International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288
- Current Issue
- Volume 22; 2026
- Volume 21; 2025
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Archive
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Results and Discussion
Conclusion
Materials and Methods
Abbreviations
Acknowledgements
References
Introduction
Results and Discussion
Conclusion
Materials and Methods
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(7):784-790. doi:10.7150/ijbs.22489 This issue Cite
Short Research Communication
Recalling Cohnheim's Theory: Papillary Renal Cell Tumor as a Model of Tumorigenesis from Impaired Embryonal Differentiation to Malignant Tumors in Adults
Daniel Banyai1, Donat P. Sarlos1, Anetta Nagy1, Gyula Kovacs1,2 ![]()
1. Department of Urology, Medical School, University of Pecs, Hungary
2. Medical Faculty, Ruprecht-Karls-University, Heidelberg, Germany
Received 2017-8-23; Accepted 2017-12-9; Published 2018-5-21
Citation:
Banyai D, Sarlos DP, Nagy A, Kovacs G. Recalling Cohnheim's Theory: Papillary Renal Cell Tumor as a Model of Tumorigenesis from Impaired Embryonal Differentiation to Malignant Tumors in Adults. Int J Biol Sci 2018; 14(7):784-790. doi:10.7150/ijbs.22489. https://www.ijbs.com/v14p0784.htm
Other stylesAbstract

We have suggested that papillary renal cell tumor (PRCT) of the kidney arises from nephrogenic rest-like lesions. To approve our hypothesis, we worked up 14 kidneys bearing papillary and 14 ones with conventional renal cell carcinoma (CRCC) histologically and found 42 papillary lesions in average per kidney bearing PRCT. PRCTs are characterized by loss of the Y chromosome and trisomy of chromosomes 7 and 17. The MET and HNF1B are localized to chromosome 7q31 and 17q21 and are frequently amplified in PRCT. We have analyzed the expression of the mutant MET in hereditary PRCTs and precursor lesions and found duplication and expression of the mutated allele. Because both genes are involved in early stage of nephron development, we have analyzed the expression of MET and HNF1B by immunohistochemistry in fetal kidneys, precursor lesions and PRCTs. We detected strong expression of MET and HNF1B in distal compartment of S-shaped body of fetal kidneys and in nephrogenic rest-like precursor lesions. Our finding suggests an association between expression of MET and HNF1B in precursor lesions and development of PRCT. We propose a model involving chromosomal clonal evolution and corresponding gene expression for development of PRCTs from embryonic rests due to impaired differentiation. Our model suggests that PRCT have a natural history distinct from that of most common CRCC.
Keywords: Cohnheim's theory, Papillary RCT, MET, HNF1B, Model of tumor development
Introduction
In 1882 Cohnheim has postulated that “ein Fehler, eine Unregelmässigkeit der embryonalen Anlage ist, in der die eigentliche Ursache der späteren Geschwulst gesucht werden muss”, e.g. an error and impaired regulation in embryonic Anlage may later lead to tumor development [1]. „Aber was wir als angeboren verlangen, ist ja nicht die Geschwulst, sondern lediglich die Anlage dazu, d.h. nach der von uns proponirten Formulirung die Existenz desjenigen über das physiologische Maass hinaus producirten Zellenquantum, aus dem eine Geschwulst sich entwickeln kann“ e.g. we expect the newborn to bring not the tumor itself but merely the superabundant cell material into the world, from the latter a tumor may develop.
Wilms' tumor (WT) has already been recognized as a useful model to explore tumor development from “not differentiated superabundant” embryonic rest cells [2]. Molecular genetic studies confirmed the connection between WT and impaired differentiation during kidney development [3]. Several genes which are expressed spatially and temporally during early nephrogenesis, especially during the mesenchyme to epithelium transition (MET) have already been implicated in the molecular biology of WT [4].
Previously, we have suggested that papillary renal cell tumor (PRCT) may also arise from nephrogenic rest-like lesions [5]. Small tubular-papillary lesions associated with clinically detected PRCT display consequently a combined trisomy of chromosomes 7 and 17, which remain the hallmark of frankly malignant tumors [6-9]. Loss of the Y chromosome has been detected in 85% of PRCTs of male patients. [10]. PRCTs develops at least 8 times more frequently in males than in females indicating the role of a yet unknown gene at the Y chromosome. Duplication and amplification of tyrosine kinase MET at chromosome 7q31 and of transcription factor HNF1B at chromosome 17q12 are characteristic genetic changes in PRCTs [11]. MET and HNF1B play a crucial role in the early stage of nephron development. Therefore, we suggest that their alteration may be responsible for impaired differentiation of embryonal kidney resulting in not or not fully differentiated remnants, which later may arise to PRCTs in adults.
To approve this hypothesis, we have searched for precursor lesions in kidneys bearing PRCT, analyzed hereditary as well as sporadic PRCT and their precursor lesions for expression of MET and HNF1B and also expression of the mutant MET.
Results and Discussion
Tubular-papillary parenchymal abnormalities are associated with PRCTs
We have detected 42 nephrogenic rest-like lesions on average in kidneys with PRCT and only 0.4 in kidneys with CRCC suggesting an association of tubular-papillary lesions with development of PRCTs. The vast majority of lesions in the size of a glomerulus displayed tubular-papillary structures of small “basophilic” cells intermingled with normal parenchymal elements. The association of multiple so-called cortical adenomas with PRCTs has noticed earlier. Keyes pointed out more than 100 years ago that “we do not regard it as extremely rare, since, owing the small size and inoffensive character of the growths, they are liable to be passed over without notice” [12]. By detailed histological analysis, Cristol et al. identified 37 “adenomas”, all but one with papillary structure in 22 kidneys removed due to cancer [13]. Apitz found 725 macroscopically detectable cortical adenomas in 305 kidneys in 4309 consecutive autopsies [14]. PRCTs occur preferentially in males, the male to female ratio is 5-10 to 1 [5]. The 10 to 1 and 4.2 to 1 male to female ratio in the two series, respectively, suggest that the small tubular-papillary lesions found by Cristol et al. [13] and Apitz [14] are associated with PRCTs. Apitz have suggested that adenoma develops due to disturbance of differentiation in embryonal kidney, and the late sclerosis is an environmental effect that increases the number and size of these lesions [13]. The strong association between nephrogenic rest-like lesions and PRCTs showed in our study are in conform with Apitz suggestion and Cohnheim's theory.
Trisomy of chromosomes 7 and 17 and expression of mutant MET allele
Array CGH revealed a gain of chromosome 7 and 17 in each of the nine papillary lesions obtained from the index patient with germ line MET mutation (Figure 1). Analysis of genomic DNA from the index patients' kidney and tumors revealed a base pair change A3954C in exon 19 leading to non-conservative amino acid substitution S1254A in auto-phosphorylation site of MET (Figure 2A). By applying SSCP technique we have detected both wild type and mutant MET alleles in index patients' kidney and papillary lesions. An increased signal of the mutant allele in the latter indicates the duplication of the mutant allele (Figure 2C). Both wild type and mutant alleles were expressed in kidney and tumor tissues obtained from the index patient whereas in a control kidney only the wild type MET sequences were detected (Figure 2B). Trisomy of chromosome 7 with duplication of the mutant allele has been described in hereditary PRCTs [7, 15]. The consequent duplication and overexpression of mutant MET allele indicates that these alterations are necessary before cells enter the tumorigenic pathway. It is likely that mutation of one MET allele does not affect the function of wild type allele during embryonal morphogenesis and the germline mutation alone is not sufficient for initiation of papillary RCTs. This is indicated by the fact that members of hereditary PRCT families carrying the activating MET mutations in their germline develop normal functioning nephrons.
Expression of MET and HNF1B proteins in fetal kidneys, precursor lesions and tumors
Immunohistochemistry revealed a strong expression of MET at the distal compartment of S-shaped body in fetal kidney as well as in all precursor lesions from hereditary and sporadic cases (Figure 3A-C). The continuous high expression of MET in all precursor lesions suggests that MET may be instrumental in early phase of development of precursor lesions. In experimental setting, activated Met oncogene induced diverse mammary carcinomas, in which similarly to PRCT, duplication or amplification of the Met locus has been found [16, 17]. Development of tumors of diverse histological phenotypes also suggests that Met initiates the tumorigenic processes in early progenitor cell. The extremely high number of over 1000 small solid-papillary precursor lesions in hereditary PRCC cases also suggests that mutational activation and duplication/overexpression of MET is instrumental in the early abrogation of nephrogenesis and retention of residual embryonic cells with tumorigenic potential [18]. Of interest, MET is also expressed in 22% of nephrogenic rest and 14% of WTs mainly in well-differentiated epithelial cells, whereas blastemal components are negative in both nephrogenic rests and WTs [19]. In spite of this similarity, the precursor lesions for WT and PRCT correspond to distinct stages of cellular differentiation [20].
Considering the consequent duplication of chromosome 17 in papillary renal cell adenomas (PRCA) and PRCCs we have analyzed the HNF1B expression. In fetal kidneys a weak expression was seen at the tip of branching ureteric bud, strong expression at the distal compartment of comma and S-shaped body and emerging distal tubules, whereas proximal tubules and loop of Henle displayed a weak nuclear expression (Figure 3D). All precursor lesions obtained from hereditary and sporadic cases displayed strong nuclear expression of HNF1B (Figure 3E&F). HNF1B is required for activation of segment specific gene expression program which is central to the development and function of the nephron [21]. Hnf1b mutant mouse embryos show a deformation of S-shaped bodies with the absence of subdomain encompassing part of the prospective proximal tubule and part of the adjacent intermediate region [22]. The HNF1B is a regulator of early tubular segmentation, it starts to be expressed during MET and it can be detected from the renal vesicle stage in all tubular parts of the nephron.
Figure 1
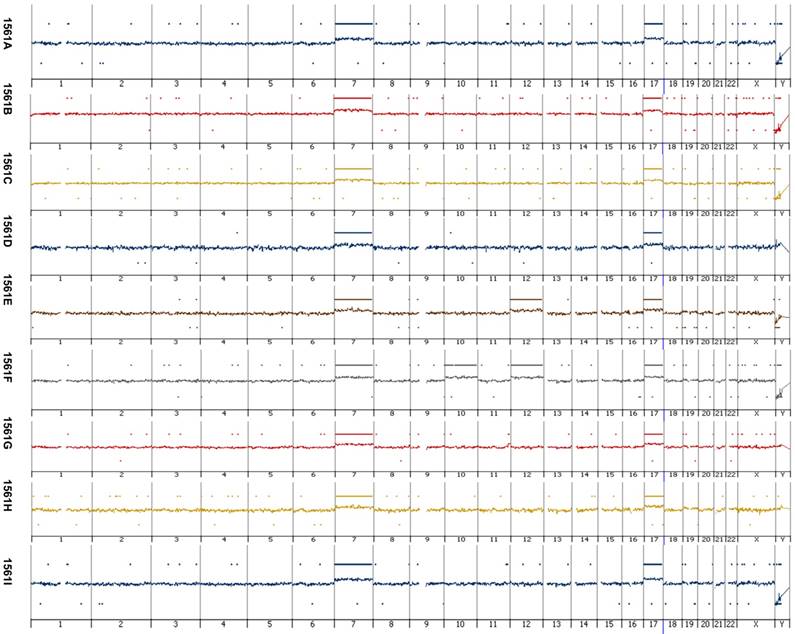
Array-based comparative genomic hybridization of papillary lesions of the patients with germ-line MET mutation. Each papillary lesion revealed copy number gains at chromosome 7 and 17. Additional, in one of the papillary RCT s (1561b) a copy number gain at chromosome 10 and 12 and in another (1561b) at chromosome 12 was seen. Loss of the Y chromosome occurred in six of the nine tumors analyzed by array CGH. No amplification or other small genomic alteration was seen by high resolution array analysis.
Figure 2
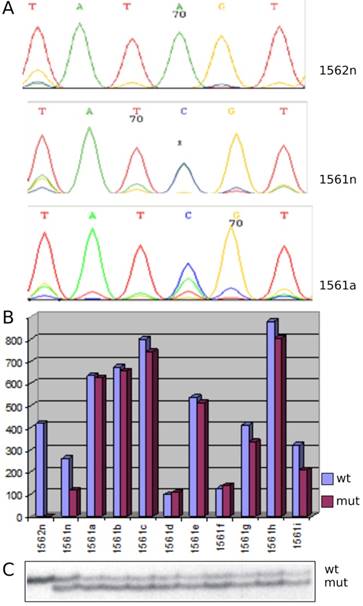
MET alteration in hereditary papillary RCTs. (A) Sequencing detected an A to C base pair change in MET in patient kidney (1561n) and tumor cells (1561a) whereas wild type sequences were seen in normal control kidney (1562n). (B) In normal control (1562n) only the wild type MET sequences were detected, whereas the kidney (1561n) and all tumors (1561a-i) of the patients expressed both the mutant and wild type sequences. (C) SSCP analysis of exon 19 displayed wild type (wt) and mutant (mut) sequences in kidney and tumors of the index patient. Note the stronger signal of the mutant allele corresponding to its duplication in tumor cells (1561a-i). Control kidney (1562n) showed only one signal corresponding to two wild type alleles.
In adult kidney MET expression was limited to the distal nephron and only a weak expression of HNF1B was seen in proximal and distal tubules. A moderate MET and HNF1B expression was detected in 43 and 41 of 76 PRCTs placed onto TMAs, respectively. Co-expression of HNF1B and MET was detected in 32 PRCCs. The strong expression of both genes in embryonal structures and precursor lesions and low expression in 54-56% of PRCTs suggest that temporally and spatially altered expression of MET and HNFB may initiate precursor lesions, but their continuous expression is not necessary for the progression of PRCTs. The latter acquire additional trisomy 8, 12, 16 and 20 and shift their gene expression profile to the increased dosage of yet (with exception of the LRRK2 gene) unknown genes which drive the malignant progression [8].
Conclusion
Termination of nephrogenesis and differentiation of specialized cell types along the nephron is a well-coordinated molecular process. Taking into account the finely tuned relationship between growth and differentiation controlling signals, changes e.g. mutation or altered gene dosage of MET may disturb the balance and favor cell proliferation. To produce the appropriate number of cells for development of approximately 200 thousand to 2 million nephron, cells in the renewing blastemal niche undergo a forced mitotic activity resulting in variable level of errors, e.g. in an embryonal genetic noise. An explanation for multiple papillary lesions may come from earlier cytogenetic studies, which revealed trisomy of chromosome 7 in up to 10% of normal kidney cells [23]. Germ line mutation of the MET gene leads to gain of function with constitutive activation of the MET protein. Duplication of mutant chromosome 7 in hereditary cases associated with over 1000 precursor lesions whereas trisomy of wild type chromosome 7 leads to increased dosage of wild type MET which has a lower penetrance resulting in an average of 42 precursor lesions per kidney [18, present paper].
It was proposed 40 years ago that neoplasm arises from a single cell and tumor progression is characterized by genetic variability within the original clone allowing sequential selection of more aggressive sub lines [24]. Previously, we have delineated three genetic developmental stages of papillary RCTs [8]. Based on the present and previous findings, we propose a genetic model of precursor lesion-adenoma-carcinoma sequence involving chromosomal clonal evolution with corresponding gene expression for development of PRCTs. The possible role of trisomy of chromosome 7 and 17 and corresponding expression of MET and HNF1B in the early stage of tumorigenesis has been discussed above. Recently, amplification of the chromosome 12q12 locus harboring the leucine-rich repeat kinase 2 (LRRK2) has been described in PRCCs [25]. It was suggested that amplification of the LRRK2 gene is a requirement for oncogenic signaling of the MET as co-amplification and/or co-expression of both genes occurred in 55% of PRCCs.
The well-defined clonal evolution, e.g. alteration of chromosomes 7, 17 and 12 and corresponding over expression of MET, HNF1B and LRRK2 genes supports our hypothesis on the developmental sequences for hereditary and sporadic papillary RCTs (Figure 4). Until the gene on Y chromosome and its involvement in the development of PRCT is not disclosed, we do not include the Y chromosome loss in this model. The spatially and temporally altered gene dosage of MET and HNF1B leads to delayed or arrested differentiation during nephron development, which also explains the morphological heterogeneity within clinically recognized PRCTs [26]. Our model strongly suggests that PRCT have a natural history distinct from that of most common CRCC which develops from cells of proximal tubules. This should be taken into account in the clinical management of the disease.
Materials and Methods
Tissue samples
We have included 14 kidneys with sporadic PRCTs (M:F=13:1) and 14 kidneys with CRCCs (M:F=9:5) in detailed histological analysis. Each kidney was cut in 3 mm thick slices and embedded in paraffin. Three to five consecutive slides per paraffin block were stained with hematoxylin and eosin. Each slide was scored for parenchymal lesions twice. Tissue micro array (TMA) was constructed from 76 clinically detected PRCTs. We have also analyzed multiplex papillary lesions obtained from a 33-year old male patient with germ line MET mutation. Normal kidney tissue (1561N) and 9 lesions (1561A-I) were obtained immediately after nephrectomy. Tumors 1561E and 1561F were 8 and 14 mm in diameter, respectively, whereas 7 lesions were approximately 2 mm in diameter (1561A-D and G-I). A part of tumors 1561E and 1561F was immediately snap frozen in liquid nitrogen and stored at -80°C, and another part was used for cell culture. From the precursor lesions a small piece of tissue was taken by a needle and used for short term culture. The remnants of tumors and precursors was fixed in 4% formalin and embedded in paraffin for histological diagnosis. Five fetal kidneys were obtained from legal aborts carried out at the Department of Gynecology, University of Pecs, Hungary. The collection and use of all tissue samples for this study was approved by the Ethics Committees of the University of Pecs (5343/2014).
Array-based comparative genomic hybridization (Array-CGH)
High molecular weight DNA was isolated from short term cell culture of the 9 papillary lesions identified in the hereditary case. A pool of sex matched DNA from five normal parenchymal kidneys was used as a reference for array-CGH. Array-CGH was carried out using an 8x60K Agilent array platform (AMADID 014950) as described previously [27].
Figure 3
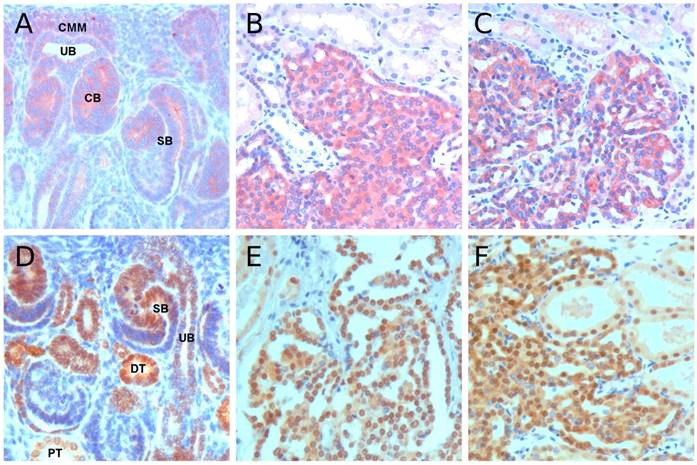
Immunohistochemistry of MET and HNF1B. (A) Positive MET staining in cells of the ureteric bud (UB), cap metanephric mesenchyme (CMM), comma-shape body (CB) and distal part of S-shape body (SB). (B&C) Strong MET staining in precursor lesions from hereditary and sporadic cases, respectively. (D) HNF1B immunohistochemistry in fetal kidney revealed a strong staining in distal compartment of S-shape body (SB) and distal tubules (DT), whereas the ureteric bud (UB) and proximal tubules (PT) showed a weak staining. (E&F) Strong nuclear staining was seen in tubular-papillary precursor lesions in hereditary and sporadic cases, respectively. Original magnification x 400.
Figure 4
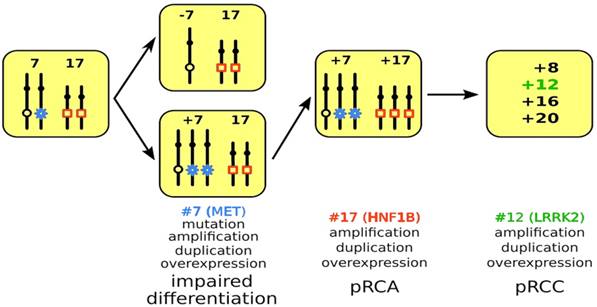
Model of PRCT development. A mitotic non-disjunction reduplication mechanism leads to duplication of chromosome 7 with the mutant MET gene (marked by blue star) in hereditary cases and the wild type MET in sporadic cases. The increased copy number (trisomy or amplification) leads to overexpression of MET and to impaired differentiation of cells affected. In the next step, copy number gain or amplification and subsequent overexpression of HNF1B (marked by red quadrat) leads to further proliferation of differentiation arrested cells yielding in pRCA. Acquiring additional chromosomal trisomies (amplification) including chromosome 12 and overexpression of LRRK2 (marked in green) may change the biological behavior of adenomas towards carcinoma (pRCC).
Mutation analysis of MET tyrosine kinase domain
Exons 16, 17, 18 and 19 of the tyrosine kinase domain of the MET gene were amplified for sequencing as described earlier [7]. The PCR products were directly sequenced with IR800 and IR700-labeled sequencing primer set using Thermosequenase cycle sequencing kit (Amersham Pharmacia, Freiburg, Germany) following the manufacturers recommendations. The products of the sequencing reaction were analyzed in both directions simultaneously on the dual laser scanner LICOR Long ReadIr 4200 sequencing machine (MWG-Biotech, Ebersberg, Germany) using the BaseImageIR software package. The read sequences were compared to the wild-type sequence (J02958) to identify mutation.
SSCP analysis of exon 19 carrying the mutation
Using the genomic MET sequence AC004416 we designed primers flanking exon 19 to amplify a 198bp fragment bearing the mutation A3954C in its central part. We combined the intronic primer K2d (nt:24839-24860) “cca cgg gta ata attt ttg tcc” with primer K1r (nt:25019-25036) “cca cat ctg act tgg tgg tgg”, which is located at the 3' end of exon 19. The 20µl PCR reactions contained 100ng DNA, 200uM dNTPs, 1.5mM MgCl2, 2pmol of each primer (the forward primer was radioactively labelled) and 0.5 unit Taq-polymerase and were subjected to a 2min lasting initial denaturation at 94°C followed by 27 cycles with 30sec 94°C, 30sec 55°C and 40sec 72°C and a final elongation of 5min at 72°C. To each completed PCR reaction 20µl of loading buffer, 2µl 1M NaOH and 2µl 20mM EDTA were added, the samples were denatured for 2min at 94°C and 3µl were loaded onto a MDE gel with 10% glycerol. Electrophoresis was performed over night at 6W and a constant temperature. Afterwards the gel was dried and exposed overnight to an X-ray film.
RT-PCR analysis of wild type and mutant MET
Total RNA was extracted by TRIzol (Invitrogen GmbH, Karlsruhe, Germany) from snap frozen tissues or tumor cells growing in short term culture according to the manufacturer's recommendations. First strand cDNA synthesis was performed with 2µg of total RNA using Superscript II reverse transcriptase (Invitrogen) and following manufacturer's protocol. Real-time PCR reaction was performed in DNA Engine Opticon system (MJ Research Inc., Watertown, USA) using Platinum SYBRGreen qPCR SuperMix-UDG (Invitrogen) and β-actin as a control. All reactions were carried out in duplicates and the results were averaged. Primers are available upon request.
Tissue multi array (TMA) and immunohistochemistry
Original paraffin blocks of foetal and adult kidneys, precursor lesions as well as a TMA containing PRCCs were used for this study. TMA was constructed by one of the authors (GK) as described earlier [11]. The 4µm sections placed onto FLEX IHC microscope slides (DAKO, Glostrup, Denmark), dewaxed in xylene and rehydrated in graded ethanol. Antigen retrieval was performed by boiling the slides in 10 µM sodium citrate buffer, pH 6.0 in 2100-Retriever (Pick-Cell Laboratories, Amsterdam, The Netherlands). Endogenous peroxidase activity and nonspecific staining were blocked by incubation with 3% hydrogen peroxide containing 1% normal horse serum for 10min at room temperature.
Slides were then incubated overnight at 4°C in moist chamber with the rabbit polyclonal anti-MET antibody (sc-12, Santa Cruz Biotechnology, Inc.) and rabbit polyclonal anti-HNF1B antibody (HPA-002083, Sigma Aldrich, Inc.), both diluted 1:100. HRP conjugated anti-rabbit secondary antibody (MACH4 Universal HRP-Polymer, Biocare Medical, Concord, CA, USA) was applied for 30min and color was developed using the AEC or DAB substrates (DAKO). Tissue sections were counterstained with Mayer's hematoxylin and cover-slipped with Faramount (DAKO) or Pertex (Medite GmbH, Burgdorf, Germany). In negative controls, the primary antibody was omitted.
Abbreviations
PRCT: papillary renal cell tumor, including adenoma and carcinoma; PRCA: papillary renal cell adenoma; PRCC: papillary renal cell carcinoma; CRCC: conventional renal cell carcinoma; WT: Wilms tumor; MET: mesenchyme to epithelium transition; CGH: comparative genomic hybridization; TMA: tissue micro array.
Acknowledgements
We thank Dr. David Semjen for providing paraffin blocks for TMA construction and Zsuzsanna Halas for preparing the TMA slides. The present scientific contribution is dedicated to the 650th anniversary of the foundation of the University of Pecs, Hungary.
Funding
This work was supported by a grant of the Medical Faculty, University of Pecs, Hungary (PTE-AOK-KA-2014/1).
Author contributions
GK conceived and designed the project and wrote the manuscript. DB, DPS, and AN performed the experiments and evaluated the data. DB and AN wrote the Methods.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cohnheim J. Vorlesungen über allgemeine Pathologie. Berlin. 1882 August Hirschwald p.737
2. Beckwith JB, Kiviat NB, Bonadio JF. Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms' tumor. Pediatr Pathol. 1990;10:1-36
3. Rivera MN, Haber DA. Wilms tumor: connecting tumorigenesis and organ development in the kidney. Nat Rev Cancer. 2005;5:699-712
4. Li CM, Guo M, Borczuk A. et al. Gene expression in Wilms” tumor mimics the earliest committed stage in the metanephric mesenchymal-epithelial transition. Am J Pathol. 2002;160:2181-2190
5. Kovacs G. Molecular cytogenetics of renal cell tumors. Adv Cancer Res. 1993;62:89-124
6. Kovacs G, Fuzesi L, Emanuel A. et al. Cytogenetics of papillary renal cell tumors. Genes Chromosomes Cancer. 1991;3:249-255
7. Fischer J, Palmedo G, von Knobloch R. et al. Duplication and overexpression of the mutated allele of the MET proto-oncogene in multiple hereditary papillary renal cell tumours. Oncogene. 1998;17:733-739
8. Szponar A, Zubakov D, Pawlak J. et al. Three genetic developmental stages of papillary renal cell tumors: duplication of chromosome 1q marks fatal progression. Int J Cancer. 2009;124:2071-2076
9. Schmidt L, Duh FM, Chen F. et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nature Genet. 1997;16:68-73
10. Kovacs G, Tory K, Kovacs A. Development of papillary renal cell tumours is associated with loss of Y-chromosome specific DNA sequences. J Pathol. 1994;173:39-44
11. Szponar A, Yusenko MV, Kuiper R. et al. Genomic profiling of papillary renal cell tumours identifies small regions of DNA amplifications: a possible role of HNF1B in tumour development. Histopathology. 2011;58:934-943
12. Keyes EL. Adenoma of the kidney. Succesful nephrectomy. Am J Med Sci. 1890;100:549-558
13. Cristol DS, McDonald FR, Immel FL. Renal adenomas in hypernephromatous kidneys: a study of their incidence, nature and relationship. J Urol. 1946;55:328-359
14. Apitz K. Die Geschwülste und Gewebsmissbildungen der Nierenrinde. Die Adenoma. Virchows Arch. 1944;311:328-359
15. Zhuang Z, Park WS, Pack S. et al. Trisomy 7-harbouring non-random duplication of the mutant MET allele in hereditary papillary renal carcinomas. Nat Genet. 1998;20:66-69
16. Graveel CR, DeGroot JD, Su Y. et al. Met induces diverse mammary carcinomas in mice and is associated with human basal breast cancer. Proc Natl Acad Sci USA. 2009;106:12909-12914
17. Ponzo MG, Lesurf R, Petkiewitz S. et al. Met induces mammary tumors with multiple pathologies and is associated with both poor outcome and basal-type breast cancers. Proc Natl Acad Sci USA. 2009;106:12903-12908
18. Ornstein DK, Lubensky IA, Venzon D. et al. Prevalence of microscopic tumors in normal appearing renal parenchyma of patients with hereditary papillary renal cancer. J Urol. 2000;163:431-433
19. Vounonvirta R, Sebire NJ, Messahl B. et al. Expression of hepatocyte growth factor and its receptor Met in Wilms tumors and nephrogenic rests reflects their roles in kidney development. Clin Cancer Res. 2009;15:2723-2730
20. Szponar A, Kovacs G. Expression of KRT7 and WT1 differentiates precursor lesions of Wilms' tumours, papillary renal cell tumours and mucinous tubular and spindle cell carcinomas. Virchows Arch. 2012;460:423-427
21. Heliot C, Desgrange A, Buisson I. et al. HNF1B controls proximal-intermediate nephron segment identity in vertebrates by regulating Notch signaling components and Irx1/2. Development. 2013;140:873-885
22. Massa F, Garbay S, Bouvier R. et al. Hepatocyte nuclear factor 1 beta controls nephron tubular development. Development. 2013;140:886-896
23. Kovacs G, Brusa P. Clonal chromosomal aberrations in normal kidney tissue from patients with renal cell carcinoma. Cancer Genet Cytogenet. 1989;37:289-290
24. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23-28
25. Looyenga BD, Furge KA, Dykema KJ. et al. Chromosomal amplification of leucine-rich repeat kinase-2 (LRRK2) is required for oncogenic MET signaling in papillary renal and thyroid carcinomas. Proc Natl Acad Sci USA. 2011;108:1439-1444
26. Balint I, Szponar A, Jauch A. et al. Trisomy 7 and 17 marks papillary renal cell tumours irrespectively of variation of the phenotype. J Clin Pathol. 2009;62:892-895
27. Yusenko MV, Kuiper RP, Boethe T. et al. High-resolution DNA copy number and gene expression analyses distinguish chromophobe renal cell carcinomas and renal oncocytomas. BMC Cancer. 2009;9:152
Author contact
![]() Corresponding author: Gyula Kovacs, g.kovacsde; gyula.kovacsuni-heidelberg.de; ORCID:0000-0003-2890-3781
Corresponding author: Gyula Kovacs, g.kovacsde; gyula.kovacsuni-heidelberg.de; ORCID:0000-0003-2890-3781
Citation styles
APA
Banyai, D., Sarlos, D.P., Nagy, A., Kovacs, G. (2018). Recalling Cohnheim's Theory: Papillary Renal Cell Tumor as a Model of Tumorigenesis from Impaired Embryonal Differentiation to Malignant Tumors in Adults. International Journal of Biological Sciences, 14(7), 784-790. https://doi.org/10.7150/ijbs.22489.
ACS
Banyai, D.; Sarlos, D.P.; Nagy, A.; Kovacs, G. Recalling Cohnheim's Theory: Papillary Renal Cell Tumor as a Model of Tumorigenesis from Impaired Embryonal Differentiation to Malignant Tumors in Adults. Int. J. Biol. Sci. 2018, 14 (7), 784-790. DOI: 10.7150/ijbs.22489.
NLM
Banyai D, Sarlos DP, Nagy A, Kovacs G. Recalling Cohnheim's Theory: Papillary Renal Cell Tumor as a Model of Tumorigenesis from Impaired Embryonal Differentiation to Malignant Tumors in Adults. Int J Biol Sci 2018; 14(7):784-790. doi:10.7150/ijbs.22489. https://www.ijbs.com/v14p0784.htm
CSE
Banyai D, Sarlos DP, Nagy A, Kovacs G. 2018. Recalling Cohnheim's Theory: Papillary Renal Cell Tumor as a Model of Tumorigenesis from Impaired Embryonal Differentiation to Malignant Tumors in Adults. Int J Biol Sci. 14(7):784-790.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.