Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(10):1175-1185. doi:10.7150/ijbs.26011 This issue Cite
Research Paper
Astemizole Inhibits mTOR Signaling and Angiogenesis by Blocking Cholesterol Trafficking
Junfang Lyu1, Eun Ju Yang1, Sarah A. Head2, Nana Ai1, Baoyuan Zhang1, Changjie Wu1, Ruo-Jing Li2, Yifan Liu1, Harapriya Chakravarty1, Shaolin Zhang1, Kin Yip Tam1, Yongjun Dang3, Ho Jeong Kwon4, Wei Ge1, Jun O. Liu2,5, Joong Sup Shim1 ![]()
1. Faculty of Health Sciences, University of Macau, Taipa, Macau SAR, China
2. Department of Pharmacology and Molecular Sciences, Johns Hopkins School of Medicine, Baltimore, MD 21205, USA; The SJ Yan and HJ Mao Laboratory of Chemical Biology, Johns Hopkins School of Medicine, Baltimore, MD 21205, USA
3. Key Laboratory of Metabolism and Molecular Medicine, Ministry of Education, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Fudan University, Shanghai 200032, China
4. Chemical Genomics Global Research Laboratory, Department of Biotechnology, College of Life Science & Biotechnology, Yonsei University, Seoul 120-749, Republic of Korea
5. Department of Oncology, Johns Hopkins School of Medicine, Baltimore, MD 21205, USA
Received 2018-3-12; Accepted 2018-5-3; Published 2018-6-23
Abstract

Cholesterol plays a key role in membrane protein function and signaling in endothelial cells. Thus, disturbing cholesterol trafficking is an effective approach for inhibiting angiogenesis. We recently identified astemizole (AST), an antihistamine drug, as a cholesterol trafficking inhibitor from a phenotypic screen. In this study, we found that AST induced cholesterol accumulation in the lysosome by binding to the sterol-sensing domain of Niemann-Pick disease, type C1 (NPC1), a lysosomal surface protein responsible for cholesterol transport. Inhibition of cholesterol trafficking by AST led to the depletion of membrane cholesterol, causing SREBP1 nuclear localization. The depletion of membrane cholesterol resulted in dissociation of mammalian target of rapamycin (mTOR) from the lysosomal surface and inactivation of mTOR signaling. These effects were effectively rescued by addition of exogenous cholesterol. AST inhibited endothelial cell proliferation, migration and tube formation in a cholesterol-dependent manner. Furthermore, AST inhibited zebrafish angiogenesis in a cholesterol-dependent manner. Together, our data suggest that AST is a new class of NPC1 antagonist that inhibits cholesterol trafficking in endothelial cells and angiogenesis.
Keywords: cholesterol trafficking, angiogenesis, astemizole, NPC1, mTOR
Introduction
Cholesterol metabolism and trafficking have recently been highlighted as emerging drug targets for anti-angiogenic agents. Cholesterol is a vital component of the membrane bilayer where it plays a key role in maintaining the membrane structure and regulating intracellular transport and signal transduction [1, 2]. In endothelial cells, plasma membrane cholesterol is important for VEGFR2-mediated signaling and its depletion in turn causes inhibition of angiogenesis [3, 4]. We and other groups recently showed that cholesterol trafficking is important for proper intracellular angiogenic signaling. Small molecule inhibitors of Niemann-Pick C1 (NPC1), a lysosomal cholesterol transport protein, or those that change lysosomal pH could block cholesterol trafficking at the lysosomes, leading to accumulation of free cholesterol inside the lysosomal lumen, inhibition of mTOR signaling in endothelial cells and thereby blocking angiogenesis [5-7]. Based on this notion, we previously screened an FDA-approved drug library (Johns Hopkins Drug Library, JHDL) to identify cholesterol trafficking inhibitors from the existing drug space. From the study, we found 13 existing drugs, including cepharanthine (CEP) and astemizole (AST), as cholesterol trafficking inhibitors in endothelial cells [8].
AST is a non-sedating selective H1-histamine receptor antagonist that has been approved in the UK and USA since the mid-1980s for the treatment of allergic rhinitis [9]. Due to rare but fatal side effects related to the inhibition of hERG potassium channel, it was withdrawn from the global market in 1999 [10]. However, AST was recently repurposed as an anti-malarial drug and is being sold in more than 30 countries [11]. The ether-a-gogo (Eag1) and Eag-related gene (hERG) potassium channels are two well-known targets of AST [12, 13]. A recent report showed that AST inhibited Eag1-dependent tumor growth by inhibiting tumor cell hypoxia inducible factor (HIF)-1α activity and the secretion of vascular endothelial growth factor (VEGF), thereby blocking tumor angiogenesis [14]. More recent studies also suggested a direct antitumor effect of AST with regard to the inhibition of Eag1 potassium channel [15, 16]. However, it is unclear whether Eag1 or hERG potassium channel is the sole target of AST for its antitumor effects. Furthermore, the Eag1 or hERG-related pharmacological effect accompanies a risk of fatal side effects, limiting further clinical development of the drug as an antitumor agent. Therefore, it is important to elaborate other possible molecular targets of AST in addition to Eag1 or hERG in its antitumor effects.
In this study, we show that AST inhibits angiogenesis by binding to NPC1 and blocking endothelial cell cholesterol trafficking. The blockade of cholesterol trafficking led to inhibition of mTOR signaling, a master regulator of cellular metabolism, protein synthesis and proliferation, in endothelial cells. Our study provides strong evidence that the NPC1-mediated cholesterol trafficking pathway is an additional target of AST for its anti-angiogenic and antitumor effects.
Materials and Methods
Cell culture
Human umbilical vein endothelial cells (HUVEC) were purchased from Thermo Fisher Scientific and grown in Medium 200 supplemented with Low serum growth supplement (LSGS) (Thermo Fisher Scientific, Waltham, MA). A549 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA). A549 cells were grown in RPMI Medium 1640 supplemented with 10% fetal bovine serum (Thermo Fisher Scientific). All the cells were cultured in a humidified incubator at 37ºC adjusted to 5% CO2.
Reagents and antibodies
Astemizole (AST) and cholesterol were purchased from Santa Cruz Biotechnology (Dallas, Texas). Methyl-β-cyclodextrin, filipin, itraconazole and N-Phenylthiourea (PTU) were purchased from Sigma-Aldrich (St. Louis, MO). Hoechst 33342 (H3570) was purchased from Thermo Fisher Scientific. Antibodies against S6 kinase (S6K, sc-8418), phospho-S6K (Thr421/Ser424, sc-7984-R), glyceraldehyde 3-phosphate dehydrogenase (GAPDH, sc-365062) and lysosomal-associated membrane protein 1 (LAMP1, sc-20011) and SRE-binding proteins (SREBP-1, sc-8984) were purchased from Santa Cruz Biotechnology. Antibodies against eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1, 9452S), protein disulfide isomerase (PDI, 3501S) and mammalian target of rapamycin (mTOR, 2983S) were purchased from Cell Signaling Technology (Danvers, MA). Antibody against GM130 (610823) was purchased from BD Biosciences (San Jose, CA). Antibody against NPC1 (13926-1-AP) was from Proteintech (Chicago, IL). Horseradish peroxidase (HRP)-conjugated secondary antibodies (sc-2005, goat anti-mouse IgG-HRP; sc-2004, goat anti-rabbit IgG-HRP) were purchased from Santa Cruz Biotechnology. Secondary antibodies conjugated with Alexa Fluor 488 (A21202, donkey anti-mouse IgG conjugate; A21206, donkey anti-rabbit IgG conjugate) and Alexa Fluor 647 (A21235, goat anti-mouse IgG conjugate; A21244, goat anti-rabbit IgG conjugate) were from Thermo Fisher Scientific.
Immunofluorescence
HUVEC were cultured in a Nunc Lab-Tek II 8-Chamber Slide (Thermo Scientific, Rockford, IL) at 2 × 104 cells/well and treated with compounds for indicated time. Cells were then fixed with 4% paraformaldehyde for 20 min at room temperature. For general immunostaining of proteins, cells were washed by PBS and permeabilized with 0.5% Triton X-100 or 0.2% saponin (for co-staining with filipin) for 20 min. Cells were blocked with 3% bovine serum albumin (BSA) in PBS containing 0.1% Tween-20 for 1 h and incubated with primary antibodies overnight at 4ºC. Cells were then incubated with secondary antibodies conjugated with Alexa Fluor 488 or Alexa Fluor 647 for 1 h at room temperature. The cellular nuclei were stained with Hoechst 33342. Cells were rinsed with PBS, mounted with Immu-mount (Thermo Fisher Scientific), and observed under a Carl Zeiss LSM 710 confocal microscope (Carl Zeiss, Thornwood, NY). For filipin staining of cholesterol, cells were fixed with 4% paraformaldehyde for 20 min, washed with PBS, and stained with filipin at a final concentration of 50 µg/ml for 1 h at room temperature in a darkroom. Cells were then washed with PBS and mounted with Immu-mount prior to observation under the confocal microscope.
NPC1 competitive binding assay
NPC1 competitive binding assay was performed as previously described [17]. Briefly, A549 cells were seeded into 6-cm dishes at the density of 3 × 106 cells overnight and treated with competitors for 30 min before addition of 200 nM itraconazole photo-affinity probe for 1 h. The cells were rinsed with cold PBS and irradiated with UV for 3 min on ice. After cell lysis with 0.4% sodium dodecyl sulfate (SDS) and sonication, proteins were denatured by boiling for 5 min followed by capture of the itraconazole photo-affinity probe by click reaction with biotin-azide (Click Chemistry Tools, Scottsdale, AZ). Proteins were then precipitated with four volumes of cooled acetone to remove unreacted biotin-azide, and after resolubilization the biotin-labeled proteins were isolated with streptavidin beads (Thermo Fisher Scientific) and subjected to Western blotting with NPC1 antibody.
Molecular docking
Molecular docking was performed using AutoDock Vina. The structure file for the protein (5U73) was obtained from protein data bank (http://www.rcsb.org/pdb) [18], and the structure files for itraconazole, U18666A, cholesterol and astemizole were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/). The configuration settings were followed as described previously [17].
Cell proliferation assay
HUVEC were seeded in 96 well plates (2 × 103 cells /well) or 12 well plates (5 × 104 cells /well) and treated with compounds for 72 hr. AlamarBlue reagent (Thermo Fisher Scientific) was added into the media at a final concentration of 10% for 2 h. The fluorescence signal was measured with an excitation wavelength of 560 nm and an emission wavelength of 590 nm using a SpectraMax M5 fluorescence microplate reader (Molecular Devices, Sunnyvale, CA). The data were plotted using the GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA).
Wound healing cell migration assay
The wound healing assay was performed as previously described [8]. HUVEC were seeded in a 96-well ImageLock Microplate (Essen BioScience, Ann Arbor, MI) at the density of 1 × 104 cells/well. The wound area was created by the WoundMakerTM (Essen BioScience). Cells were then treated with compounds for 28 h. Cell migration was measured in real-time using the IncuCyte ZOOM based on relative wound density. The data were plotted by GraphPad Prism 5.0 software.
Tube formation assay
A 15 well µ-slide plate (ibidi, Martinsried, Germany) was coated with 10 µL Matrigel (BD Biosciences) at 37ºC for 30 min until solidified. HUVEC treated with compounds were seeded on the Matrigel-coated wells at a density of 1.5 × 103 cells/well in 50 μL medium. After 20 h incubation, tubular structures were stained with Calcein-AM (2 μM, Thermo Fisher Scientific) and observed under a Carl Zeiss Axio Observer fluorescence microscope (Carl Zeiss). Area of tube formation was quantified using the Angiogenesis Analyzer for Image J [19].
Western blotting
HUVEC seeded in 6-well plates at a density of 1 × 105 cells/well were treated with compounds for 24 h. Cells were lysed by 2× Laemmli buffer (Bio-Rad, Hercules, CA) and run on the SDS-PAGE gel prior to transfer to a nitrocellulose membrane. The membrane was blocked with 5% non-fat dried milk at room temperature for 1 h and incubated with primary antibodies at 4ºC overnight. The membrane was then incubated with HRP-conjugated secondary antibody at room temperature for 1 h. The protein was detected with enhanced chemiluminescence (ECL, Bio-Rad, Hercules, CA) under a ChemiDoc MP Imaging system (Bio-Rad).
Zebrafish angiogenesis assay
The transgenic zebrafish line Tg(fli1a:EGFP)y1 was used to evaluate in vivo angiogenesis as previously described [20]. Zebrafish embryos were generated by natural pairwise mating and maintained at 28°C in embryo water (0.2 g/L of Instant Ocean® Salt in distilled water). AST (100 and 200 μM in PBS containing 1% DMSO) with or without cholesterol-cyclodextrin complex (800 μg/mL cholesterol in 10% methyl-β-cyclodextrin) was directly injected into embryos at 8 hour-post-fertilization (hpf) with Nanoject II Auto-Nanoliter Injector (Drummond Scientific Company, Broomall, PA) at the injection volume of 4.6 nL. After 24 h, N-phenylthiourea (PTU, 3 mg/mL, Sigma-Aldrich) was added into the embryos. All the treatment groups were incubated at 28°C for 72 h. The formation of subintestinal vessels (SIVs) were observed under a Carl Zeiss LSM 710 confocal microscope. The number of SIV branch points were used to assess the zebrafish angiogenesis and plotted with the GraphPad Prism 5.0 software.
Statistical analysis
Statistical differences of the data between control and treatment groups were tested by two-sided Student's t-test or One Sample t-test with the GraphPad Prism 5.0 software. The P values less than 0.05 was considered significant.
Results
AST induces NPC-like phenotype in endothelial cells
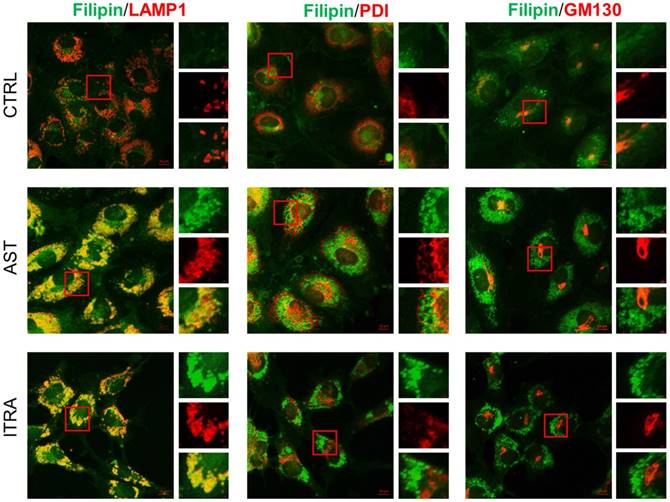
We previously conducted a phenotypic screen to identify endothelial cell cholesterol trafficking inhibitors from a FDA-approved drug library and AST was amongst the candidate drugs identified from the screening [8]. In this study, we further investigated the pharmacological effects of AST at the cellular and molecular levels in endothelial cells. We have shown that all the cholesterol trafficking inhibitors identified, including AST, induced a significant accumulation of cholesterol in the perinuclear region in endothelial cells, a phenomenon called NPC-like phenotype [8]. We then analyzed the precise location of the cholesterol accumulation in endothelial cells using the subcellular organelle markers, including LAMP1 (lysosomal-associated membrane protein 1, a marker of late endolysosomes), PDI (protein disulfide isomerase, a marker of the endoplasmic reticulum) and GM130 (a marker of cis-Golgi). Immunofluorescence analysis of intracellular cholesterol and organelle markers revealed that cholesterol accumulation by AST was nicely colocalized with the LAMP1-positive late endolysosomes, but not with that of endoplasmic reticulum or Golgi (Fig. 1). Itraconazole, a small molecule inhibitor of NPC1, showed a similar effect on endothelial cell cholesterol. These data demonstrated that AST inhibited cholesterol trafficking in the late endolysosomes and induced NPC-like phenotype in endothelial cells.
Astemizole induces cholesterol accumulation in the late endolysosomes in HUVEC. HUVEC were treated with indicated compounds for 8 h and processed for immunofluorescence staining of filipin (cholesterol, green), LAMP1 (endolysosome marker, red), PDI (endoplasmic reticulum marker, red), and GM130 (Golgi marker, red). Scale bar = 10 μm. The image inlets (red square) were magnified and shown on the right at each figure. Scale bar = 1μm.
AST binds to the sterol-sensing domain of NPC1
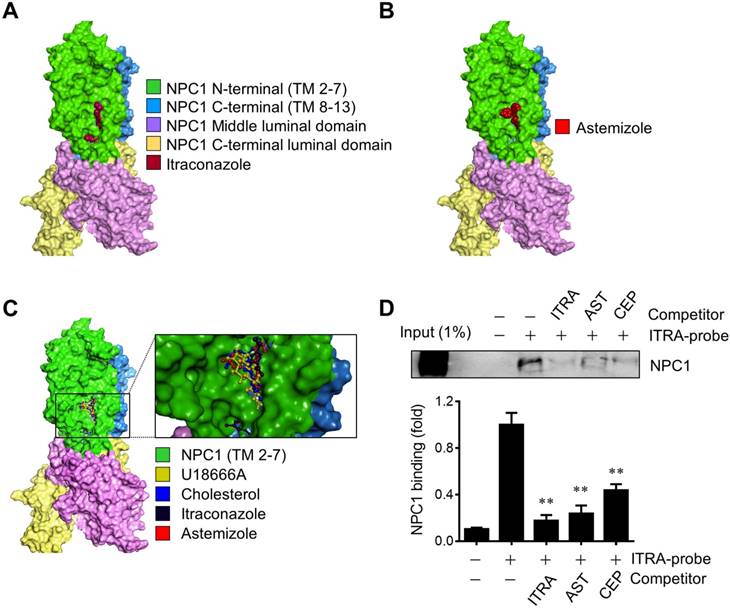
NPC1 is a lysosomal surface protein that exports free cholesterol out of lysosomes through the N-terminal cholesterol-binding pocket and the sterol sensing domain (SSD) in the integral transmembrane portion [18, 21]. Small molecule cholesterol trafficking inhibitors, U18666A and itraconazole are known to bind to the SSD of NPC1 and induce cholesterol accumulation in the lumen of the late endolysosomes [17, 22]. Previously we used an itraconazole photo-affinity probe that enables capture of NPC1 protein in living cells to test the screening hits for NPC1 binding. Among all the screening candidates, two drugs including cepharanthine and AST were able to compete with the itraconazole photo-affinity probe for the binding to NPC1 [8]. Here we further verified the binding of AST to NPC1 using a molecular docking and the itraconazole-NPC1 competitive binding assay. We first conducted a molecular docking analysis with the full length NPC1 structure (5U73) [18] to test whether AST shares a common binding site with other known NPC1 ligands. Itraconazole nicely fit to the SSD pocket of the transmembrane segment in NPC1, which was in agreement with the previous report [17] (Fig. 2A). AST bound to the same site in NPC1 (Fig. 2B). A superimposed docking simulation suggested that the binding site of AST overlapped with all other 3 ligands, including itraconazole, cholesterol and U18666A (Fig. 2C). The binding affinities for AST, itraconazole, cholesterol and U18666A were determined to be -9.2, -9.9, -8.9 and -8.7 kcal/mol, respectively. Next, to verify whether astemizole shares the same binding site with itraconazole in NPC1, we employed the itraconazole-NPC1 competitive binding assay. Intracellular NPC1 protein was successfully captured and pulled down by itraconazole photo-affinity probe and this capture was competed away by the pre-incubation of the cells with AST (Fig. 2D). Itraconazole or cepharanthine used as positive controls also competed away the probe binding to NPC1. These data suggested that AST occupied the SSD of NPC1 and prevented the itraconazole photo-affinity probe from binding to the SSD. These data further implied that, similar to itraconazole, AST occupied the SSD of NPC1, a cholesterol binding pocket, and blocked cholesterol export from the lysosomes, leading to the high accumulation of cholesterol in the lysosomal lumen.
Astemizole binds to the sterol-sensing domain of NPC1. (A, B) Structure of human NPC1 with ligands docked into the sterol-sensing domain (SSD). Surface representation of NPC1 with itraconazole (A) and astemizole (B) docked using AutoDock Vina is shown. (C) Close-up of the SSD binding pocket with docked astemizole, cholesterol, U18666A and itraconazole is shown. Transmembrane segments 2-7 (TM 2-7) of the SSD are colored green. Surface Diagram was created using Discovery Studio 2017. (D) A549 cells were used for the live-cell NPC1 competitive binding assay. The cells were incubated with or without competitors, including itraconazole (ITRA), astemizole (AST) and cepharanthine (CEP) at 20 μM, and NPC1 was captured and pulled down with itraconazole-photoaffinity probe (ITRA-probe). The upper panel is a representative Western blot of the binding assay and the lower panel is a quantitative data from three independent experiments of the binding assay. **P<0.01 vs ITRA-probe control.
AST causes depletion of cholesterol from intracellular membrane
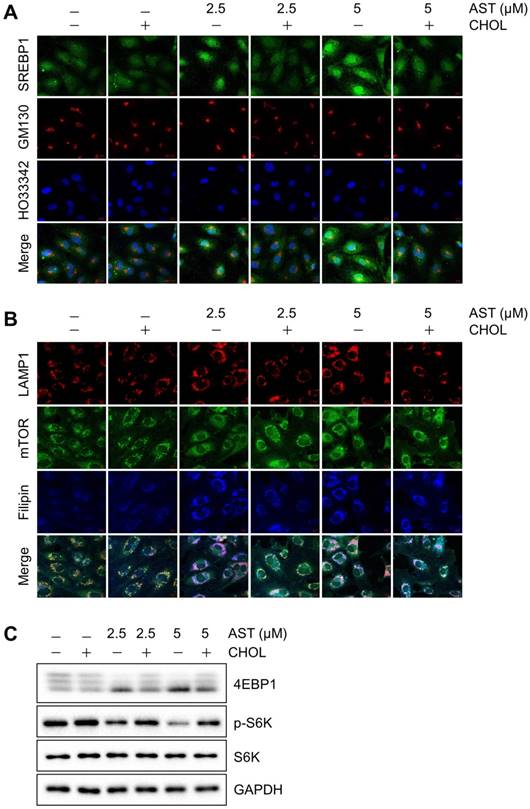
Since AST blocks cholesterol transport from the lysosomes, it could lead to the depletion of intracellular cholesterol content. We thus analyzed the membrane cholesterol sensor, sterol regulatory element-binding protein 1 (SREBP1). When cells are rich in cholesterol, SREBP1 is trapped in the endoplasmic reticulum (ER) membrane by a sterol-mediated protein-protein interaction with SCAP (SREBP cleavage-activating protein) and INSIG-1 [23]. The lack of cholesterol promotes the release of the SREBP1-SCAP complex from the ER. SREBP1 will then be matured in the Golgi and transported into the nucleus for transcriptional activation of target genes including those involved in cholesterol biosynthesis [24]. In control cells or cells treated with cholesterol, SREBP1 was mainly located in the cytosol. However, in cells treated with AST, a large portion of SREBP1 translocated to the Golgi and nuclei, suggesting an activation of SREBP1 (Fig. 3A). Once cellular cholesterol was replenished by cholesterol-cyclodextrin complex, the AST-induced nuclear localization of SREBP1 was reversed back to the cytosol (Fig. 3A). These data suggested that AST caused depletion of intracellular membrane cholesterol and led to the nuclear localization of SREBP1 in a cholesterol dependent manner.
Astemizole causes depletion of intracellular cholesterol and inhibition of mTOR signaling in a cholesterol-dependent manner. (A) Immunofluorescence analysis of the intracellular cholesterol sensor, SREBP1 is shown. HUVEC were treated with astemizole (AST) with or without cholesterol-cyclodextrin complex (CHOL) for 24 h and were subjected to immunofluorescence analysis of SREBP1 (green), GM130 (red) and Hoescht33342 (HO33342, blue). Scale bar = 10 μm. (B) Analysis of mTOR and LAMP1 subcellular localization is shown. HUVEC were treated with AST with or without CHOL for 24 h and were subjected to immunofluorescence analysis of LAMP1 (red), mTOR (green) and filipin (blue). Scale bar = 10 μm. (C) HUVEC were treated with AST with or without CHOL for 24 h and the lysates were subjected to Western blotting for 4EBP1, phospho-S6 kinase (p-S6K) and total S6 kinase (S6K). Note the multiple bands of 4EBP1, representing upper bands as hyper-phosphorylated forms and lower bands as hypo-phosphorylated forms. GAPDH was used as an internal loading control.
AST dissociates mTOR from lysosomes in a cholesterol-dependent manner
Cellular cholesterol content is important for proper membrane protein signaling in endothelial cells. In particular, mTOR plays an important role in sensing intracellular membrane cholesterol [5, 8]. We therefore analyzed the effect of the depletion of intracellular membrane cholesterol by AST on mTOR localization in endothelial cells. Immunofluorescence analysis showed that mTOR was nicely co-localized with the late endolysosome marker, LAMP1 in control endothelial cells. However, the treatment of cells with AST dose-dependently dissociated mTOR from the LAMP-1-positive endolysosomes (Fig. 3B). The dissociation of mTOR from endolysosomes was significantly reversed by cholesterol replenishment (Fig. 3B), suggesting a cholesterol-dependent association of mTOR with the lysosomal surface and its inhibition by AST. We further showed that AST dose-dependently inhibited mTOR signaling, as shown by reduced phosphorylation of the eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) and S6 kinase (Fig. 3C). Cholesterol replenishment also reversed the inhibition of mTOR signaling by AST, further demonstrating that AST-induced intracellular cholesterol depletion led to an inhibition of mTOR signaling in endothelial cells.
AST inhibits mTOR signaling and endothelial cell proliferation
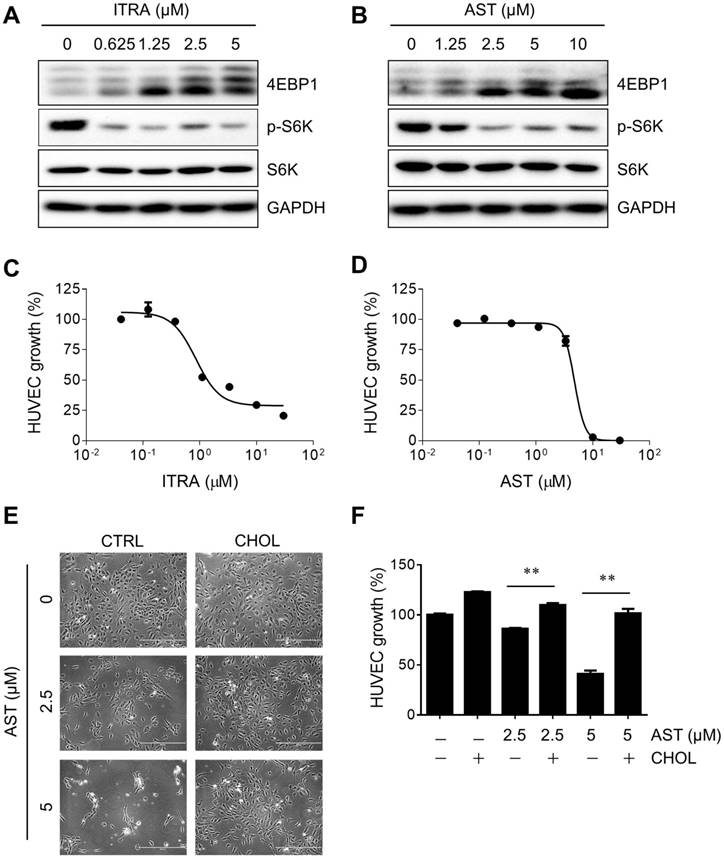
mTOR is a central signaling pathway in endothelial cells regulating cell proliferation, cell cycle progression, protein synthesis, cell migration and angiogenic sprout formation [25, 26]. To investigate the causal relationship between AST-mediated inhibition of mTOR and endothelial cell proliferation, we analyzed a dose-response curve of the inhibition and cholesterol rescue effects. Treatment of cells with itraconazole or AST dose-dependently inhibited mTOR signaling as determined with the reduction of hyper-phosphorylated forms of 4EBP1 and S6 kinase (Fig. 4A and B). The inhibition of mTOR by itraconazole began from 0.625 µM treatment, while AST showed inhibition at 2.5 µM treatment. In cell proliferation assay, half-maximal inhibitory concentrations (IC50) of itraconazole and AST were determined to be 0.86 and 4.7 µM, respectively (Fig. 4C and D). In both assays, AST inhibition was seen in the low-micromolar range and was 4-5 times less potent than itraconazole. Therefore, there is a correlation between inhibition of mTOR and endothelial cell proliferation. We then examined the cholesterol-dependency of the anti-proliferative effect of AST on endothelial cells. While AST dose-dependently inhibited the endothelial cell proliferation, cholesterol replenishment almost completely rescued the inhibition (Fig. 4E and F). This effect was again well correlated with the cholesterol rescue of the AST-mediated mTOR inhibition.
Astemizole inhibits HUVEC proliferation in a cholesterol-dependent manner. (A, B) HUVEC were treated with itraconazole (ITRA) or astemizole (AST) for 24 h and analyzed for Western blotting of 4EBP1, phospho-S6 kinase (pS6K) and total S6 kinase (S6K). GAPDH was used as an internal loading control. (C, D) HUVEC were treated with ITRA or AST for 72 h and subjected to AlamarBlue cell viability assay. (E, F) HUVEC were treated with AST with or without cholesterol-cyclodextrin complex (CHOL) for 72 h and the cell viability was determined. Representative cell images from the cell viability assay (E) and AlamarBlue quantitation (F) are shown. Scale bar = 400 μm. **P<0.01 between two groups.
AST inhibits angiogenesis in a cholesterol-dependent manner
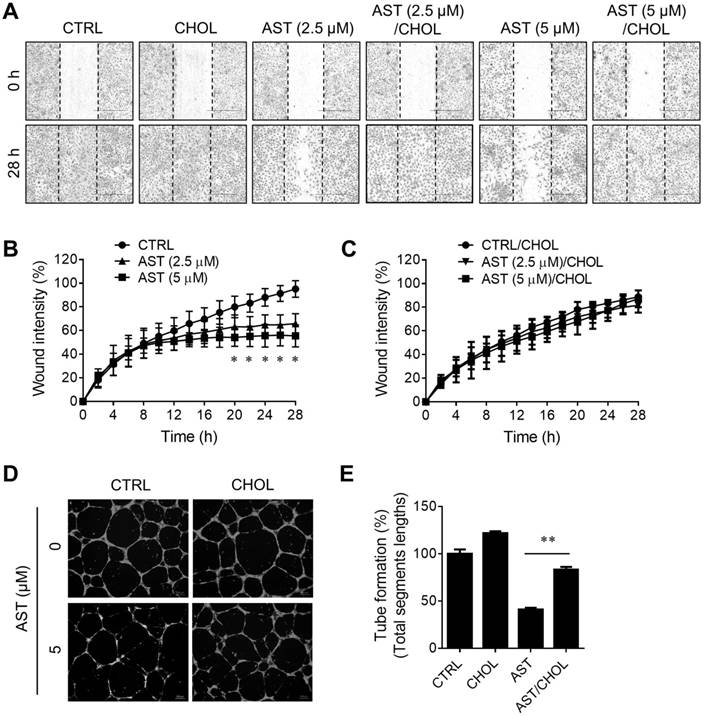
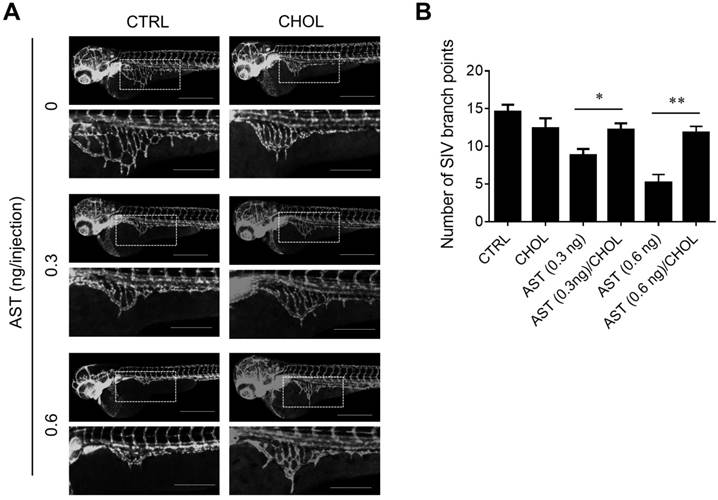
Next, we examined the effect of AST and cholesterol rescue on angiogenesis in vitro using endothelial cell migration and tube formation assays. We used AST concentrations up to 5 µM for all angiogenesis assays as it did not cause any cytotoxicity in endothelial cells (Supplementary Fig. S1). A real-time, wound-healing migration assay showed that AST dose-dependently inhibited endothelial cell migration (Fig. 5A and B). However, the inhibition was nearly completely rescued by the addition of cholesterol (Fig. 5A and C). In tube formation assays, AST again inhibited endothelial cell tube formation on Matrigel and the inhibition was significantly rescued by the addition of cholesterol (Fig. 5D and E). We further evaluated effect of AST and cholesterol on angiogenesis in vivo using the Tg (fli1a:EGFP)y1 transgenic zebrafish line. During the zebrafish embryo development, vasculogenesis is characterized by dorsal aorta (DA) and intersegmental vessel (ISV) formation within 12-48 hpf and angiogenic sprout is characterized by the formation of subintestinal vessels (SIV) at around 72 hpf [27, 28]. Due to a solubility and absorption issue of cholesterol into zebrafish embryo from media, we employed a microinjection of AST and cholesterol/cyclodextrin complex into the yolk, allowing flow and diffusion to bring the compound solution into the embryo body. AST treatment resulted in a dose-dependent inhibition of angiogenic sprout formation of SIV at 72 hpf without significant effect on ISV vasculogenic vessel formation (Fig. 6A). Co-treatment of the zebrafish with cholesterol significantly rescued the anti-angiogenic effect of AST on SIV formation (Fig. 6A and B). These results demonstrated that AST inhibited angiogenesis in a cholesterol-dependent manner.
Astemizole inhibits angiogenesis in vitro in a cholesterol-dependent manner. (A-C) Real-time wound healing assay in HUVEC treated with astemizole (AST) in the presence or absence of cholesterol-cyclodextrin complex (CHOL) is shown. (A) Representative cell migration images at 0 and 28 h. Scratch wound areas are marked with broken lines. Scale bar = 600 μm. (B, C) Real-time wound quantitation was performed using the IncuCyte ZOOM. *P<0.05 between AST (5 μM) and control (CTRL). (D) HUVEC were seeded on Matrigel and treated with AST (5 μM) with or without CHOL for 20 h for tube formation assay. The tubes were fluorescently labeled with Calcein-AM. Representative tube images are shown. Scale bar = 200 μm. (E) The total segments lengths of the tubes were quantitated using the Angiogenesis Analyzer for Image J. **P<0.01 between two groups.
Astemizole inhibits zebrafish angiogenesis in a cholesterol-dependent manner. The Tg(fli1:EGFP)y1 zebrafish embryos (8 hpf) were microinjected with astemizole (AST, final dosage of 0.3 ng or 0.6 ng/embryo) with or without cholesterol-cyclodextrin complex (CHOL, final dosage of 3.7 ng in 10% cyclodextrin/embryo). (A) The representative zebrafish images were taken at 72 hpf. Scale bar = 500 μm. The image inlets (white square) magnifying sub-intestinal vessels (SIV) are shown at the bottom of each image. Scale bar = 200 μm. (B) Zebrafish SIV angiogenesis was quantified by counting the number of SIV branch points. *P<0.05; **P<0.01 between two groups.
Discussion
AST was originally developed as an anti-histamine for the treatment of allergy symptoms [9]. It was however withdrawn from market by the manufacturer in 1999 due to the potential risk of ventricular arrhythmias at high doses, especially when used together with cytochrome P450 inhibitors [29]. It was later revisited for its promising anti-malarial and anticancer properties [11]. The molecular target of AST for its anti-malarial and anticancer effects were believed to be Eag1 and hERG potassium channels [12, 13]. As most cancer studies with AST have focused on Eag1 or hERG inhibition, to our knowledge there are no reports of additional molecular targets of AST involved in its anticancer effect.
In this study, we showed that AST binds to NPC1 and inhibits intracellular cholesterol trafficking, causing a high accumulation of free cholesterol in the lumen of late endolysosomes. NPC1 is a transmembrane protein located in the membrane of endolysosomes, whereas NPC2 is a lysosomal luminal protein. Mutations in either NPC1 or NPC2 cause a rare genetic disease, called Niemann-Pick Type C (NPC). NPC is a lysosomal storage disease characterized by a high accumulation of free cholesterol and sphingolipids in the lysosomes [30, 31]. Cholesterol is delivered into the cells in the form of low-density lipoprotein (LDL) via receptor-mediated endocytosis. The LDL-containing endosomes are then matured and fused with lysosomes where cholesterol is unesterified by lysosomal acid lipases and captured by NPC2 in the lysosomal lumen. NPC1 then accepts the free cholesterol from NPC2 through its N-terminal cholesterol binding pocket and transfers cholesterol to the SSD before transporting out from lysosomes [18, 21]. U18666A and itraconazole are well known cholesterol trafficking inhibitors that bind to the SSD of NPC1 and inhibit cholesterol transport out of lysosomes in a mutually competitive manner [17, 22]. Our live-cell NPC1 competitive binding assay and molecular docking analyses revealed that AST shares a common binding site with itraconazole, cholesterol and U18666A within the SSD of NPC1. Its strong binding affinity to NPC1 and clear phenotypes on cholesterol trafficking suggest that AST is a new member of the NPC1 antagonist class.
We further showed anti-angiogenic phenotypes of AST using endothelial cell and zebrafish models and its potential mechanism of action in inhibiting angiogenesis. Firstly, AST blockade of cholesterol trafficking leads to a depletion of intracellular cholesterol, especially membranous cholesterol, as evidenced by SREBP1 activation by AST and its reversal by cholesterol replenishment. Second, cholesterol depletion leads to dissociation of mTOR, a cholesterol sensor, from the lysosomal surface and inhibition of mTOR signaling. Third, inhibition of mTOR signaling leads to inhibition of endothelial cell proliferation, migration and tube formation, as evidenced by a dosage correlation and cholesterol rescue effects in both phenotypes. The anti-angiogenic phenotype and cholesterol rescue effect of AST was also shown in the transgenic zebrafish angiogenesis model. These data suggest that AST is a new anti-angiogenic agent blocking cholesterol trafficking and mTOR signaling in endothelial cells. Karapetyan and coworkers recently reported that astemizole induced autophagy, a hitherto unreported mode of action of the drug [32]. It can be postulated that autophagy induction by astemizole is likely to be mediated by cholesterol-dependent mTOR inhibition.
AST inhibition on Eag1 or hERG potassium channel is a double-edged sword. A number of cancer cells/patients showed high expression of Eag1 and targeting Eag1 has shown a good anticancer response in several cancer types, including gastric cancer, colorectal cancer, soft tissue sarcoma, and hepatocellular carcinoma [15, 33-35]. On the other hand, as the fatal side effects (arrhythmias) of AST come from the inhibition of the potassium channel, antagonism of Eag1 or hERG limits further clinical development of the drug for anticancer applications [36]. We observed very low expression levels of Eag1 and hERG in HUVEC compared to several tumor cell lines tested (Supplementary Fig. S2). This suggests that NPC1 could be more critical target protein of AST in endothelial cells than the potassium channels. Furthermore, our finding that AST inhibits cholesterol trafficking via targeting NPC1 in endothelial cells is crucial as this study could facilitate chemical modification of AST toward the NPC1 binding devoid of Eag1 or hERG binding. Further studies on the molecular recognition between AST and the SSD of NPC1 are warranted.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by the Science and Technology Development Fund (FDCT) of Macau SAR (FDCT/119/2013/A3 and FDCT/024/2015/A1 to J.S.S); Multi-Year Research Grant of the University of Macau (MYRG2015-00181-FHS and MYRG2017-00176-FHS to J.S.S); NRF grant (2015K1A1A2028365 and 2015M3A9B6027818 to H.J.K). Support from National Cancer Institute (R01CA184103 to J.O.L.), the Flight Attendant Medical Research Institute (J.O.L.), and the Johns Hopkins Institute for Clinical and Translational Research, which is funded in part by Grant UL1 TR 001079 from the National Center for Advancing Translational Sciences (NCATS) is also gratefully acknowledged.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612-21
2. Incardona JP, Eaton S. Cholesterol in signal transduction. Curr Opin Cell Biol. 2000;12:193-203
3. Noghero A, Perino A, Seano G, Saglio E, Sasso GL, Veglio F. et al. Liver X receptor activation reduces angiogenesis by impairing lipid raft localization and signaling of vascular endothelial growth factor receptor-2. Arterioscler Thromb Vasc Biol. 2012;32:2280-8
4. Fang L, Choi S-H, Baek JS, Liu C, Almazan F, Ulrich F. et al. Control of angiogenesis by AIBP-mediated cholesterol efflux. Nature. 2013;498:118-22
5. Xu J, Dang Y, Ren YR, Liu JO. Cholesterol trafficking is required for mTOR activation in endothelial cells. Proc Natl Acad Sci U S A. 2010;107:4764-9
6. Shim JS, Li R-J, Lv J, Head SA, Yang EJ, Liu JO. Inhibition of angiogenesis by selective estrogen receptor modulators through blockade of cholesterol trafficking rather than estrogen receptor antagonism. Cancer Lett. 2015;362:106-15
7. Shim JS, Li RJ, Bumpus NN, Head SA, Kumar Pasunooti K, Yang EJ. et al. Divergence of Antiangiogenic Activity and Hepatotoxicity of Different Stereoisomers of Itraconazole. Clin Cancer Res. 2016;22:2709-20
8. Lyu J, Yang EJ, Head SA, Ai N, Zhang B, Wu C. et al. Pharmacological blockade of cholesterol trafficking by cepharanthine in endothelial cells suppresses angiogenesis and tumor growth. Cancer Lett. 2017;409:91-103
9. Janssens MM-L. Astemizole. Clin Rev Allergy Immunol. 1993;11:35-63
10. Gottlieb S. Antihistamine drug withdrawn by manufacturer. Brit Med J. 1999;319:7 -
11. Lotharius J, Gamo-Benito FJ, Angulo-Barturen I, Clark J, Connelly M, Ferrer-Bazaga S. et al. Repositioning: the fast track to new anti-malarial medicines? Malar J. 2014;13:143
12. Lamothe SM, Guo J, Li W, Yang T, Zhang S. The Human Ether-a-go-go-related Gene (hERG) Potassium Channel Represents an Unusual Target for Protease-mediated Damage. J Biol Chem. 2016;291:20387-401
13. Zhou Z, Vorperian VR, Gong Q, Zhang S, January CT. Block of HERG potassium channels by the antihistamine astemizole and its metabolites desmethylastemizole and norastemizole. J Cardiovasc Electrophysiol. 1999;10:836-43
14. Downie BR, Sánchez A, Knötgen H, Contreras-Jurado C, Gymnopoulos M, Weber C. et al. Eag1 expression interferes with hypoxia homeostasis and induces angiogenesis in tumors. J Biol Chem. 2008;283:36234-40
15. de Guadalupe Chávez-López M, Pérez-Carreón JI, Zuñiga-García V, Díaz-Chávez J, Herrera LA, Caro-Sánchez CH. et al. Astemizole-based anticancer therapy for hepatocellular carcinoma (HCC), and Eag1 channels as potential early-stage markers of HCC. Tumor Biol. 2015;36:6149-58
16. Sales TT, Resende FFB, Chaves NL, Titze-De-Almeida SS, Báo SN, Brettas ML. et al. Suppression of the Eag1 potassium channel sensitizes glioblastoma cells to injury caused by temozolomide. Oncol Lett. 2016;12:2581-9
17. Head SA, Shi WQ, Yang EJ, Nacev BA, Hong SY, Pasunooti KK. et al. Simultaneous Targeting of NPC1 and VDAC1 by Itraconazole Leads to Synergistic Inhibition of mTOR Signaling and Angiogenesis. ACS Chem Biol. 2016;12:174-82
18. Li X, Wang J, Coutavas E, Shi H, Hao Q, Blobel G. Structure of human Niemann-Pick C1 protein. Proc Natl Acad Sci U S A. 2016;113:8212-7
19. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671-5
20. Yu X, Tong Y, Han XQ, Kwok HF, Yue GGL, Lau CBS. et al. Anti-Angiogenic Activity of Herba Epimedii on Zebrafish Embryos In Vivo and HUVECs In Vitro. Phytother Res. 2013;27:1368-75
21. Kwon HJ, Abi-Mosleh L, Wang ML, Deisenhofer J, Goldstein JL, Brown MS. et al. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 2009;137:1213-24
22. Lu FR, Liang QR, Abi-Mosleh L, Das A, De Brabander JK, Goldstein JL. et al. Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection. Elife. 2015:4
23. Yang T, Espenshade PJ, Wright ME, Yabe D, Gong Y, Aebersold R. et al. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489-500
24. Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X. et al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell. 1993;75:187-97
25. Guba M, von Breitenbuch P, Steinbauer M, Koehl G, Flegel S, Hornung M. et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: involvement of vascular endothelial growth factor. Nat Med. 2002;8:128-35
26. Farhan MA, Carmine-Simmen K, Lewis JD, Moore RB, Murray AG. Endothelial cell mTOR complex-2 regulates sprouting angiogenesis. PLoS One. 2015;10:e0135245
27. Serbedzija GN, Flynn E, Willett CE. Zebrafish angiogenesis: a new model for drug screening. Angiogenesis. 1999;3:353-9
28. He ZH, Ge W, Yue GGL, Lau CBS, He MF, But PPH. Anti-angiogenic effects of the fruit of Alpinia oxyphylla. J Ethnopharmacol. 2010;132:443-9
29. de Abajo FJ, Rodriguez LA. Risk of ventricular arrhythmias associated with nonsedating antihistamine drugs. Br J Clin Pharmacol. 1999;47:307-13
30. Chang TY, Reid PC, Sugii S, Ohgami N, Cruz JC, Chang CC. Niemann-Pick type C disease and intracellular cholesterol trafficking. J Biol Chem. 2005;280:20917-20
31. Shamburek RD, Pentchev PG, Zech LA, Blanchette-Mackie J, Carstea ED, VandenBroek JM. et al. Intracellular trafficking of the free cholesterol derived from LDL cholesteryl ester is defective in vivo in Niemann-Pick C disease: insights on normal metabolism of HDL and LDL gained from the NP-C mutation. J Lipid Res. 1997;38:2422-35
32. Karapetyan YE, Sferrazza GF, Zhou M, Ottenberg G, Spicer T, Chase P. et al. Unique drug screening approach for prion diseases identifies tacrolimus and astemizole as antiprion agents. Proc Natl Acad Sci U S A. 2013;110:7044-9
33. Ding XW, Luo HS, Jin X, Yan JJ, Ai YW. Aberrant expression of Eag1 potassium channels in gastric cancer patients and cell lines. Med Oncol. 2007;24:345-50
34. Ding XW, Yan JJ, An P, Lu P, Luo HS. Aberrant expression of ether a go-go potassium channel in colorectal cancer patients and cell lines. World J Gastroenterol. 2007;13:1257-61
35. Mello de Queiroz F, Suarez-Kurtz G, Stuhmer W, Pardo LA. Ether a go-go potassium channel expression in soft tissue sarcoma patients. Mol Cancer. 2006;5:42
36. Izumi-Nakaseko H, Nakamura Y, Cao X, Wada T, Ando K, Sugiyama A. Possibility as an anti-cancer drug of astemizole: Evaluation of arrhythmogenicity by the chronic atrioventricular block canine model. J Pharmacol Sci. 2016;131:150-3
Author contact
![]() Corresponding author: Joong Sup Shim, PhD, Faculty of Health Sciences, University of Macau, Av. Universidade, Taipa, Macau SAR, China. Email: jsshimmo; Tel: +853-8822-4990
Corresponding author: Joong Sup Shim, PhD, Faculty of Health Sciences, University of Macau, Av. Universidade, Taipa, Macau SAR, China. Email: jsshimmo; Tel: +853-8822-4990