Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Chromatin changes in the...
Pathological hypertrophy-related...
Interplay between non-coding...
Future considerations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(9):1133-1141. doi:10.7150/ijbs.26215 This issue Cite
Review
Non-coding RNA-linked epigenetic regulation in cardiac hypertrophy
Yanhan Dong ![]() , Sheng Xu, Jing Liu, Murugavel Ponnusamy, Yanfang Zhao, Yanhui Zhang, Qi Wang, Peifeng Li
, Sheng Xu, Jing Liu, Murugavel Ponnusamy, Yanfang Zhao, Yanhui Zhang, Qi Wang, Peifeng Li ![]() , Kun Wang
, Kun Wang ![]()
1. Institute for Translational Medicine, Qingdao University, Deng Zhou Road 38, Qingdao 266021, China
Received 2018-3-21; Accepted 2018-6-4; Published 2018-6-22
Abstract

Cardiac hypertrophy is an adaptive enlargement of myocardium in response to pressure overload caused various pathological insults, which is accompanied by alteration of a complex cascade of signaling pathways. During the hypertrophy process, many changes occur at cellular level including gene reprogramming by turning off chromatin regulators. Studies from the past decade have demonstrated that the abnormal epigenetic modifications, such as DNA methylation, histone modification, and oxidative modification of nucleic acid, could lead to changes in chromosome structure and cardiac dysfunction. Increasing evidence indicates that non-coding RNAs (ncRNAs) have functional significance in modulating the gene expression during those pathological events in the heart. Emerging evidences have highlighted that ncRNAs might serve as a signal for changing the state of chromatin, however, the knowledge about the ncRNA-linked epigenetic regulatory mechanisms in cardiac pathologies is still largely unexplored. In this review, we summarize the current information on association between ncRNAs and epigenetic modifications in cardiac hypertrophy, and we have discussed their crosstalk. In addition, this review provides insights into their therapeutic and diagnostic potential for treating hypertrophic heart disease.
Keywords: Noncoding RNAs, Epigenetics, Cardiac hypertrophy, miRNAs, LncRNAs
Introduction
Cardiovascular diseases (CVDs) are the predominant causes of morbidity and mortality worldwide [1]. Among them, cardiac hypertrophy is one of the most common CVDs that is characterized by an increase in heart size resulting from increased volume of cardiomyocyte cells [2, 3]. It occurs in response to a variety of physiological and pathophysiological stimuli and its onset is believed to represent a compensatory response in order to meet the altered functional demands of the cardiac tissue [4]. However, this adaptive response turns out to be maladaptive and irreversible hypertrophy under pathological stress, which is accompanied by cardiomyocyte loss, proliferation of interstitial fibroblasts and collagen deposition. This ultimately leads to decreased compliance and increases the risk of heart failure and sudden death in human [5-8]. Pathological cardiac hypertrophy is linked with many critical alterations in cardiomyocytes including modulation of genetic and epigenetic signaling [9]. Several molecular signaling pathways, including PI3K/Akt, calcineurin/NFAT and MAPK/ERK pathways are implicated in cardiac hypertrophy [10].
In the past few decades, many research groups have investigated the association between genetic changes and cardiac hypertrophy. In recent years, a growing number of studies have revealed that changes in epigenetic status are also involved in the cardiac development, disorder and regeneration without altering DNA sequences [11]. The epigenetic modifications occur at genomic (DNA methylation), or nucleosomal and chromatin (histone modification, chromatin remodeling complexes) levels [12]. Interestingly, non-coding RNAs (ncRNAs) are often accompanied with these epigenetic events that serve as signals for decoding gene transcription, protein translation, genomic stability and chromatin modifications [13-15]. NcRNAs originate from genome but generally not translated into proteins, which are classified into two subgroups based on their length: long ncRNAs contain more than 200 nucleotides (lncRNAs); and short ncRNAs (<200 nucleotides), including microRNAs (miRNAs), piwi-interacting RNAs (piRNAs), short interfering RNAs (siRNAs), and others [16]. Despite their functional impact on various pathological conditions are well defined, the underlying ncRNA-linked epigenetic regulatory mechanisms in heart pathologies are still at the early stage. In this review, we will discuss the current knowledge of association between ncRNAs and covalent modifications of chromatin through controling epigenetic machinery during the development of cardiac hypertrophy, which reveals novel therapeutic strategies for the clinical settings.
Chromatin changes in the pathogenesis of cardiac hypertrophy
DNA methylation is an essential epigenetic modification that often occurs at the fifth carbon position of cytosine residues located within CpG sites in the mammalian genome, which is implicated in X-chromosome inactivation, gene imprinting, and the inhibition of transposons [17]. There are three DNA methyltransferases (DNMTs) and three ten-eleven-translocation (TET) enzymes responsible for the DNA methylation and demethylation, respectively [18, 19] (Fig. 1A). Among them, DNMT1 maintains stable DNA methylation during replication, whereas DNMT3a and DNMT3b are responsible for the process of de novo DNA methylation [19]. Recently, some studies pointed out that there are 60~80% CpG sites are methylated in the human genome [17] and global DNA methylation status is closely linked to cardiovascular diseases. In general, DNA methylation modulates gene expression by blocking transcription-factor binding or recruiting methyl-binding proteins [20, 21]. However, knowledge of DNA methylation with its associated cofactors in cardiac hypertrophy is still infancy. A consistent global DNA methylation profile in end-stage failing hearts and normal human hearts revealed that DNA methylation differences are present in promoters and body regions of genes along with demethylation in up-regulated gene regions [22]. Movassagh et al found that double-homeobox protein 4 (DUX4) were heavily methylated in failing hearts, which implicates a mechanism by which DNA methylation changes can be functionally relevant to alterations in gene expression found in the failing heart [22]. In addition, Movassagh and colleagues identified three angiogenic factors (PECAM1, ARHGAP24, AMOTL2), whose expression is altered through DNA methylation in heart failure. Recently, Gilsbach et al also addressed the dynamic changes in DNA methylation in mouse model subjected to pressure overload for 3 weeks [23]. They have established DNA methylation as a highly dynamic process during the adaptation of cardiomyocytes under pathological stress, a process tightly linked to gene regulation and activity. Regardless, the underlying mechanisms of DNA methylation in dysregulation of gene expression linked to cardiac hypertrophy needs further elucidated.
Chromatin modifications in gene-expression regulation. A: DNA methylation and demethylation processes are mediated by DNMTs and TET enzymes. B: Histone modifications by post-translational changes control gene expression by regulating the dynamics of chromatin that involves the N-terminal tails of histones. The acetylation and demethylation lead to a decondensed and transcriptionally active chromatin, while histone methylation and deacetylation are usually associated with transcriptional repression. C: Under excessive ROS, S-nitrosylation is the most important form of oxidative modification of proteins at sulfhydryl group; 8-OHdG in DNA and 8-OHG in RNA are two common forms of oxidative modification of nucleotides, respectively. Abbreviations: DNMT, DNA methyltransferase; HAT, histone acetyltransferase; HDAC, histone deacetylase; HDM, histone demethylase; HMT, histone methyltransferase; TET, ten-eleven translocation.
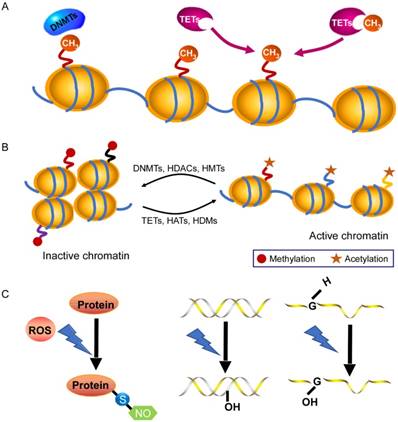
In the nucleus of cell, DNA wraps around a histone octamer (2 copies each of the core histones H2A, H2B, H3 and H4) to form the nucleosome cores, which is the fundamental primary unit of chromatin [24, 25]. Histones are able to confer plasticity to chromatin packaging or gene transcription through their post-translational modifications [26]. Study of Papait et al identified various changes of histone marks using ChIP-seq after heart transverse aortic constriction and underlying cardiac hypertrophy [27]. Histone modifications usually take place on the N-terminal tails that include acetylation, phosphorylation, methylation, sumoylation, biotinylation, ubiquitination and ADP-ribosylation [26, 28] (Fig. 1B). Histone acetylation/deacetylation are the most common modifications, which are catalyzed by histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively [29, 30]. There are eleven HDAC members, which are divided into four classes (I-IV) and they play a prominent role in cardiac hypertrophy and failure. For instance, class II histone deacetylase HDAC4 become oxidized in cardiac hypertrophy, which leads to HDAC4 shuttling out of nucleus in a Trx1-sensitive manner, and thereby dismissing these pro-hypertrophy genes [31]. Furthermore, Hohl and colleagues found that an acute increase of cardiac preload induced HDAC4 nuclear export, H3K9 (lysine 9 of histone 3) demethylation, HP1 (heterochromatin protein 1) dissociation from the promoter region, and activation of the ANP gene in isolated working murine hearts [32]. Pathological cardiac hypertrophy causes an increase in the expression of HDAC6, and the HDAC6-null mice exhibited cardiac dysfunction and fibrosis upon treatment with angiogensin II (Ang II), which probably occurs through acetylating sarcomere protein [33]. In addition, deficiency in another class II HDAC, HDAC9, caused the sensitivity of the mice heart to hypertrophy by repressing MEF2 activity [34]. On the other hand, the class I deacetylase HDAC2 accelerates cardiac hypertrophy associated with the Akt/GSK-3β pathway [35]. Taken together, these studies illustrate that deacetylases have key roles as signal-responsive of the transcriptional program governing the cardiac hypertrophy and heart failure process. In fact, drugs acting as HDAC inhibitors have been developed for decreasing myocardial hypertrophy [36], which provides a direction for the treatment of cardiovascular diseases.
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are referred as critical cellular oxidants, which leads to the cell damage through oxidative modification of macromolecules including proteins and nucleotides when it is excessive [37]. S-nitrosylation at the sulfhydryl group of cysteine is most firmly implicated in the oxidative signaling [37, 38], and its dysregulation has been shown to closely related to heart diseases, including cardiac hypertrophy. For instance, Sirtuin 6 (Sirt6), a site-specific histone deacetylase that prevents development of cardiac hypertrophy and heart failure that can be oxidatively modified. In detail, tyrosine 257 of Sirt6 is nitrated upon oxidative stress, which leads to its decreased activity and mutation of tyrosine 257 abolished the stimulation- induced nitration and reduction of its activity [39]. Ago et al reported that Cys-667/Cys-669 in HDAC4 are oxidized and that form intramolecular disulfide bonds during ROS-generating hypertrophic stimuli. Trx1 recruits a stable complex with TBP-2 and DnaJb5 and subsequently reduces HDAC4 activity and its nuclear export. In this series of events, electrons are transported from NADPH to HDAC4 through the anti-hypertrophic signaling complex and multiple dithiol-disulfide exchange reactions [31, 40]. Besides, oxidative modifications of DNA and RNA are also involved in cardiovascular physiology and pathophysiology. The generation of 8-dihydro-2'-deoxyguanosine (8-OHdG) in DNA and 8-oxo-7, 8-dihydroguanosine (8-OHG) in RNA are initiated by ROS, which are two common forms of oxidative modification of nucleotides (Fig. 1C). They are commonly used as biomarkers for monitoring oxidative damage [41, 42]. A growing evidence has demonstrated that high levels of 8-OHdG in blood are associated with cardiovascular diseases. It is well documented that the concentration of 8-OHdG is very high in the heart failure patients with dilated cardiomyopathy compared to healthy subjects [43, 44]. In heart failure, a decreased number of mitochondrial DNA as well as a decline in its protein level is in agreement with increased 8-OHdG concentrations, which indicates that 8-OHdG probably promotes heart failure through targeting mitochondrial DNA [45, 46]. Additionally, oxidative modifications of RNA participate in the pathogenesis of cancers, aging and neurodegenerative diseases [47, 48]. According to a recent report from Wang et al, the oxidative modification of miR-184 by ROS forms 8-OHG, which leads to the mismatch of miR-184 with Bcl-xL and Bcl-w. This modification increases cardiomyocyte apoptosis and ischemia/ reperfusion injury [49]. However, it remains largely unknown as to how 8-OHG initiates cardiac hypertrophy? Further investigations are required to examine it.
Pathological hypertrophy-related ncRNAs
NcRNAs are recognized to play key roles in cellular differentiation, development and diseases by reprogramming gene expression through modulating RNA transcription, stability, and translation. Among them, miRNAs are the most extensively studied ncRNA type in the cardiac hypertrophic pathways [50]. They typically regulate gene expression at the post-transcriptional level by hybridizing with the 3'UTR of target mRNA and thereby inhibiting their translation or stability [51]. For example, over-expression of miR-1, one of the most abundantly expressed miRNAs in the heart, attenuates cardiomyocyte hypertrophy by negatively regulating the calcium signaling components calmodulin, Mef2a, and Gata4 [52]. Li et al reported that downregulation of miR-1 induces increased Twf1 (Twinfilin 1) expression, which in turn evokes hypertrophy through the regulation of cardiac cytoskeleton [53]. MiR-1 also acts on IGF-1 (insulin-like growth factor) and it forms a feedback loop to modulate the PI3K/Akt pathway [54]. Calcineurin signaling is a critical regulator of cardiomyocyte growth, and its downstream target NFATc3 (nuclear of activated T-cells) directly activates miR-23a expression. Meanwhile, miR-23a inhibits the translation of muscle-specific ring finger protein 1, which is an anti-hypertrophic protein [55]. With regard to the MAPK pathway, the increased activity of this signaling pathway promotes fibrosis. MiR-21 level is selectively increased in fibroblasts of the failing heart. The silencing of miR-21 by antagomir in a mouse pressure-overload-induced disease model inhibits cardiac ERK-MAP kinase activity, which reduces interstitial fibrosis and attenuates cardiac dysfunction [56]. Afterwards, Ganesan et al demonstrated that miR-378 targets four key components of the MAPK signaling: MAPK1, IGF-1 receptor, growth factor receptor-bound protein 2, and kinase suppressor of Ras 1 [57]. Overexpression of miR-378 significantly attenuated thoracic aortic constriction-induced cardiac hypertrophy and improved cardiac function in animal model. There have been so many reviews summarizing cardiovascular diseases associated miRNAs in detail (e.g. [10, 58, 59]). Figure 2 illustrates known miRNAs involved in three classical hypertrophic signaling pathways. Apart from them, many other miRNAs, such as miR-18 [60], miR-146a [61], miR-297 [62], miR-497 [63], and miR-672 [64], etc, are found to influence other pathways associated with cardiomyocyte hypertrophy. Together, these studies indicate that many functional miRNAs are being investigated for their role in pathological conditions, however, the intensive mechanisms by which miRNAs are involved in cardiac hypertrophy remains to be discovered. Further challenge lies in exploring these miRNAs as diagnostic and potential therapeutic tools for heart failure.
Noncoding RNAs involved in cardiac hypertrophic pathways. Three signaling pathways, including PI3K/Akt, MAPK and cGMP/PKG, control the hypertrophic response of cardiomyocytes under various stresses. NcRNAs marked in blue indicate anti-hypertrophic function; ncRNAs colored in red indicate pro-hypertrophic function. IGF-1, insulin-like growth factor 1; TGF-β, transforming growth factor-b; NPs, natriuretic peptides; PI3K, phosphatidylinositol 3 kinase; AKT, protein kinase B; Ras, small G-protein; MAPK, Mitogen-activated protein kinase; cGMP, cyclic guanosine monophosphate; PKG, protein kinase G.
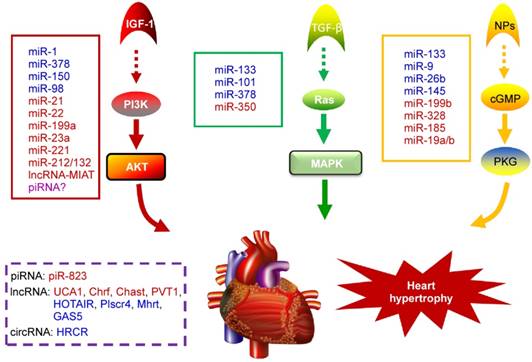
PiRNAs, are another intriguing class of small non-coding RNAs, that are involved in the regulation of epigenetic state, maintenance of genomic integrity and germ cell functions [65, 66]. It has been recently indicated that piRNAs are also expressed in cardiomyocytes and they act as new mediators of heart pathophysiology [67, 68]. In the earlier research, Rajan et al suggested that PIWI-piRNA pathway could be activated during Akt-mediated cell survival [67]. Afterwards, Rajan and his colleagues identified the abundance and altered expression of piRNAs during cardiac hypertrophy [69]. Studies from Yan et al provided evidence that piRNA-823 is directly relevant to DNMT3a/3b [70]. Meanwhile, loss of DNMT3a/3b alters the global methylation and it is responsible for various cardiac pathologies by regulating the RASSF1A-ERK1/2 signaling pathway [71, 72]. Based on these observations, we speculate that the level of DNA methylation and correlated piRNA expression could be an additional factor to determine cardiac hypertrophy [73]. Nevertheless, the role of piRNAs in cardiovascular diseases remains largely unknown and needs to be studied in detail.
Besides small ncRNAs, lncRNAs are usually defined as non-protein-coding transcripts that are involved in many biological processes. Regarding their genomic localization, lncRNAs can be derived from intergenic regions, a genomic interval between 2 genes, or from the antisense strand of a target gene [74]. LncRNAs are classified into 7 main categories based on their diverse molecular functions: cell fate specification, chromatin modification, enhancer functions, RNA splicing, nuclear architecture and compartmentalization, X chromosome inactivation, genomic imprinting [75]. The lncRNA profiles of normal adult and hypertrophied adult hearts have revealed that the expression of myocardial lncRNAs are altered upon heart failure [76, 77], which indicates new insights into the influence of lncRNAs in the pathogenesis of cardiac diseases. Mhrt (myosin heavy-chain-associated RNA transcripts), which originates from MYH7 loci, is the firstl characterized cardiac-specific lncRNA in the adult heart [78]. It is abundantly expressed under normal physiological condition and it is down-regulated under pressure overload. The inhibition of Mhrt transcription mediated by Brg1-Hdac-Parp chromatin repressor complex is an essential step for the development of cardiomyopathy, and restoration of Mhrt expression can protect the heart from the development of hypertrophy and failure. Another lncRNA that is linked to cardiac hypertrophy is Chrf, which is elevated during Ang II-induced hypertrophic response in cardiomyocytes [79]. In vivo studies confirmed that Chrf is also significantly increased in the mice heart after transverse aortic constriction and in human heart failure sample. In fact, Chrf serves as a sponge of miR-489, thereby derepressing the Myd88 suppression by miR-489 in order to regulate cardiomyocyte hypertrophy. In recent years, different research groups have identified several novel lncRNAs as critical regulators of cardiac hypertrophy by modulating complex biological processes. For instance, Chast and PVT1 are sufficient to promote cardiac hypertrophy [80, 81]. Lai et al found that lncRNA-HOTAIR is down-regulated in heart tissues from transverse aortic constriction (TAC)-operated mice, and HOTAIR acts as a competing endogenous RNA for miR-19, thereby increases the expression of its endogenous target PTEN, which is a negative modulator of cardiac hypertrophy progress [82]. In another study, a new lncRNA-Plscr4/miR-214/Mfn2 axis pathway is proposed as a blocker of hypertrophic response [83]. In this signaling pathway, Plscr4 acts as an endogenous sponge of miR-214. The forced expression of Plscr4 downregulates miR-214 expression to promote Mfn2 and attenuates hypertrophy. A recent study found that lncRNA UCA1 assembles a regulatory axis consisting of UCA1, miR-184, and mRNA HOXA9 and this axis promotes the progression of cardiac hypertrophy [84]. It is notable that lncRNA UCA1 is a well-known oncogenic molecule in cancers [85]. These findings suggest that lncRNAs are key regulators in diverse biological behaviors. Similarly, lncRNA-MIAT facilitates the development of pathological AngII-induced cardiac hypertrophy by regulating the PI3K/Akt/mTOR pathway via sponging miR-93 [86]. In addition, Wei et al found that the lncRNA GAS5/miR-23a/Foxo3a axis plays an inhibitory role in the development of cardiac hypertrophy through targeting Wnt/β-catenin signal pathway [87]. Together with above evidences, it is confirmed that the lncRNA-based interaction is mainly through the competing endogenous RNA (ceRNA) mechanism in which lncRNA adsorbs its corresponding miRNA, thus releasing the target mRNA of miRNA [88].
Circular RNA (circRNA) is a novel type of ncRNA with its 3' and 5' ends joined together to form a closed continuous loop, which was discovered decades ago [89]. In recent years, it has been demonstrated that circRNAs participate in multiple cellular processes by regulating gene expression [90], especially acting as competing endogenous RNAs to miRNAs through complementary base pairing [91]. Li et al investigated a circRNA (termed heart-related circRNA, HRCR), which acts as an endogenous miR-223 sponge to suppress its activity and thereby increasing the expression of anti-apoptotic protein ARC and inhibiting cardiac hypertrophy [92]. Following that, Li and his colleagues also found a mitochondrial fission and apoptosis-related circRNA (MFACR) in cardiomyocytes, which blocks mitochondrial fission by sequestering miR-652-3p [93]. Figure 2 summarizes the mechanism of action of cardiac hypertrophy associated piRNAs, lncRNAs and circRNAs. As circRNAs mostly serve as miRNA sponges and many miRNAs involved in the pathogenesis of cardiovascular diseases, it could be speculated that circRNAs might play important roles in cardiac pathological processes. Moreover, another circRNA MICRA is associated with heart failure after acute myocardial infarction, providing incremental values of circRNAs as prognostic biomarker for risk stratification in patients with MI [94]. Thus, circRNAs may supply great potential therapeutic avenue for cardiovascular diseases in the future.
Interplay between non-coding RNAs and chromatin modifications in regulating cardiac hypertrophy
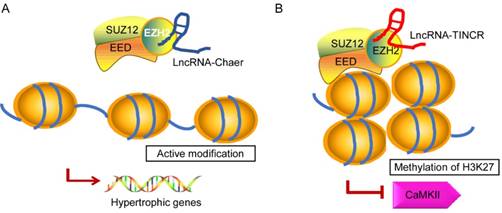
In recent years, emerging evidence implies that ncRNAs serves as a signal to modulate heart physiology and pathology by directly interacting with several epigenetic modifiers. It is of note that lncRNAs show broader associations with the chromatin modification and it is probably due to its biochemical abilities to interact with a wide range of molecules to form RNA-RNA, RNA-DNA, or RNA-protein complexes [95, 96]. Brg1 is a chromatin remodeler that is induced under pathological stress to assemble Brg1/HDAC/PARP (poly (ADP ribose) polymerases) chromatin complex, which triggers hypertrophy and heart failure [97]. LncRNA-Mhrt is predominantly expressed in the cardiac tissue that can protect heart from pathological hypertrophy [78]. Mhrt antagonizes the function of Brg1 by binding to its helicase domain, which is crucial for tethering Brg1 to chromatinize DNA targets, thus inhibiting chromatin targeting and gene regulation during stress. The Mhrt-Brg1 feedback circuit is also a conserved mechanism in human cardiomyopathy. However, the expression of Mhrt was able to reverses Brg1-mediated pathological Myh switch by directly binding to the helicase domain of Brg1 and this interaction results in removal of Brg1 from its target genes [95]. Another typical example is the interaction between heart-enriched lncRNA-Chaer and PRC2 [98] (Fig. 3A). PRC2 (polycomb repressor complex 2) is a histone methyltransferase that catalyzes the di- and trimethylation of histone H3 at H3K27, which was identified to have a hypertrophic signaling-dependent interaction with lncRNA Chaer. Chaer contributes to the timing and specificity of cardiac epigenetic reprogramming during hypertrophy by targeted modulation of histone methylation and de-methylation [98, 99]. On the other hand, lncRNA TINCR can attenuate myocardial hypertrophy by targeting EZH2, which binds to CaMKII promoter region and mediates H3K27me3 modification [100] (Fig. 3B). In addition, PRC2-interacting lncRNAs, such as Fendrr and ANRIL, play an essential role in the regulatory networks controlling the heart development and diseases [101, 102].
Proposed models for the interaction of lncRNA-Chaer/TINCR and PRC2. In normal cases, PRC2 inhibits gene activation by trimethylation of H3K27 at the promoters. EZH2, EED, and SUZ12 are core components of PRC2 complex. A: Under stress stimulation, lncRNA-Chaer directly interacts with the catalytic subunit (EZH2) of PRC2 and interferes with PRC2 genomic targeting, thereby activating hypertrophic gene expression. B: LncRNA-TINCR binds to PRC2 and provokes EZH2-mediated H3K27me3 modification by recruiting it to the promoter of CaMKII, which functions as an inducer of cardiac hypertrophy.
Growing number of researches in recent years have demonstrated that piRNAs also perform critical functions in the regulation of epigenetic status. For instance, piwi/piRNA complex facilitates serotonin-dependent methylation of a conserved CpG island in the promoter of CREB2 and consequently leads to enhanced long-term memory [103]. It is well known that piRNAs silence transposons and their nearby gene expression through recruiting piwi proteins and altering H3K9 tri-methylation [104]. In addition, the expression of some piRNAs are highly enriched in the heart [105], implying unique advantages of piRNAs combined with epigenetic modifications in cardiovascular diseases. However, the underlying mechanisms by which piRNAs regulating epigenetic status and cardiac hypertrophy have not yet been completely clarified.
Future considerations
Over the past decade, epigenetic modifications and ncRNAs have been emerged as critical components in the regulation of myocardial function in health and disease, including cardiac hypertrophy. Taken together with their emerging role in chromatin modifications, the ncRNAs would offer novel perspectives and new approaches to restore or reverse the effects of pathological hypertrophy in the failing heart. Recently, circulating miRNAs have emerged as new biomarkers in patients with heart diseases due to the fact that miRNAs in serum or plasma samples can be easily detected [106]. For instance, elevated plasma levels of miR-29a are associated with hemolysis in patients with hypertrophic cardiomyopathy [107]. MiR-7-5p and miR-26b-5p are upregulated in the circulation of patients with left ventricular hypertensive compared with healthy subjects, which entail them as novel biomarkers for this disease [108]. Given the fact that ncRNA mimics or inhibitors can be easily synthesized and delivered to cells or organs with low toxicity [109], ncRNAs will also be a promising and important modifiers to prevent cardiac hypertrophy. A study of Santaris Pharma's LNA-modified anti-miR-122 for the treatment of hepatitis C virus infection has completed phase 1 and phase 2 clinical trials without causing viral resistance [110]. The inhibition of miR-208a also improved survival and cardiac function during HF progression in rat model [111]. However, there are still many challenges to be resolved in the development of ncRNA-based diagnostics or therapy, e.g. the difficulty in miRNA quantitation, the off-target effects, and so on. Up to now, some inhibitors targeting DNMTs or HDACs have shown significant benefits in the treatment of cardiovascular diseases [112]. For example, DNA methylation inhibitor zebularine has gained attention in the treatment of cardiac injury and HF as a result of its positive effect on cardiomyocyte differentiation from embryonic stem cells [113]. Curcumin is a natural compound from turmeric, which exerts its anti-fibrotic and anti-hypertrophic effects by suppressing activity of HDAC1, 3, and 8, respectively [114]. However, it remains to be determined whether these agents could be transformed in the development of new therapies for cardiac diseases. In this review, we have addressed DNA methylation as well as other epigenetic mechanisms such as histone modifications, oxidative modification, and non-coding RNA-mediated mechanisms, which are fascinating and pose challenges to the development of cardiac-specific therapeutic applications and unique biomarkers for cardiac hypertrophy. However, further works must be conducted to elucidate the complex mechanisms of ncRNA-mediated cardiovascular epigenetics in myocardial pathological conditions.
Acknowledgements
This work was supported by the Natural Science Foundation of China (Grant No: 31701733, 81470522, 31430041), Applied Basic Research Programs of Qingdao, China (Grant No: 17-1-1-46-jch), and Shandong Provincial Natural Science Foundation, China (Grant No: ZR2016CQ31).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Go AS, Mozaffarian D, Roger VL. et al. Heart disease and stroke statistics-2014 update: a report from the American Heart Association. Circulation. 2014;129:e28-e292
2. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. Journal of Molecular and Cellular Cardiology. 2016;97:245-62
3. Dadson K, Hauck L, Billia F. Molecular mechanisms in cardiomyopathy. Clin Sci (Lond). 2017;131:1375-92
4. Shirakabe A, Ikeda Y, Sciarretta S. et al. Aging and Autophagy in the Heart. Circ Res. 2016;118:1563-76
5. Rachmin I, Tshori S, Smith Y. et al. Erbin is a negative modulator of cardiac hypertrophy. P Natl Acad Sci USA. 2014;111:5902-7
6. Mehta G, Kumarasamy S, Wu J. et al. MITF interacts with the SWI/SNF subunit, BRG1, to promote GATA4 expression in cardiac hypertrophy. Journal of Molecular and Cellular Cardiology. 2015;88:101-10
7. Rosca MG, Tandler B, Hoppel CL. Mitochondria in cardiac hypertrophy and heart failure. Journal of Molecular and Cellular Cardiology. 2013;55:31-41
8. Yang Y, Del Re DP, Nakano N. et al. miR-206 Mediates YAP-Induced Cardiac Hypertrophy and Survival. Circ Res. 2015;117:891-904
9. Schiano C, Vietri MT, Grimaldi V. et al. Epigenetic-related therapeutic challenges in cardiovascular disease. Trends Pharmacol Sci. 2015;36:226-35
10. Wang J, Yang X. The function of miRNA in cardiac hypertrophy. Cell Mol Life Sci. 2012;69:3561-70
11. Dulac C. Brain function and chromatin plasticity. Nature. 2010;465:728-35
12. Martinez SR, Gay MS, Zhang L. Epigenetic mechanisms in heart development and disease. Drug Discov Today. 2015;20:799-811
13. Bagga S, Bracht J, Hunter S. et al. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005;122:553-63
14. Folco HD, Pidoux AL, Urano T. et al. Heterochromatin and RNAi are required to establish CENP-A chromatin at centromeres. Science. 2008;319:94-7
15. Varambally S, Cao Q, Mani RS. et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science. 2008;322:1695-9
16. Boon RA, Dimmeler S. MicroRNAs in myocardial infarction. Nat Rev Cardiol. 2015;12:135-42
17. Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204-20
18. Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156:45-68
19. Reik W, Dean W, Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089-93
20. Hendrich B, Tweedie S. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 2003;19:269-77
21. Greco CM, Condorelli G. Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat Rev Cardiol. 2015;12:488-97
22. Movassagh M, Choy MK, Knowles DA. et al. Distinct epigenomic features in end-stage failing human hearts. Circulation. 2011;124:2411-22
23. Gilsbach R, Preissl S, Gruning BA. et al. Dynamic DNA methylation orchestrates cardiomyocyte development, maturation and disease. Nat Commun. 2014;5:5288
24. Luger K, Mader AW, Richmond RK. et al. Crystal structure of the nucleosome core particle at 2.8 angstrom resolution. Nature. 1997;389:251-60
25. Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074-80
26. Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41-5
27. Papait R, Cattaneo P, Kunderfranco P. et al. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc Natl Acad Sci U S A. 2013;110:20164-9
28. Vaquero A, Loyola A, Reinberg D. The constantly changing face of chromatin. Sci Aging Knowledge Environ. 2003;2003:RE4
29. Verdone L, Caserta M, Di Mauro E. Role of histone acetylation in the control of gene expression. Biochem Cell Biol. 2005;83:344-53
30. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381-95
31. Ago T, Liu T, Zhai P. et al. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell. 2008;133:978-93
32. Hohl M, Wagner M, Reil JC. et al. HDAC4 controls histone methylation in response to elevated cardiac load. J Clin Invest. 2013;123:1359-70
33. Demos-Davies KM, Ferguson BS, Cavasin MA. et al. HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling. Am J Physiol Heart Circ Physiol. 2014;307:H252-8
34. Zhang CL, McKinsey TA, Chang SR. et al. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479-88
35. Trivedi CM, Luo Y, Yin Z. et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat Med. 2007;13:324-31
36. Gallo P, Latronico MV, Grimaldi S. et al. Inhibition of class I histone deacetylase with an apicidin derivative prevents cardiac hypertrophy and failure. Cardiovasc Res. 2008;80:416-24
37. Rasmussen HH, Hamilton EJ, Liu CC. et al. Reversible oxidative modification: implications for cardiovascular physiology and pathophysiology. Trends Cardiovasc Med. 2010;20:85-90
38. Janssen-Heininger YM, Mossman BT, Heintz NH. et al. Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic Biol Med. 2008;45:1-17
39. Hu S, Liu H, Ha Y. et al. Posttranslational modification of Sirt6 activity by peroxynitrite. Free Radic Biol Med. 2015;79:176-85
40. Oka S, Ago T, Kitazono T. et al. The role of redox modulation of class II histone deacetylases in mediating pathological cardiac hypertrophy. J Mol Med (Berl). 2009;87:785-91
41. Shan X, Lin CL. Quantification of oxidized RNAs in Alzheimer's disease. Neurobiol Aging. 2006;27:657-62
42. Lee CY, Isaac HB, Wang H. et al. Cautions in the use of biomarkers of oxidative damage; the vascular and antioxidant effects of dark soy sauce in humans. Biochem Biophys Res Commun. 2006;344:906-11
43. Watanabe E, Matsuda N, Shiga T. et al. Significance of 8-hydroxy-2'-deoxyguanosine levels in patients with idiopathic dilated cardiomyopathy. J Card Fail. 2006;12:527-32
44. Kono Y, Nakamura K, Kimura H. et al. Elevated levels of oxidative DNA damage in serum and myocardium of patients with heart failure. Circ J. 2006;70:1001-5
45. Karamanlidis G, Nascimben L, Couper GS. et al. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. 2010;106:1541-8
46. Kroese LJ, Scheffer PG. 8-hydroxy-2'-deoxyguanosine and cardiovascular disease: a systematic review. Curr Atheroscler Rep. 2014;16:452
47. Zhan Y, Dhaliwal JS, Adjibade P. et al. Localized control of oxidized RNA. J Cell Sci. 2015;128:4210-9
48. Ding Q, Markesbery WR, Chen Q. et al. Ribosome dysfunction is an early event in Alzheimer's disease. J Neurosci. 2005;25:9171-5
49. Wang JX, Gao J, Ding SL. et al. Oxidative Modification of miR-184 Enables It to Target Bcl-xL and Bcl-w. Mol Cell. 2015;59:50-61
50. Greco S, Gorospe M, Martelli F. Noncoding RNA in age-related cardiovascular diseases. J Mol Cell Cardiol. 2015;83:142-55
51. Joladarashi D, Thandavarayan RA, Babu SS. et al. Small Engine, Big Power: MicroRNAs as Regulators of Cardiac Diseases and Regeneration. International Journal of Molecular Sciences. 2014;15:15891-911
52. Ikeda S, He A, Kong SW. et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol Cell Biol. 2009;29:2193-204
53. Li Q, Song XW, Zou J. et al. Attenuation of microRNA-1 derepresses the cytoskeleton regulatory protein twinfilin-1 to provoke cardiac hypertrophy. J Cell Sci. 2010;123:2444-52
54. Elia L, Contu R, Quintavalle M. et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation. 2009;120:2377-85
55. Lin Z, Murtaza I, Wang K. et al. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci U S A. 2009;106:12103-8
56. Thum T, Gross C, Fiedler J. et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980-4
57. Ganesan J, Ramanujam D, Sassi Y. et al. MiR-378 controls cardiac hypertrophy by combined repression of mitogen-activated protein kinase pathway factors. Circulation. 2013;127:2097-106
58. Wang J, Liew OW, Richards AM. et al. Overview of MicroRNAs in Cardiac Hypertrophy, Fibrosis, and Apoptosis. Int J Mol Sci. 2016:17
59. Da Costa Martins PA, De Windt LJ. MicroRNAs in control of cardiac hypertrophy. Cardiovasc Res. 2012;93:563-72
60. Huang CY, Pai PY, Kuo CH. et al. p53-mediated miR-18 repression activates HSF2 for IGF-IIR-dependent myocyte hypertrophy in hypertension-induced heart failure. Cell Death Dis. 2017;8:e2990
61. Heggermont WA, Papageorgiou AP, Quaegebeur A. et al. Inhibition of MicroRNA-146a and Overexpression of Its Target Dihydrolipoyl Succinyltransferase Protect Against Pressure Overload-Induced Cardiac Hypertrophy and Dysfunction. Circulation. 2017;136:747-61
62. Bao Q, Zhao M, Chen L. et al. MicroRNA-297 promotes cardiomyocyte hypertrophy via targeting sigma-1 receptor. Life Sci. 2017;175:1-10
63. Xiao Y, Zhang X, Fan S. et al. MicroRNA-497 Inhibits Cardiac Hypertrophy by Targeting Sirt4. PLoS One. 2016;11:e0168078
64. Lu Y, Wu F. A new miRNA regulator, miR-672, reduces cardiac hypertrophy by inhibiting JUN expression. Gene. 2018;648:21-30
65. Brennecke J, Aravin AA, Stark A. et al. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007;128:1089-103
66. Moyano M, Stefani G. piRNA involvement in genome stability and human cancer. Journal of Hematology & Oncology. 2015:8
67. Rajan KS, Velmurugan G, Pandi G. et al. miRNA and piRNA mediated Akt pathway in heart: antisense expands to survive. Int J Biochem Cell Biol. 2014;55:153-6
68. Vella S, Gallo A, Lo Nigro A. et al. PIWI-interacting RNA (piRNA) signatures in human cardiac progenitor cells. Int J Biochem Cell Biol. 2016;76:1-11
69. Rajan KS, Velmurugan G, Gopal P. et al. Abundant and Altered Expression of PIWI-Interacting RNAs during Cardiac Hypertrophy. Heart Lung Circ. 2016;25:1013-20
70. Yan H, Wu QL, Sun CY. et al. piRNA-823 contributes to tumorigenesis by regulating de novo DNA methylation and angiogenesis in multiple myeloma. Leukemia. 2015;29:196-206
71. Nuhrenberg TG, Hammann N, Schnick T. et al. Cardiac Myocyte De Novo DNA Methyltransferases 3a/3b Are Dispensable for Cardiac Function and Remodeling after Chronic Pressure Overload in Mice. PLoS One. 2015;10:e0131019
72. Tao H, Yang JJ, Chen ZW. et al. DNMT3A silencing RASSF1A promotes cardiac fibrosis through upregulation of ERK1/2. Toxicology. 2014;323:42-50
73. Rajan KS, Ramasamy S, George-William JN. et al. Emerging cardiac non-coding landscape: The importance of meta-analysis. Biochimie. 2017;133:87-94
74. Moran VA, Perera RJ, Khalil AM. Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 2012;40:6391-400
75. Thum T, Condorelli G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ Res. 2015;116:751-62
76. Matkovich SJ, Edwards JR, Grossenheider TC. et al. Epigenetic coordination of embryonic heart transcription by dynamically regulated long noncoding RNAs. Proc Natl Acad Sci U S A. 2014;111:12264-9
77. Lee JH, Gao C, Peng GD. et al. Analysis of Transcriptome Complexity Through RNA Sequencing in Normal and Failing Murine Hearts. Circulation Research. 2011;109:1332-41
78. Han P, Li W, Lin CH. et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature. 2014;514:102-6
79. Wang K, Liu F, Zhou LY. et al. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res. 2014;114:1377-88
80. Viereck J, Kumarswamy R, Foinquinos A. et al. Long noncoding RNA Chast promotes cardiac remodeling. Sci Transl Med. 2016;8:326ra22
81. Yu YH, Hu ZY, Li MH. et al. Cardiac hypertrophy is positively regulated by long non-coding RNA PVT1. Int J Clin Exp Pathol. 2015;8:2582-9
82. Lai Y, He S, Ma L. et al. HOTAIR functions as a competing endogenous RNA to regulate PTEN expression by inhibiting miR-19 in cardiac hypertrophy. Mol Cell Biochem. 2017;432:179-87
83. Lv L, Li T, Li X. et al. The lncRNA Plscr4 Controls Cardiac Hypertrophy by Regulating miR-214. Mol Ther Nucleic Acids. 2018;10:387-97
84. Zhou G, Li C, Feng J. et al. lncRNA UCA1 Is a Novel Regulator in Cardiomyocyte Hypertrophy through Targeting the miR-184/HOXA9 Axis. Cardiorenal Med. 2018;8:130-9
85. Zhou Y, Chen Y, Ding W. et al. LncRNA UCA1 impacts cell proliferation, invasion, and migration of pancreatic cancer through regulating miR-96/FOXO3. Iubmb Life. 2018;70:276-90
86. Li Y, Wang J, Sun L. et al. LncRNA myocardial infarction-associated transcript (MIAT) contributed to cardiac hypertrophy by regulating TLR4 via miR-93. Eur J Pharmacol. 2018;818:508-17
87. Wei W, Chen Y, Gao J. et al. WITHDRAWN: The lncRNA-GAS5/miR-23a/Foxo3a axis regulates cardiac hypertrophy by Wnt/beta-catenin signal pathway. Biochem Biophys Res Commun. 2017;494:424
88. Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152:1298-307
89. Hsu MT, Coca-Prados M. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature. 1979;280:339-40
90. Memczak S, Jens M, Elefsinioti A. et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333-8
91. Hansen TB, Jensen TI, Clausen BH. et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384-8
92. Wang K, Long B, Liu F. et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J. 2016;37:2602-11
93. Wang K, Gan TY, Li N. et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death and Differentiation. 2017;24:1111-20
94. Salgado-Somoza A, Zhang L, Vausort M. et al. The circular RNA MICRA for risk stratification after myocardial infarction. Ijc Heart Vasc. 2017;17:33-6
95. Chang CP, Han P. Epigenetic and lncRNA regulation of cardiac pathophysiology. Biochim Biophys Acta. 2016;1863:1767-71
96. Quinn JJ, Ilik IA, Qu K. et al. Revealing long noncoding RNA architecture and functions using domain-specific chromatin isolation by RNA purification. Nat Biotechnol. 2014;32:933-40
97. Hang CT, Yang J, Han P. et al. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature. 2010;466:62-7
98. Wang ZH, Zhang XJ, Ji YX. et al. The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med. 2016;22:1131-9
99. Shi L, Sun LY, Li Q. et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. P Natl Acad Sci USA. 2011;108:7541-6
100. Shao M, Chen G, Lv F. et al. LncRNA TINCR attenuates cardiac hypertrophy by epigenetically silencing CaMKII. Oncotarget. 2017;8:47565-73
101. Grote P, Wittler L, Hendrix D. et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev Cell. 2013;24:206-14
102. Yap KL, Li SD, Munoz-Cabello AM. et al. Molecular Interplay of the Noncoding RNA ANRIL and Methylated Histone H3 Lysine 27 by Polycomb CBX7 in Transcriptional Silencing of INK4a. Mol Cell. 2010;38:662-74
103. Rajasethupathy P, Antonov I, Sheridan R. et al. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell. 2012;149:693-707
104. Sienski G, Donertas D, Brennecke J. Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell. 2012;151:964-80
105. Rajan KS, Velmurugan G, Gopal P. et al. Abundant and Altered Expression of PIWI-Interacting RNAs during Cardiac Hypertrophy. Heart Lung Circ. 2016;25:1013-20
106. Creemers EE, Tijsen AJ, Pinto YM. Circulating microRNAs: novel biomarkers and extracellular communicators in cardiovascular disease? Circ Res. 2012;110:483-95
107. Ntelios D, Meditskou S, Efthimiadis G. et al. Elevated plasma levels of miR-29a are associated with hemolysis in patients with hypertrophic cardiomyopathy. Clin Chim Acta. 2017;471:321-6
108. Kaneto CM, Nascimento JS, Moreira MCR. et al. MicroRNA profiling identifies miR-7-5p and miR-26b-5p as differentially expressed in hypertensive patients with left ventricular hypertrophy. Braz J Med Biol Res. 2017;50:e6211
109. Dong Y, Liu C, Zhao Y. et al. Role of noncoding RNAs in regulation of cardiac cell death and cardiovascular diseases. Cell Mol Life Sci. 2018;75:291-300
110. Janssen HL, Reesink HW, Lawitz EJ. et al. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685-94
111. Montgomery RL, Hullinger TG, Semus HM. et al. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation. 2011;124:1537-47
112. Zhang Y, Ren J. Epigenetics and obesity cardiomyopathy: From pathophysiology to prevention and management. Pharmacol Ther. 2016;161:52-66
113. Horrillo A, Pezzolla D, Fraga MF. et al. Zebularine regulates early stages of mESC differentiation: effect on cardiac commitment. Cell Death Dis. 2013;4:e570
114. Tao H, Shi KH, Yang JJ. et al. Histone deacetylases in cardiac fibrosis: current perspectives for therapy. Cell Signal. 2014;26:521-7
Author contact
![]() Corresponding authors: Yanhan Dong, Email: yanhandongedu.cn; Peifeng Li, Email: peifliedu.cn; Kun Wang, Email: wangk696edu.cn; Tel: 86-532-82991791
Corresponding authors: Yanhan Dong, Email: yanhandongedu.cn; Peifeng Li, Email: peifliedu.cn; Kun Wang, Email: wangk696edu.cn; Tel: 86-532-82991791