in transgenic embryos but was virtually absent in control embryos injected with the empty vector (Figure 2B, Figure S7). These observations suggest that the two QTNs act as an enhancer to up-regulate the expression of Vrtn in structures along the anterior-posterior (AP) axis for the formation of somites and in regions involved in the development of other organs like heart and head.
To obtain direct evidence for the effect of the two QTNs on VRTN expression in pigs, we measured the quantity of VRTN mRNA in full-sib pig embryos representing three QTN genotypes (QQ, Qq and qq) at 17.5 dpc. VRTN transcript levels were increased by over 9-fold in QQ embryos and 5-fold in Qq embryos relative to the levels in the qq embryos (Figure 2C). It is known that the formation of somites along the AP axis is under the control of a set of cyclic genes expressed within the paraxial mesoderm during early embryogenesis [31]. To test whether VRTN is also expressed in the paraxial mesoderm, we investigated the location of pig 17.5 dpc embryos using immunofluorescence staining of an anti-VRTN antibody. We detected VRTN in the paraxial mesoderm where somites are formed, while the staining was relatively ubiquitous in these embryos. The expression level of VRTN tended to gradually decrease along the AP axis. Staining was strong in the cranial and cardiac regions and was weak in the tail region (Figure 2D). We also quantified the relative protein levels of VRTN in frozen sections of pig embryos at this stage. A pattern similar to that of VRTN mRNA expression was observed, i.e., a higher level of fluorescence intensity was detected in the thoracic somite, head, heart and tail (PSM) regions of QQ embryos in comparison with Qq embryos (Figure 2E). Thus, we conclude that the mutant alleles at the two QTN sites increase the expression of VRTN at both the transcription and translation levels in pigs.
VRTN is a novel transcription factor
To characterize the molecular features of VRTN, we first determined the subcellular location of this protein within cells. The C-terminal region has been predicted to contain three nuclear localization signals via SeqNLS analysis [32], indicating that VRTN probably localizes to the nucleus. To test this possibility, we constructed a green fluorescent protein (GFP)-VRTN fusion cassette and then conducted transient transfection assays in pig kidney (PK-15) cells. We found that the GFP-VRTN fusion protein localized exclusively to the nucleus (Figure 3A). To determine whether VRTN binds to DNA, we next examined the nuclear diffusional mobility of GFP-VRTN in pig PK-15 cells using confocal fluorescence recovery after photobleaching. We found that GFP-VRTN exhibited a slower rate of diffusion compared with that of GFP alone (Figure 3B), suggesting that VRTN is a DNA-binding protein.
VRTN is a novel transcription factor. (A) VRTN localizes to the nucleus. PK-15 cells were transfected with pEGFP-C1-VRTN plasmids. Subcellular localization of VRTN was determined via GFP fluorescence at 24 hours post-transfection. Cell nuclei were counterstained with DAPI. Scale bar: 10 µm. (B) VRTN binds to DNA. PK-15 cells expressing GFP or the GFP-VRTN fusion protein were imaged before and during recovery after briefly photobleaching the fluorescence in a small strip spanning the nucleus. The recovery rate of fluorescence was recorded over time. The GFP-VRTN showed a slower recovery of fluorescence compared with GFP alone. (C) Genomic distribution of VRTN binding peaks identified via ChIP sequencing. The schematic diagram (lower panel) shows the definitions of the intergenic, upstream, promoter, exon and intron regions. TSS, transcription start site. (D) VRTN binding motif revealed by an enrichment analysis of ChIP sequencing. (E) VRTN regulates the expression of target genes. VRTN peak proximity scores for each of 514 differentially expressed genes (DEGs) were calculated and plotted together with the log2 values of fold changes in DEGs. Blue dots indicate negative and the red dots positive significant fold changes. The horizontal dashed line at the peak proximity score of 10 represents the chosen cut-off for defining a gene likely to be a direct target of VRTN.
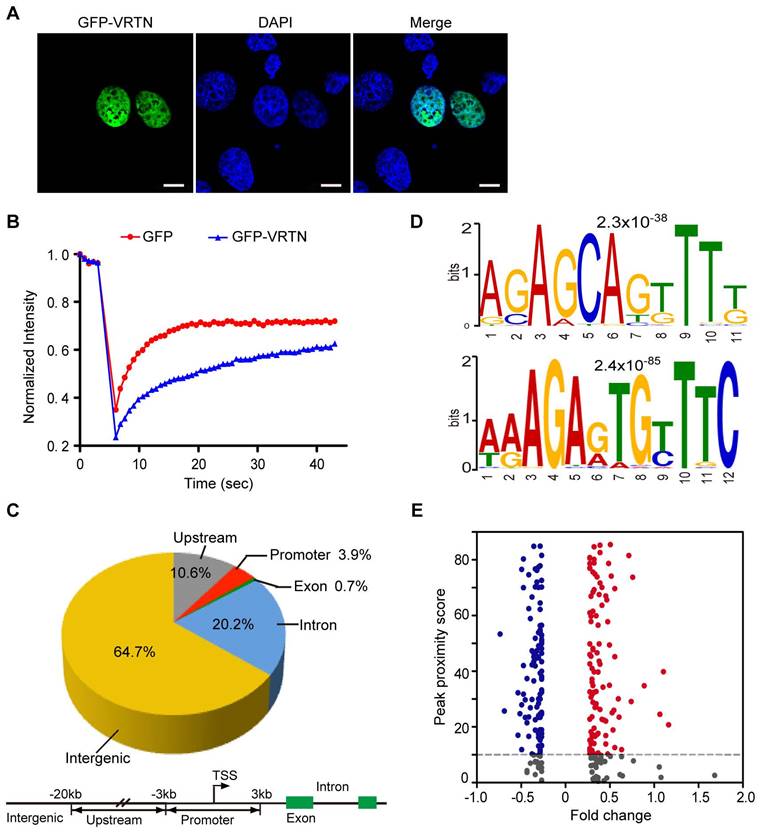
To uncover the binding sites of VRTN across the genome, we then performed a chromatin immunoprecipitation (ChIP) sequencing assay using anti-FLAG agarose beads. Considering that an anti-VRTN ChIP-grade antibody is not currently available and the annotation of the pig reference genome (Sscrofa 10.2) is far from perfect, we constructed a FLAG-pig-VRTN fusion cassette and then transfected them into human embryonic kidney 293T (HEK 293T) cells. The anti-FLAG immunoprecipitation system allowed us to effectively pull down VRTN bound to various DNA fragments. An Illumina HiSeq 2500 system was used to sequence the ChIP-enriched DNA fragments, resulting in 31,633,260 reads that mapped uniquely to the human genome (GRCh38.p5). An in-depth analysis of these data revealed 1,730 peak signals with a minimum of 50 overlapping extended reads (Table S2). We further determined the locations of these reads relative to the transcriptional start sites (TSSs) of the annotated genes. Only 0.7% and 3.9% of the peak signals were located in exons and proximal promoters, respectively, within 3 kb from the TSSs, whereas most enriched reads were found in upstream, introns and intergenic regions (Figure 3C). To test whether human VRTN binding sites share conserved primary sequence patterns, we evaluated VRTN binding features on a genome-wide scale using a MEME analysis [33]. Three distinct motifs were identified in the peak regions, two of which comprise a strictly defined sequence element (Figure 3D). This is in accordance with the assumption that VRTN is a DNA-binding transcription factor.
To further investigate whether VRTN is a transcription factor, we examined the relationship between differentially expressed genes (DEGs) and VRTN binding signals as previously described [22, 34]. We measured global transcriptomic changes in human HEK293T cells after treating them with VRTN using whole-genome microarrays containing 28,869 oligonucleotide probes. In total, 514 genes notably differed in expression levels (with a false discovery rate of less than 5%) between the VRTN-treated cells and the untreated cells (Table S3). An integrative analysis of the ChIP-seq and RNA-chip data revealed that 214 (41.6%) of the DEGs contained strong proximal VRTN binding sites (peak proximity score > 10), suggesting that VRTN directly regulates the expression of specific target genes (Figure 3E). Taken together, our findings indicate that VRTN is a novel transcription factor. It should be mentioned that our ChIP-seq and RNA-chip studies were conducted in HEK293T cells, an immortalized human embryonic kidney cell. Transcription factors always bind to DNA in a site-specific manner, and the binding sites can vary considerably between different cells [35]. Hence, the array of VRTN binding sites and the targets of VRTN may change in different pig embryonic cells.
Vrtn is required for thoracic somitogenesis in mice
To further uncover the role of VRTN in determining TVN, we generated Vrtn-knockout mice using TAL-effector endonuclease (TALEN) technology (see Materials and Methods). We intercrossed the heterozygous (Vrtn+/-) mice and generated 101 two-week-old mice (Table 1). According to the Mendelian law, the expected number of Vrtn-deficient homozygous (Vrtn-/-) animals in the progeny of this intercross was 25. However, no null mutant survived to this stage (Table 1). The ~2:1 ratio of Vrtn+/- to Vrtn+/+ progeny strongly indicates that homozygous Vrtn deficiency in mice results in embryonic lethality.
Two-week-old offspring of intercrosses between Vrtn+/- mice
| No. of offspring of genotype | ||||
|---|---|---|---|---|
| +/+ | +/- | -/- | Total | |
| Predicted | 25 | 50 | 25 | 100 |
| Observed | 35 | 66 | 0 | 101 |
We further examined the embryonic stage at which Vrtn-/- embryos discontinue the development. For this, we collected embryos at different developmental points (9.5-12.5 dpc) from an intercross between Vrtn+/- individuals. We observed a normal Mendelian distribution of Vrtn+/+, Vrtn+/- and Vrtn-/- embryos (Table S4) at 9.5 and 10.5 dpc. However, Vrtn-/- embryos were not viable at 11.5 dpc (Figure S8), indicating that Vrtn-/- embryos cease to develop between E10.5 and E11.5.
We noticed that Vrtn-null embryos displayed developmental retardation since 9.5 dpc. At E9.5, Vrtn+/+ and Vrtn+/- embryos reached Theiler stage 14-15 (3 branchial bars, 13-29 somites), while null mutants arrested at Theiler stage 13 (2 arches, 8-12 somites) (Figure 4A). All Vrtn-/- embryos did not show axial rotation, which is normally completed in embryos at ~E9.0 (Theiler stage 14). At 10.5 dpc, null mutants still fail to turn the embryo (Figure 4A). In additional to the developmental arrest at Theiler stage 13, Vrtn-/- embryo at 9.5 and 10.5 dpc showed morphological defects in heart and neural tubes (Figure 4B). However, all Vrtn-/- embryo at 8.5 dpc displayed normal developmental characteristics without gross defects (data not shown). These results indicate that Vrtn is essential for the development of mice embryo at Theiler stage 14 (13-20 somite), corresponding to the developmental stage of thoracic somites.
Vrtn is essential for the development of thoracic veterbrae in mice. (A) At 9.5 and 10.5 dpc, Vrtn-null embryos (Vrtn-/-, right) cannot complete axial rotation and have 8-12 somites. Embryos were all imaged at the same magnification (4×). Scale bar: 100 µm. (B) Compared with the Vrtn wild-type (Vrtn+/+, left) individual, Vrtn-/- mouse embryos at 9.5 dpc (middle and right) show thoracic somite defects (abnormal and fewer (less than 13) somite segments) and delays in axial turning and neural tube closure. (C) Compared with Vrtn+/+ mice (left), some Vrtn+/- animals had one less thoracic vertebrae (middle) or were missing a rib from the last thoracic element on one side (right). The positions of the first (R1) and last (R12 or R13) rib are labeled. (D) Comparison of the number of thoracic vertebrae and ribs between Vrtn+/+ and Vrtn+/- mice (n = 26). NTV, number of thoracic vertebrae. Data are expressed as means ± standard deviations. Student t-test was used to evaluate phenotypic difference between the two groups. A P value of smaller than 0.05 was considered statistically significant. (E) Phenotypic variation in the number of thoracic vertebrae and ribs in Vrtn+/- mice (n = 26). 12/12, 12 thoracic vertebrae with 12 pairs of ribs; 13/12, 13 thoracic vertebrae with a rib missing from the last thoracic element on one side; 13/13, 13 thoracic vertebrae with 13 pairs of ribs.
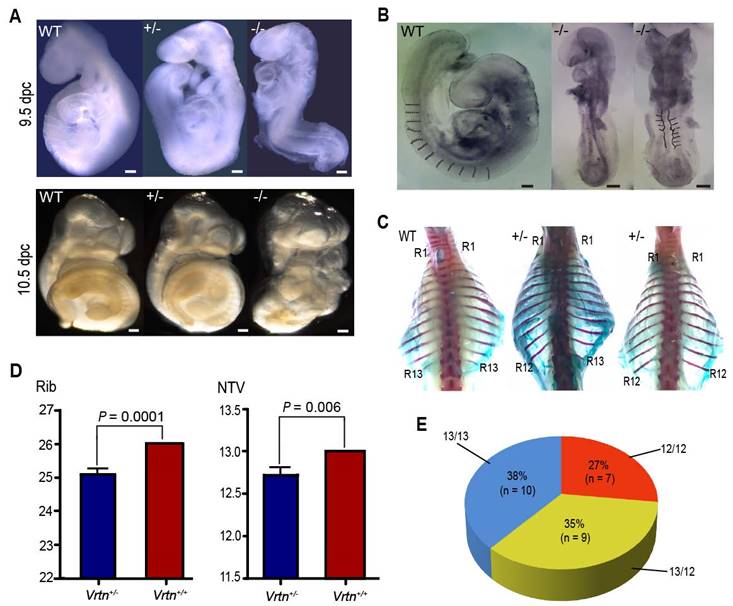
Of note, more than half (16/26) of the Vrtn+/- animals exhibited abnormal spinal development, with prominent kyphosis at 2-months old - although they did survive, and they were generally healthy. We examined the complete skeletons of 26 adult Vrtn+/- mice (Figure S9 and Figure 4C). Vrtn heterozygous mice have a statistically significant difference in the number of thoracic vertebrate (P = 0.006) and ribs (P = 0.0001) compared with wild-type (Vrtn+/+) mice (Figure 4D). Of the 26 Vrtn+/- mice, approximate one-fourth of animals (7/26) had one less thoracic vertebrae and one-third (9/26) of animals were missing a rib from the last thoracic element on one side (randomly on the right or left), and the remaining individuals had normal TVNs and ribs (Figure 4E).
In summary, these findings show that (i) Vrtn is highly expressed at Theiler stage 11-15 (7.5-9.5 dpc), which includes the thoracic-somite stage (Theiler stage 14, ~9.0 dpc), (ii) Vrtn-deficient embryos have fewer (less than 13) somites, cannot complete axial rotation and cease to develop over Theiler stage 13 (8.5 dpc), (iii) more than half of adult Vrtn+/- animals show abnormal spinal development and (iv) Vrtn+/- mice have significantly fewer thoracic vertebra and ribs than wild type animals. These findings provide the first biological evidence that Vrtn is indispensable for the formation of thoracic somites and, hence, for the determination of TVN.
The possible molecular mechanism for the regulation of TVN by VRTN
Given that VRTN is a transcription factor, we hypothesized that VRTN regulates the TVN by modulating the transcript abundance of its downstream targets, including well-known genes for the formation of somites.
To test this hypothesis, we quantified the transcriptomes of six full-sib pig embryos at 17.5 dpc that corresponds to 9.5 dpc in mice, of which three were homozygous for the two QTNs (QQ) and three were wild type (qq). These embryos were harvested from a VRTN heterozygous Qq sow that was mated with a heterozygous Qq boar. Using RNA-seq techniques, we obtained over 20 million single-end clean reads of 50 bp from each of the six samples, approximately 78% and 65% of which unambiguously mapped to the genome reference assembly and the reference gene database (Sscrofa 10.2), respectively. We then compared the expression profiles of these genes between the mutant (QQ) and wild-type (qq) embryos. In total, 42 differentially expressed genes (DEGs) were identified as targets of VRTN, including 39 up-regulated and 3 down-regulated genes with a > 2-fold change at a false discovery rate of less than 0.001. These DEGs were enriched in genes involved in developmental processes (Table S5), and the top 20 KEGG-enriched pathways (Figure S10) included the Notch signaling pathway and body axis formation, which are closely related to segment number in vertebrate embryos. Of note, two of the DEGs - the genes encoding NOTCH2 and heparan sulfate proteoglycan 2 (HSPG2) - have been implicated in abnormal vertebral segmentation in human and animal models (Table S6) [36]. Intriguingly, NOTCH2 has a similar expression pattern to that of VRTN as it is expressed at a high level in prospective and recently formed somites and its expression gradually weakens throughout the PSM and primitive streak [37]. The activity of Notch molecules are known to be absolutely essential for the formation of the somites [38, 39]. Mice carrying loss-of-function mutations in any one of the genes encoding ligands, receptors or downstream effectors of Notch display severe segmentation defects in the thoracic and lumbar skeleton, with minimal effects observed in the sacral and caudal vertebrae [1, 40-42]. Therefore, we focused on the role of VRTN in the Notch signaling pathway in our subsequent analyses.
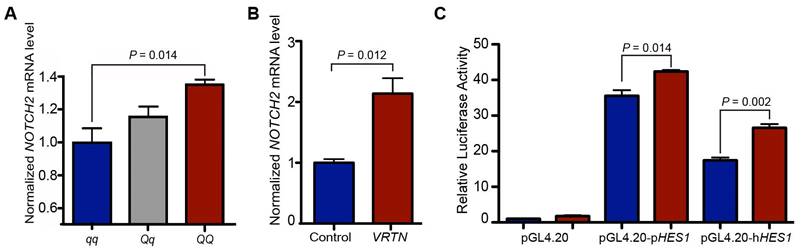
First, we measured the quantity of NOTCH2 mRNA in full-sib pig embryos (17.5 dpc) representing three VRTN QTN genotypes (QQ, Qq and qq) via RT-qPCR. Consistent with the RNA-seq results, NOTCH2 transcript levels were higher in the QQ embryos (P = 0.014) than in the qq embryos (Figure 5A). To provide direct evidence that VRTN regulates the expression of NOTCH2, we then conducted an overexpression assay by transfecting an expression vector harboring the full-length coding sequence of pig VRTN driven by a CMV promoter into pig PK-15 cells. The overexpression of VRTN led to a two-fold (P = 0.012) increase in NOTCH2 transcripts in the cells (Figure 5B). Next, we constructed two luciferase reporter plasmids with fragments derived from the promoters of the human and pig HES1 gene, a target gene of Notch [43]. The luciferase reporter plasmids with the HES1 promoter were cotransfected into pig PK-15 cells with the VRTN expression vector. The overexpression of VRTN resulted in significantly higher (P < 0.05) reporter activities compared with the controls (Figure 5C). Altogether, our data indicate that VRTN mediates the expression of NOTCH2 and, consequently, regulates its downstream genes in the Notch signaling pathway.
VRTN regulates the Notch pathway. (A) NOTCH2 mRNA levels in VRTN mutant (QQ, n = 3), heterozygous (Qq, n = 18) and wild-type (qq, n = 7) pig embryos at E17.5. Values were determined via RT-qPCR and are expressed as the mean ± standard error (S.E) of triplicate experiments after normalization to BACT mRNA levels. The expression level of the qq samples was set as 1. (B) VRTN up-regulates the expression of NOTCH2. VRTN expression and control vectors were transfected into pig PK-15 cells. Values were determined via RT-qPCR and are expressed as the mean ± S.E. of triplicate experiments after normalization to BACT mRNA levels. (C) Transcriptional activation of HES1 by VRTN. PK-15 cells were co-transfected with luciferase reporter plasmids driven by a pig (pGL4.20-pHES1) / human (pGL4.20-hHES1) promoter and VRTN expression plasmids (red) or control vectors (blue). The ratio of firefly luciferase to Renilla activity was defined as the relative luciferase activity. The data represent fold increases (mean ± S.E. of triplicate experiments) relative to empty vector-transfected values. Student t-test was performed to determine the significance of difference between each paired group. A P value of smaller than 0.05 was considered statistically significant.
It has been well established that the Notch signaling pathway regulates the segmentation clock, which is a molecular oscillator controlling the periodicity of somite formation [44-46]. A faster segmentation clock gives rise to a reduced periodicity of somitogenesis and more but shorter segments in mice [46]. Therefore, we expected that the increased NOTCH2 levels driven by the VRTN causative variants may regulate the gene expression that in the Notch signaling pathway, resulting in more but shorter somites in pig. During fetal development, each segment of the vertebral column is formed by fusing the posterior portion of one somite with the anterior portion of the adjacent somite. Therefore, we predicted that the total length of the thoracic vertebrae would increase by half the size of one thoracic vertebra when TVN was increased by one in VRTN mutant pigs compared to wild-type pigs. To test this prediction, we measured the number and total length of thoracic vertebrae in 304 DLY pigs. Compared with wild-type homozygotes (qq), the number of thoracic vertebrae was increased by one in homozygous mutant (QQ) animals, and the total length of the thoracic vertebrae in homozygous mutant DLY pigs was increased by 1.56 cm (P < 0.001 in both cases), which is nearly half (1.56/3.23) the size of one thoracic vertebra (Table 2). The findings indicate that a faster segmentation clock gives rise to a reduced periodicity of somitogenesis and more but shorter segments, which support our prediction that VRTN controls the number of vertebrae possibly by regulating segmentation clock via Notch signaling pathway.
Comparisons of thoracic vertebral parameters between VRTN mutant (QQ), heterozygous (Qq), and wild-type (qq) pigs
| QQ (n = 136) | Qq (n = 139) | qq (n = 29) | P-value | |
|---|---|---|---|---|
| Vertebral number | 21.62 ± 0.20a | 21.00 ± 0.05b | 20.57 ± 0.09c | 2.20E-16 |
| Vertebral length | 58.17 ± 1.27a | 57.20 ± 0.33b | 56.61 ± 0.56c | 2.20E-16 |
| Carcass length | 102.96 ± 0.37a | 101.25 ± 0.52b | 100.24 ± 0.88c | 4.93E-04 |
| 7th thoracic vertebral length | 3.19 ± 0.02 | 3.24 ± 0.02 | 3.32 ± 0.04 |
Note: Phenotypic values are shown as means ± standard deviations. One-way analysis of variance (ANOVA) was used to evaluate the significance of difference in phenotypic data among the three groups. Values with different superscripts in the same line are significantly different (P < 0.05).
Discussion
VRTN has been associated with variation in TVN and TVN-related traits, including body length and teat number in pigs [10], but the causal relation between VRTN and TVN remains to be established, and the developmental role of VRTN in the formation of thoracic vertebrae were previously unknown. Here we provide further genetic evidence that VRTN is the causative gene for the QTL on SSC7 affecting TVN, and that g.19034A>C and g.20311_20312ins291 are the two causative mutations which additively modulate the transcript abundance of VRTN. Furthermore, we show that VRTN is a novel DNA-binding transcription factor as (i) VRTN localizes exclusively in the nucleus, (ii) VRTN binds to DNA on a genome-wide scale, and (iii) VRTN regulates the transcription of a set of genes that harbor VRTN binding motifs. Our study also establishes the critical role of VRTN in the development of thoracic vertebrae. (i) Vrtn temporarily expressed during a period (7.5-9.5 dpc) covering the thoracic somite stage (~9.0 dpc) in mice, (ii) Somitogenesis ceases at the thoracic somite stage in Vrtn-null embryos that discontinue axial turning and show a reduced number and abnormal shape of somites. (iii) Vrtn+/- mice have fewer thoracic vertebrae and ribs than wild-type mice and more than half of Vrtn+/- individuals show defects in spinal development. Together, these findings demonstrate that VRTN is an essential gene for the development of thoracic vertebrae in pigs and mice, and presumably in all mammals considering developmental constraint of the vertebral formula in these species [47].
The identity and number of thoracic vertebrae is determined when somites form along the embryonic axis via sequential segmentation from the PSM [42]. Hox genes are known to determine somatic segmental identity [48, 49]. Hoxc-4 and Hoxc-5 are both required to determine the proper identities of the second through tenth or eleventh thoracic vertebrae [50]. Hoxc8 gain-of-function mutation increases one more thoracic vertebrae in mice [51]. The upstream regulatory genes of these Hox genes also play a role in the segmentation of thoracic vertebrae, such as Cdxs, ActRIIB and ActRIIA [52-54]. Moreover, mutations impairing Notch, WNT and FGF signaling pathways cause abnormal thoracic somitogenesis [55-58]. Indeed, the Notch, WNT and FGF signaling gradients along PSM determine somitogenesis, which is known as a 'clock-and-wavefront' model in which the wavefront represents the anterior-to-posterior progression of the embryonic development [55, 59]. The two intersecting activities (the clock and the wavefront) precisely define the number of somites for a given species in a synchronized and rhythmic fashion [60]. When the balance between the segmentation clock rate and the embryonic development rate is altered, the number of somites varies [61]. If the clock rate is increased relative to the development rate, more somites of smaller size will develop in mice [46]. Interestingly, VRTN homozygous mutant (QQ) pigs typically have one more thoracic vertebra and are 1.5 cm longer than wide-type (qq) animals. This difference in length is approximately half the size of a normal vertebra, so each of the thoracic vertebrae in QQ animals is slightly shorter than thoracic vertebrae of qq animals. It thus raises the possibility that VRTN causative variants may alter the balance between the segmentation clock rate and the embryonic development rate. Here we show that VRTN QTN increases the expression of NOTCH2 and HES1, one of downstream cyclical gene of Notch pathway. The Notch mediates intercellular communication within the PSM through Notch-Delta signal transduction pathway, the activity of which couples neighboring cells to facilitate synchronized oscillation of the segmentation clock [58, 60, 62, 63]. Hence, we speculate that VRTN QTN could accelerate the segmentation clock via the notch signaling pathway, and consequently affect the number of thoracic somites and ribs as well, but it has no effect on embryonic development.
In summary, we show that VRTN is a transcription factor indispensable for the formation of thoracic vertebra in mammals. This finding provides insight into the mechanisms underlying the specification and determination of segment number in mammals. Moreover, the VRTN QTNs provide a robust tool to improve pork production by increasing TVN and body length.
Supplementary Material
Supplementary figures and tables.
Abbreviations
VRTN: pig vertnin; Vrtn: mouse vertnin; TVN: thoracic vertebral number; PSM: presomitic mesoderm; QTL: quantitative trait locus; QTN: quantitative trait nucleotide; GWAS: genome-wide association study; DLY: Duroc × (Landrace × Large White); SNP: single nucleotide polymorphism; LOD: logarithm of the odds ratio; PK-15: pig kidney cells; HEK 293T: human embryonic kidney 293T; PCR: Polymerase Chain Reaction; qPCR: quantitative PCR; ddPCR: digital droplet PCR; DAPI: 4',6-diamidino-2-phenylindole;FRAP: fluorescence recovery after photobleaching; RVM: random variance model; FDR: false-discovery rate; DEG: differentially expressed genes; ChIP: chromatin immunoprecipitation; MACS: model-based analysis of ChIP-seq; TALEN: transcription activator-like effector nuclease; RPKM: reads per kilo bases per million reads; GO: Gene ontology; KEGG: kyoto encyclopedia of genes and genomes; AP: anterior-posterior; TSSs: transcriptional start sites.
Acknowledgements
We acknowledge funding from the National Natural Science Foundation of China (31525023, 31560304 and 31671288) that supported this study.
Disclaimers
The views expressed in the submitted article are our own and not an official position of the institution or funder.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sparrow DB, Chapman G, Turnpenny PD, Dunwoodie SL. Disruption of the somitic molecular clock causes abnormal vertebral segmentation. Birth Defects Research. 2007;81:93-110
2. Narita Y, Kuratani S. Evolution of the vertebral formulae in mammals: a perspective on developmental constraints. Journal of Experimental Zoology Part B, Molecular and Developmental Evolution. 2005;304:91-106
3. Asher RJ, Lin KH, Kardjilov N, Hautier L. Variability and constraint in the mammalian vertebral column. Journal of Evolutionary Biology. 2011;24:1080-90
4. Zhang ZG LB, Chen XH. Pig breeds in China. Shanghai Scientific and Technical Publisher, Shang, China. 1986
5. Borchers N. RN, Kalm E. The number of ribs and vertebrae in a Pie´train cross: variation, heritability and effects on performance traits. Jouranl of Animal Breeding and Genetics. 2004;121:392-403
6. Rohrer GA, Nonneman DJ, Wiedmann RT, Schneider JF. A study of vertebra number in pigs confirms the association of vertnin and reveals additional QTL. BMC Genetics. 2015;16:129
7. Yin Fan, Yuyun Xing, Zhiyan Zhang, Huashui Ai, Zixuan Ouyang, Jing Ouyang. et al. A Further Look at Porcine Chromosome 7 Reveals VRTN Variants Associated with Vertebral Number in Chinese and Western Pigs. PloS One. 2013;8:e62534
8. Mikawa S, Morozumi T, Shimanuki S, Hayashi T, Uenishi H, Domukai M. et al. Fine mapping of a swine quantitative trait locus for number of vertebrae and analysis of an orphan nuclear receptor, germ cell nuclear factor (NR6A1). Genome Research. 2007;17:586-93
9. Mikawa S, Sato S, Nii M, Morozumi T, Yoshioka G, Imaeda N. et al. Identification of a second gene associated with variation in vertebral number in domestic pigs. BMC Genetics. 2011;12:5
10. Yang J, Huang L, Yang M, Fan Y, Li L, Fang S. et al. Possible introgression of the VRTN mutation increasing vertebral number, carcass length and teat number from Chinese pigs into European pigs. Scientific Reports. 2016;6:19240
11. Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294-6
12. Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263-5
13. Solecki DJ, Liu XL, Tomoda T, Fang Y, Hatten ME. Activated Notch2 signaling inhibits differentiation of cerebellar granule neuron precursors by maintaining proliferation. Neuron. 2001;31:557-68
14. Ludlow AT, Robin JD, Sayed M, Litterst CM, Shelton DN, Shay JW. et al. Quantitative telomerase enzyme activity determination using droplet digital PCR with single cell resolution. Nucleic Acids Research. 2014;42:e104
15. Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser-a database of tissue-specific human enhancers. Nucleic Acids Research. 2007;35:D88-92
16. von Knethen A, Tzieply N, Jennewein C, Brune B. Casein-kinase-II-dependent phosphorylation of PPARgamma provokes CRM1-mediated shuttling of PPARgamma from the nucleus to the cytosol. Journal of Cell Science. 2010;123:192-201
17. Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19:2448
18. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841-2
19. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biology. 2008;9:R137
20. Bailey TL, Johnson J, Grant CE, Noble WS. The MEME Suite. Nucleic Acids Research. 2015;43:W39-49
21. Salmon-Divon M, Dvinge H, Tammoja K, Bertone P. PeakAnalyzer: genome-wide annotation of chromatin binding and modification loci. BMC Bioinformatics. 2010;11:415
22. Norton L, Chen X, Fourcaudot M, Acharya NK, DeFronzo RA, Heikkinen S. The mechanisms of genome-wide target gene regulation by TCF7L2 in liver cells. Nucleic Acids Research. 2014;42:13646-61
23. Mallo M, Brandlin I. Segmental identity can change independently in the hindbrain and rhombencephalic neural crest. Developmental Dynamics. 1997;210:146-56
24. Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K. et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966-7
25. Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 2008;5:621-8
26. Gotz S, Garcia-Gomez JM, Terol J, Williams TD, Nagaraj SH, Nueda MJ. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Reseach. 2008;36:3420-35
27. Mikawa S, Hayashi T, Nii M, Shimanuki S, Morozumi T, Awata T. Two quantitative trait loci on Sus scrofa chromosomes 1 and 7 affecting the number of vertebrae. Journal of Animal Science. 2005;83:2247-54
28. Ren DR, Ren J, Ruan GF, Guo YM, Wu LH, Yang GC. et al. Mapping and fine mapping of quantitative trait loci for the number of vertebrae in a White Duroc x Chinese Erhualian intercross resource population. Animal Genetics. 2012;43:545-51
29. Uemoto Y, Nagamine Y, Kobayashi E, Sato S, Tayama T, Suda Y. et al. Quantitative trait loci analysis on Sus scrofa chromosome 7 for meat production, meat quality, and carcass traits within a Duroc purebred population. Journal of Animal Science. 2008;86:2833-9
30. Edwards DB, Ernst CW, Raney NE, Doumit ME, Hoge MD, Bates RO. Quantitative trait locus mapping in an F2 Duroc x Pietrain resource population: II. Carcass and meat quality traits. Journal of Animal Science. 2008;86:254-66
31. Aulehla A, Pourquié O. Signaling gradients during paraxial mesoderm development. Cold Spring Harbor Perspectives in Biology. 2010;2:a000869
32. Lin JR, Hu J. SeqNLS: nuclear localization signal prediction based on frequent pattern mining and linear motif scoring. PloS One. 2013;8:e76864
33. Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Research. 2006;34:W369-73
34. Ouyang Z, Zhou Q, Wong WH. ChIP-Seq of transcription factors predicts absolute and differential gene expression in embryonic stem cells. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:21521-6
35. Arvey A, Agius P, Noble WS, Leslie C. Sequence and chromatin determinants of cell-type-specific transcription factor binding. Genome Research. 2012;22:1723-34
36. Turnpenny PD, Alman B, Cornier AS, Giampietro PF, Offiah A, Tassy O. et al. Abnormal vertebral segmentation and the notch signaling pathway in man. Developmental Dynamics. 2007;236:1456-74
37. Barrantes IB, Elia AJ, Wunsch K, Hrabe de Angelis MH, Mak TW, Rossant J. et al. Interaction between Notch signalling and Lunatic fringe during somite boundary formation in the mouse. Current Biology. 1999;9:470-80
38. Ferjentsik Z, Hayashi S, Dale JK, Bessho Y, An H, Strooper BD. et al. Notch is a critical component of the mouse somitogenesis oscillator and is essential for the formation of the somites. PloS Genetics. 2009;5:e1000662
39. Bone RA, Bailey CSL, Wiedermann G, Ferjentsik Z, Appleton PL, Murray PJ. et al. Spatiotemporal oscillations of Notch1, Dll1 and NICD are coordinated across the mouse PSM. Development. 2014;141:4806-16
40. Shifley ET, Vanhorn KM, Perez-Balaguer A, Franklin JD, Weinstein M, Cole SE. Oscillatory lunatic fringe activity is crucial for segmentation of the anterior but not posterior skeleton. Development. 2008;135:899-908
41. Julian L, Anja H, Maxine H. Notch signaling, the segmentation clock, and the patterning of vertebrate somites. Journal of Biology. 2009;8:1-7
42. Gibb S, Maroto M, Dale JK. The segmentation clock mechanism moves up a notch. Trends in Cell biology. 2010;20:593-600
43. Jouve C, Palmeirim I, Henrique D, Beckers J, Gossler A, Ish-Horowicz D. et al. Notch signalling is required for cyclic expression of the hairy-like gene HES1 in the presomitic mesoderm. Development. 2000;127:1421-9
44. Riedel-Kruse IH, Muller C, Oates AC. Synchrony dynamics during initiation, failure, and rescue of the segmentation clock. Science. 2007;317:1911-5
45. Jiang YJ, Aerne BL, Smithers L, Haddon C, Ish-Horowicz D, Lewis J. Notch signalling and the synchronization of the somite segmentation clock. Nature. 2000;408:475-9
46. Herrgen L, Ares S, Morelli LG, Schroter C, Julicher F, Oates AC. Intercellular coupling regulates the period of the segmentation clock. Current Biology. 2010;20:1244-53
47. Galis F, Carrier DR, van Alphen J, van der Mije SD, Van Dooren TJM, Metz JAJ. et al. Fast running restricts evolutionary change of the vertebral column in mammals. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:11401-6
48. Carapuço M, Nóvoa A, Bobola N, Mallo M. Hox genes specify vertebral types in the presomitic mesoderm. Genes & Development. 2005;19:2116
49. Mallo M, Vinagre T, Carapuço M. The road to the vertebral formula. International Journal of Developmental Biology. 2009;53:1469
50. Mak TW, Penninger J, Roder J, Rossant J, Saunders M. Hoxc-5. Gene Knockout Factsbook. 1998;28:502-3
51. Griffiths AJ, Miller JH, Suzuki DT, Lewontin RC. An Introduction to Genetic Analysis, 7th edition. Whfreeman. 2000
52. Oh SP, Li E. The signaling pathway mediated by the type IIB activin receptor controls axial patterning and lateral asymmetry in the mouse. Genes & Development. 1997;11:1812-26
53. Oh SP, Yeo CY, Lee Y, Schrewe H, Whitman M. and Li E. Activin type IIA and IIB receptors mediate Gdf11 signaling in axial vertebral patterning. Genes & Development. 2002;16:2749
54. Van dAE, Forlani S, Chawengsaksophak K, De GW, Beck F, Meyer BI. et al. Cdx1 and Cdx2 have overlapping functions in anteroposterior patterning and posterior axis elongation. Development. 2002;129:2181
55. Dequéant ML, Pourquié O. Segmental patterning of the vertebrate embryonic axis. Nature Reviews Genetics. 2008;9:370
56. Dao DY, Yang X, Flick LM, Chen D, Hilton MJ, O'Keefe RJ. Axin2 regulates chondrocyte maturation and axial skeletal development. Journal of Orthopaedic Research. 2010;28:89-95
57. Naiche LA, Holder N, Lewandoski M. FGF4 and FGF8 comprise the wavefront activity that controls somitogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:4018-23
58. Okubo Y, Sugawara T, Abe-Koduka N, Kanno J, Kimura A, Saga Y. Lfng regulates the synchronized oscillation of the mouse segmentation clock via trans-repression of Notch signalling. Nature Communications. 2012;3:1141
59. Palmeirim I, Henrique D, Ish-Horowicz D, Pourquié O. Avian hairy gene expression identifies a molecular clock linked to vertebrate segmentation and somitogenesis. Cell. 1997;91:639
60. Horikawa K, Ishimatsu K, Yoshimoto E, Kondo S, Takeda H. Noise-resistant and synchronized oscillation of the segmentation clock. Nature. 2006;441:719-23
61. Gomez C, Ozbudak EM, Wunderlich J, Baumann D, Lewis J, Pourquie O. Control of segment number in vertebrate embryos. Nature. 2008;454:335-9
62. Ozbudak EM, Lewis J. Notch signalling synchronizes the zebrafish segmentation clock but is not needed to create somite boundaries. PloS Genetics. 2008;4:e15
63. Rida PC, Le MN, Jiang YJ. A Notch feeling of somite segmentation and beyond. Developmental Biology. 2004;265:2-22
Author contact
![]() Corresponding authors: renjunedu.cn or renjunjxaucom; lushenghuangcom.
Corresponding authors: renjunedu.cn or renjunjxaucom; lushenghuangcom.