ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2018; 14(10):1389-1398. doi:10.7150/ijbs.24739 This issue Cite
Research Paper
A novel FGFR1-binding peptide exhibits anti-tumor effect on lung cancer by inhibiting proliferation and angiogenesis
Qiaoyan Tan1#, Zuqiang Wang1#, Quan Wang1, Yuanqiang Wang2, Zhifeng Huang3, Nan Su1, Min Jin1, Liang Kuang1, Huabing Qi1, Zhenhong Ni1, Can Li1, Ying Zhu1, Wanling Jiang1, Hangang Chen1, Chuxia Deng4, Xiaolan Du1 ![]() , Yangli Xie1
, Yangli Xie1 ![]() , Lin Chen1
, Lin Chen1 ![]()
1. Department of Rehabilitation Medicine, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, Chongqing, China.
2. College of Bioengineering, Chongqing Institute of Technology, Chongqing, China.
3. School of Pharmacy, Wenzhou Medical University, Wenzhou, China.
4. Faculty of Health Sciences, University of Macau, Macau SAR, China.
# These authors contributed equally to this work.
Abstract

It has been reported that overactivation of fibroblast growth factor receptor 1 (FGFR1) is an important characteristic found in most non-small cell lung cancer (NSCLC) samples. Here, we identified a FGFR1 inhibitory peptide R1-P2 and investigated its effects on the lung cancer cells growth and angiogenesis in vitro and in vivo. Our results demonstrate that R1-P2 bound to human FGFR1 protein, and efficiently blocked the binding of FGF2 to FGFR1 in A549 and NCI-H460 cells. Moreover, this peptide significantly decreased the proliferation, migration and invasion, but promoted the apoptosis in both cell lines. In addition, R1-P2 treatment effectively inhibited the tumor growth and neovascularization in nude mice with xenografted A549 cells, and R1-P2 also significantly inhibited the FGF2-induced angiogenesis in tube formation experiment and CAM model. We further demonstrated that R1-P2 suppressed lung tumor growth through anti-angiogenic and anti-proliferative activity. Our data may provide a novle leading molecule with potential application in the treatment of FGFR1 activation related lung cancers.
Keywords: Phage display, FGFR1, Peptide, Cancer therapy, Lung cancer
Introduction
Lung cancer, a highly invasive prevalent cancer with strong metastasis capacity, is one of major cancers leading to death worldwide. There are multiple types of lung cancer including NSCLC. NSCLC accounts for 85% of all lung cancers, and the majority (60-80%) of patients with NSCLC were in unresectable advanced or metastatic stage when their lung cancer were at diagnosed. The prognosis of NSCLC patients remains very poor with a 5-year survival of 15% (1). During the recent decades, a variety of targeting therapies such as small molecule inhibitors, monoclonal antibodies and multiple target tyrosine kinase inhibitors have been used in clinical trials (2), the therapeutic effects, however, poor for lung cancers.
FGFR1 belongs to receptor tyrosine kinase (RTK) that participates the regulation of various cellular processes including proliferation, migration, survival and angiogenesis (3). Overactivation of FGFR1 has been reported in diverse human cancers including ovarian cancer, prostate cancer and NSCLC (4-6), and so on. In addition, overactivation of FGFR1 promotes the epithelial-mesenchymal transition (EMT) and is associated with lung cancer grades and stages (7). The important roles of FGFR1 in the pathogenesis of a variety of cancers make it a potential therapeutic target for these cancers. Silencing the expression of FGFR1 by siRNA or inhibition of FGFR1 by multiple tyrosine kinase inhibitors, such as PD173074, PD166866 and SU5402 have been demonstarted to suppress the growth of NSCLC (7-9). However, PD173074 and SU5402 failed to enter phase II clinical trials due to their high toxicities (10). Therefore, the exploration of novel FGFR1 inhibitors with low toxicity and high efficiency has attracted extensive attention in recent years.
In this study, we found a novel peptide (R1-P2) with high binding affinity to FGFR1 by using phage display-based screening. We found that peptide R1-P2 inhibited the tyrosine kinase activity of FGFR1 and its downstream ERK/MAPK pathway in A549 and NCI-H460 cells. Peptide R1-P2 also inhibited the proliferation, migration, invasion and promoted the apoptosis of A549 and NCI-H460 cells in vitro. In addition, peptide R1-P2 can effectively suppress tumor growth in xenograft mouse model carrying A549 cells.
Materials and methods
Biopanning of a 12-peptide phage display library with FGFR1
The Ph.D.-12TM Peptide Library Kit (New England Biolabs) was used for biopanning as described previously (11). Briefly, 96-well plates were coated with 20 µg/ml FGFR1 (Sigma) in 150 µl coating buffer overnight at 4°C. The eluted phages were amplified in early-log E. coli ER2738 strain cells.
ELISA assay for selected phage clones
Phage clones (2×1011 pfu/well) and control phage vcsM13 were incubated at room temperature (RT) for 1 h. For competitive elution, 2 µg/ml FGF2 (PeproTech) was added for incubation at RT for 1 h. Anti-M13 monoclonal antibody (New England Biolabs) was added and incubated at 37°C for 1 h. Next goat anti-mouse immunoglobulin G (IgG)-HRP was added and incubated at 37°C for 30 min. Final the substrate 3, 3', 5, 5'-tetramethylbenzidine (TMB; ZSGB-BIO) was added and the absorbance of the colored product was measured at 450 nm.
Peptide R1-P2 and FGFR1 binding assays
Peptide R1-P2 were synthesized at Shanghai China Peptides Co., Ltd.. 96-well plates were coated with 100 µg/ml peptide R1-P2 in 100 µl coating buffer (PBS) overnight at 4°C. 150 µl of 20 µg/ml of FGFR1 was added and incubated at 37°C for 1 h. Plates were washed and incubated with FGFR1 antibody (Santa Cruz), followed by a secondary anti-rabbit antibody conjugated with HRP (DAB kit; ZSGB-BIO), and 5-amino-2-hydroxybenzoic acid as a substrate, with absorbance measured at 450 nm.
Molecular docking
Molecular dynamics simulation was performed as follow: (1) Energy minimize simulation (1500 step, sessile receptor molecular 1-280 residue); (2) Heating (NVE, 500 ps, 0K-300 K, sessile receptor molecular 1-280 residue); (3) Density adjustment (NPT, 500 ps, 300 K, sessile receptor molecular 1-280 residue); (4) Equilibrium (NPT, 500 ps, 300 K); (5) Outcome (NPT, 5 ns, 300 K). Then binding free-energy of ligand-receptor was estimated using MM-PBSA. Averaged the final 500 ps structure of the stable complex, and then analyze the interaction of ligand-receptor by LigPlus module.
Surface plasmon resonance (SPR) analysis of peptide R1-P2
SPR analysis was performed as described previously (12). Briefly, the sample was treated with a running buffer containing different concentrations of the human FGFR1 protein (0, 0.0625, 0.125, 0.25, 0.5, 1.0 and 2.0 μM) in PBS. The injection time was set to 120 s, the dissociation time was set to 600 s.
Cell culture and reagents
Human NSCLC cell lines with high expression of FGFR1 including A549 and NCI-H460 were purchased from Cell Bank of Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in DMEM/high glucose medium (Hyclone) with 10% fetal bovine serum (FBS, Gibco) at 37°C in 5% CO2 incubator. PD173074 (Selleck), an inhibitor of FGFR1, was used as the positive control. Peptide R1-P2 is diluted with PBS.
Western blotting
A549 and NCI-H460 cells were treated with PD173074 (100 nM) or peptide R1-P2 (15 and 30 mM) for 2 h. Equal amount of protein samples (30 ug) were dissolved by 10% SDS-PAGE gels and transferred onto a polyvinylidene difluoride membrane (Millipore). After being blocked with 8% nonfat milk in Tris buffered saline Tween buffer, the membrane was probed with primary antibodies specific for pFGFR1 (1:1000 dilution; Santa Cruz) and pERK1/2 (1:1000 dilution; Cell Signaling Technology) followed by secondary antibodies. The antibody specific for β-actin (1:10000 dilution; Sigma) was applied to normalize the protein expression levels.
Cell proliferation viability assay
A549 and NCI-H460 cells were treated with peptide R1-P2 (1, 5, 15 and 30 mM, respectively) for 48 h. The viability of cells was determined by MTT assay (13).
Analysis of cell apoptosis
A549 and NCI-H460 cells were treated with peptide R1-P2 (15 and 30 mM) for 24 h. Cell apoptosis was detected with in situ cell death detection kit (Roche) according to the manufacturer's instructions.
Wound-healing assay
The migration of A549 and NCI-H460 were measured by wound-healing assay. Briefly, after the growing cell layers reached confluence, we inflicted a uniform wound in each plate using a pipette tip and washed the wounded layers with PBS to remove all cell debris. Then, cells were treated with peptide R1-P2 (15 and 30 mM) or vehicle for 24 h. The images were captured at the beginning and 24 h, and the migration rate of the cells was quantified.
Invasion assay
The bottoms of the upper chamber (on top of the membrane) were coated with 100 μl volume of matrigel (BD Biosciences). A549 and NCI-H460 cells with 200 μl serum-free medium were added into the upper chamber, and complete medium with peptide R1-P2 (15 and 30 mM) or vehicle was added into the lower chamber. After incubation for 16 h, the cells invaded to the lower surface of the membrane were stained with 1% crystal violet. Cell counting was then carried out by photographing the membrane through the microscope, and five random fields were taken.
In vivo anti-tumor study
All mice were maintained in the Animal Facility (specific pathogen free) of the Daping Hospital (Chongqing, China). All procedures were approved by the Institutional Animal Care and Use Committee of Daping Hospital (Chongqing, China). Athymic nu/nu female BALB/cA mice were purchased from the Beijing HFK Bioscience Co. Ltd. Xenograft model for anti-tumor was used according to previously study (14). Briefly, A549 cells were harvested and mixed with matrigel (BD Biosciences) at proportions of 1:1. Then, the cells (2 × 106 cells in 200 μL of saline) were injected subcutaneously into the upper and lower back of 7-week-old, BALB/cA nude mice, respectively. Two days after the A549 cells were injected, the mice were injected intraperitoneally (i.p.) with peptide R1-P2 in saline at a dosage of 5 mg/kg/day for 28 days, whereas the control mice were injected with the same volume of saline (n = 5 in each group). The volume of the tumors were determined by measuring their length (l) and width (w) and calculated using the formula; V = 0.52 × l × w2. The weights of the tumors were recorded on the day the mice were sacrificed.
Hematoxylin and eosin (H&E) staining
H&E staining was performed as manufacturer's instructions.
Immunofluorescence analysis (IFC)
IFC was performed as the manufacturer's instructions. The following primary antibodies were used: mouse anti-α-SMA polyclonal antibody (1:1000 dilution; Sigma), rabbit anti-PCNA polyclonal antibody (1:1000 dilution; BioVision), Alexa Fluor 488 goat anti-mouse IgG (1:1000 dilution; Molecular Probes) and Alexa Fluor 568 goat anti-rabbit IgG (1:1000 dilution; Molecular Probes) were used as a secondary antibody. Nuclei were counterstained with Hoechst 33258 (1:5000 dilution; Biotime Biotech). Fluorescent cells were observed and photographed under a laser scanning confocal microscope (ZEISS, LSM 880).
Tube formation assay
48-well plates coated with 100 μl matrigel basement membrane matrix (BD) per/well and polymerized at 37°C for 30 min. A549 cells were plated on the matrigel at a density of 2 × 105 cells/well. Peptide R1-P2 (15 and 30 mM) was then added together with FGF2 (10 ng/ml). After 6 h, photographing and the extent of capillary tube formation was evaluated by measuring the total tube length per field.
Chick chorioallantoic membrane (CAM) assay
CAM assay was performed as described previously (15). Briefly, on day 8 of incubation, pipe the peptide R1-P2 (15 and 30 mM) with FGF2 (10 ng/ml) onto the CAM surface for the evaluation of their angiogenic potential. After incubation for 24 h, photograph the CAM surface.
Statistical analysis
Data were presented as mean values ± SD and statistical differences between groups were assessed by Student's t-test. Analysis of variance (ANOVA) was used for comparisons of 3 or more groups followed by Tukey post hoc test (SPSS program version 13.0). P < 0.05 was considered statistically significant.
Results
Identification of specific FGFR1 binding peptide using phage display technique
Phage recovery rates of the second and third round of screening were much higher than the first round (9.6×10-7% and 5.52×10-6% compared with 1×10-8%) (Table 1), indicating that after three rounds of selection, phages with high affinities to FGFR1 were effectively enriched. A total of 23 individual phage clones were obtained after three rounds of panning. Total DNA of the phages were isolated and sequenced using -96gШ primers, then three different amino acid sequences were obtained.
Three positive phage clones with different sequence were used to examine the binding affinity for FGFR1 by ELISA (Figure 1A). OD values of three selected clones with different amino acid sequences were ten times higher than that of the control, which indicates that these three clones had high binding affinity to FGFR1. Specific ligand of FGFR1, FGF2, was used to test their binding affinity through competitive elution with FGFR1. Our data showed that FGF2 had high elution efficiency for these clones (Figure 1B). Since FGF2 exerts its biological activities via binding to the extracellular domain of FGFR1 (16,17), we deduced that these phage clones may bind to the extracellular domain of FGFR1 and mimic the biological effetcs of FGF2 binding to FGFR1.
Enrichment of phages with a high FGFR1 binding affinity
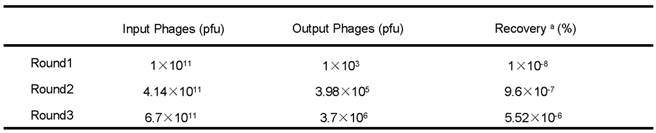
pfu, plaque forming unit
a Recovery(%) = (Output Phages/Input Phages) x 100 %
Specific binding of the positive phage clones and peptide to FGFR1. (A) The binding affinities to FGFR1 of three selected positive phage clones and the control vcsM13 were determined by ELISA assay. (B) Detection of FGF2 elution efficiency to the three selected positive phage clones. (C-D) Molecular docking of peptide R1-P2 binding to the extracellular region of FGFR1. C, The extracellular region of FGFR1 contain 211 amine acid (as blue), which have two sites binding to FGF ligand (as orange, named upper is A site, lower is B site ); D, Molecular docking of peptide R1-P2 binding to A and B site of FGFR1, respectively. (E) Affinity measured by Biacore X100. The affinity constant was calculated using the following formula: affinity constant (KD) = dissociation constant (Kd)/binding constant (Ka), which reflected the degree of antigen-antibody reaction; the smaller of the value, the stronger of its binding affinity. (F) Affinity detection of different concentration of peptide R1-P2 binding to FGFR1 by ELISA. The error bars indicate the ± SD from three separate experiments, ***P < 0.001,versus VCSM13.
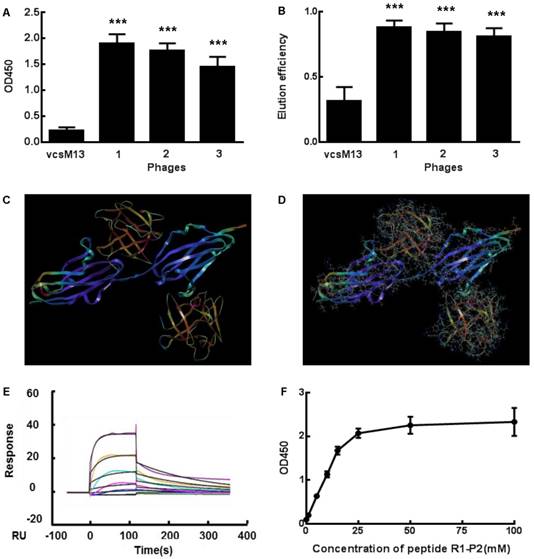
Molecular docking of peptide R1-P2 and the extracellular region of FGFR1
Free energy of peptide R1-P2 and the extracellular region of FGFR1

Property prediction and SPR analysis of peptide R1-P2
All eight clones shared the “FHDAWPNLSKSS” amino acid sequences, named as peptide R1-P2. ProtParam programs (http://web.expasy.org/protparam/) were then applied to analyze the sequence and predict its properties. Molecular weight of the peptide is 1388.50, theoretical isoelectric point of the peptide is 6.74, grand average of hydropathicity of the peptide is -0.883 (Additional file 1: Table S1).
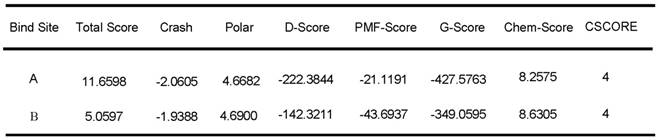
We further evaluated the molecular dynamics of the binding between peptide R1-P2 and FGFR1 protein (extracellular region, transmembrane region and intracellular region of human FGFR1 protein, respectively), the results of molecular docking and free energy indicate that peptide R1-P2 can not specifically binding to the transmembrane region and intracellular region of human FGFR1 protein (Data not shown). In order to evaluate the molecular dynamics of the binding between peptide R1-P2 and the extracellular region of FGFR1 (5AM6) protein, we analyzed the surface of the 5AM6 by MOLCAD (Figure 1C). Molecular docking showed that peptide R1-P2 can bind to A and B site of the extracellular region of FGFR1, which is similar with FGF ligand (Figure 1D). Molecular docking and free energy assay, as shown in Table 2 and Table 3, indicates that peptide R1-P2 can specifically bind to the extracellular region of FGFR1 protein. Furthermore, the binding affinity of peptide R1-P2 to human FGFR1 protein was about 1.538×10-7M (Additional file 2: Table S2) as measured by Biacore X100 (Figure 1E).
In addition, we found that peptide R1-P2 can bind to FGFR1 protein in a dose-response manner (Figure 1F). Taken together, these results demonstrated that peptide R1-P2 could specifically bind to human FGFR1 protein with high affinity.
Peptide R1-P2 significantly inhibits the activity of FGFR1 in vitro
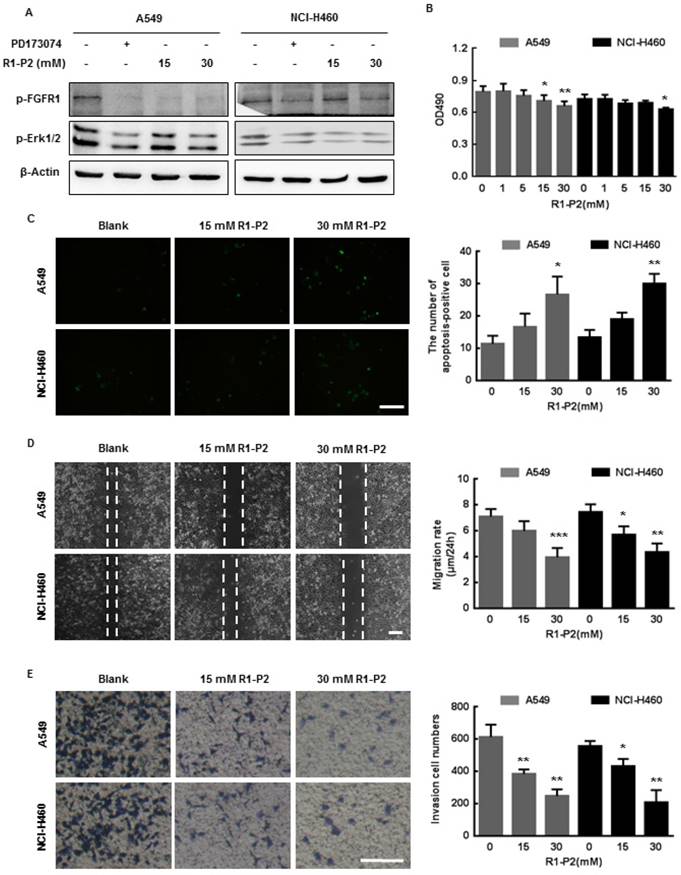
To determine the effect of peptide R1-P2 on FGFR1 signaling pathway, we investigated whether treatment with peptide R1-P2 could affect FGFR1 tyrosine kinase activity. Following stimulation of FGF2, activation of FGFR1-ERK1/2 MAPK signaling pathway was evident, while in the presence of peptide R1-P2 (15 and 30 mM), the degrees of the increase in the protein levels of phosphorylated FGFR1 and ERK1/2 were attenuated (Figure 2A).
We next examined the effects of peptide R1-P2 on cell activities of A549 and NCI-H460 cells. These cells were initially treated with different concentrations of peptide R1-P2 for 48 h, and the cell viability was evaluated by MTT assay. The results showed that peptide R1-P2 inhibited the proliferation of A549 and NCI-H460 cells in 30 mM (Figure 2B). Further, TUNEL staining analysis showed that there was no significant difference between the 15 mM peptide group and control, while peptide R1-P2 in 30 mM significantly increased the number of apoptosis-positive cells, indicating that peptide R1-P2 could induce apoptosis of A549 and NCI-H460 cells after a 24 h treatment (Figure 2C). Cell migration and invasion are essential step in neoplasm metastasis (7). We sought to evaluate the effect of peptide R1-P2 on the motility of A549 and NCI-H460 cells by wound healing assay and transwell invasion assay. The results showed that 30 mM of R1-P2 significantly inhibited the migration rate in both of cell lines (Figure 2D). Further experiments revealed that 15 and 30 mM of R1-P2 evidently suppressed the number of invasion cells, in both lung cancer cells (Figure 2E). Taken together, these results suggest that peptide R1-P2 can significantly inhibit the activity of FGFR1 in vitro.
Peptide R1-P2 inhibits the activity of A549 and NCI-H460 cells in vitro. (A) Peptide R1-P2 inhibited phosphorylation of FGFR1 and ERK1/2 induced by FGF2 (n = 3). (B) Peptide R1-P2 inhibited the vabilility of A549 and NCI-H460 cells measured by MTT assay (n = 3). (C) Peptide R1-P2 induced apoptosis in A549 and NCI-H460 cells measured by TUNEL (n = 3). (D) Peptide R1-P2 remarkably inhibited A549 and NCI-H460 cells migration (n = 3). (E) Peptide R1-P2 strongly suppressed A549 and NCI-H460 cells invasion measured by transwell assay (n = 3). Bar = 100 μm, the error bars indicate the ± SD from three separate experiments, *P < 0.05, **P < 0.01, ***P < 0.001, versus control.
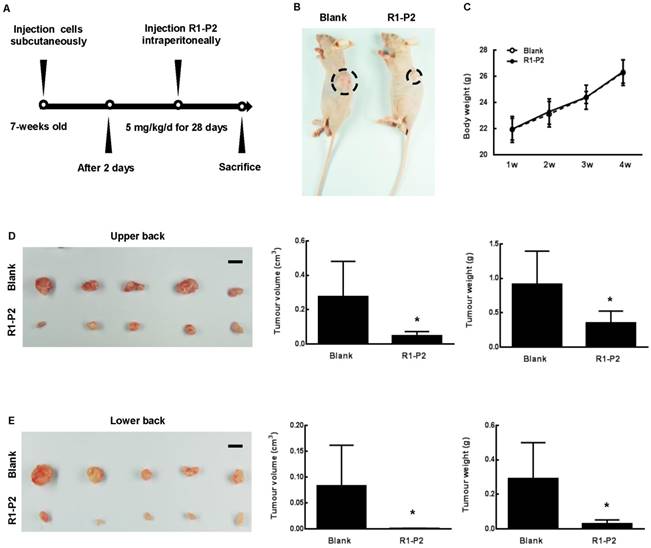
Peptide R1-P2 significantly suppresses tumor growth in xenograft mouse model
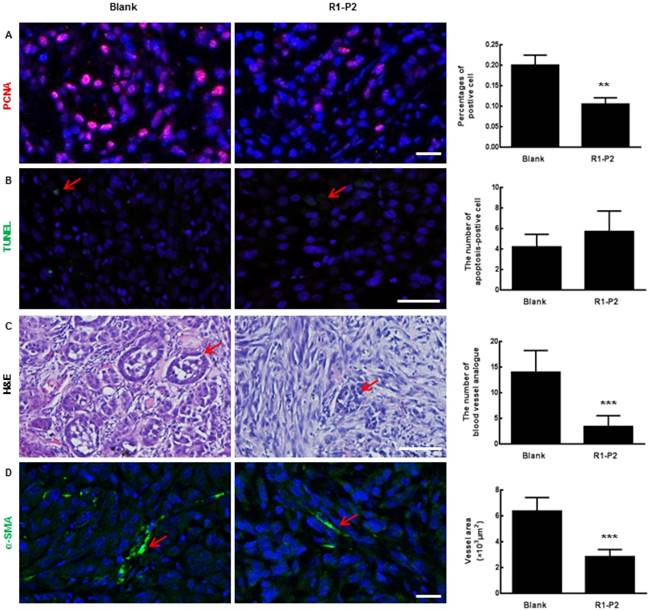
To further assess the anti-tumor activities of peptide R1-P2, xenografts were established in BALB/cA nude mice using A549 cell lines (Figure 3A). At day 30, the nude mice showed visible tumor formation at the upper and lower back (Figure 3B). Safety always is the top concern for the therapeutic study, there is no remarkable toxicity observed in the peptide R1-P2 treatment group, as evidenced by no signs of abnormal gross phenotypes including weight loss between the two groups during observation period (Figure 3C). Images of solid tumor isolated from upper (Figure 3D) and lower back of the sacrificed nude mice (Figure 3E) showed that peptide R1-P2 inhibited the growth of tumor generated by xenografted A549 in mouse model. Next we quantified the efficacies of peptide R1-P2 by measuring the volume (cm3) and weight (g) of the tumors from sacrificed nude mice. Our results showed that treatment with peptide R1-P2 for 28 days significantly reduced the tumor volume and tumor weight (Figure 3D and 3E). Previous studies have revealed that phosphorylation of FGFR1 can lead to the activation of downstream signaling cascades including ERK, leading to the proliferation and survival of cancer cells (17). The proliferation of cells was detected by PCNA using IHC. Peptide R1-P2 reduced the percentage of PCNA positive cells by 47.5% (Figure 4A). Further, peptide R1-P2 slightly induced cell apoptosis, although there was no statistical significancy (Figure 4B), indicating that the major anti-tumor effect of peptide R1-P2 is its anti-proliferative activity in vivo.
Anti-tumor effects of peptide R1-P2 in A549 xenograft model. (A) Experimental schedule for xenograft experiment. (B) Representative images of tumor 30 days after cell injection (black dotted line marking neoplasm). (C) Body weight of each group was recorded weekly (n = 5). (D) Images of tumor from sacrificed nude mice from upper back, tumor volume (cm3) and tumor weight (g) were recorded (n = 5). (E) Images of tumor from lower back of sacrificed nude mice, tumor volume (cm3) and tumor weight (g) were recorded (n = 5). Bar = 1 cm, the error bars indicate the ± SD from five separate experiments, *P < 0.05, versus blank.
Peptide R1-P2 potently suppresses the neovascularization
Angiogenic effect can be assessed by analyzing the formation and organization of tubular networks. The sections of tumor tissue harvested from nude mice were analyzed by H&E staining. Our results showed that peptide R1-P2 inhibited blood vessel analogue formation (as shown in red arrows, Figure 4C). Similarly, immunofluorescence staining results showed that peptide R1-P2 decreased the expression of α-SMA (a marker of pericytes) (Figure 4D), SM22 and CD31 in tumor tissue (Additional file 3: Figure S1).
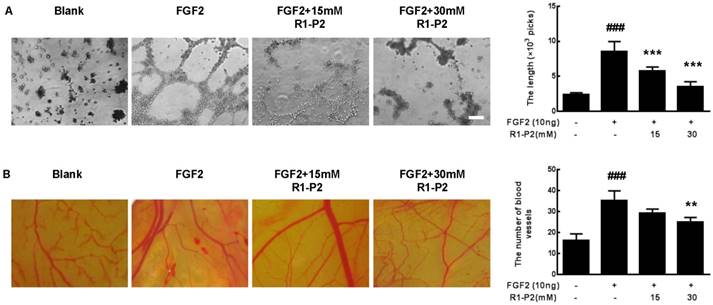
To further understand the mechanism underlying the antiangiogenic activity of peptide R1-P2, we detected the antiangiogenic activity of peptide R1-P2 in vitro and in vivo. As illustrated in Figure 5A, there was no evident tube formation in control, but FGF2 can significantly induce tube formation. At a concentration of 15 and 30 mM, peptide R1-P2 suppressed the tube formation induced by FGF2 by 32.6% and 58.4%, respectively.
We further tested the antiangiogenic activity of peptide R1-P2 using a CAM assay. The result showed that FGF2 can significantly induce angiogenesis, treatment with 15 mM peptide R1-P2 can partly inhibit the FGF2-induced neovascularization (P > 0.05). However, at a concentration of 30 mM, peptide R1-P2 potently suppressed the angiogenesis by 29.1% (Figure 5B). Together, these results indicate that as a novel inhibitory peptide for FGFR1 signaling, peptide R1-P2 potently inhibits angiogenesis both in vitro and in vivo.
Histological analysis of tumor sections. (A) The expression of PCNA in tumor sections were detected by immunofluorescence staining. (B) Apoptosis in tumor sections were detected by TUNEL (red arrows represents apoptosis cell). (C) Sections were stained with H & E (red arrows represents blood vessel analogue). (D) The expression of α-SMA in tumor sections were detected by immunofluorescence staining (red arrows represents blood vessels). Bar = 100 μm, the error bars indicate the ± SD from three separate experiments, **P < 0.01, ***P < 0.001, versus blank.
Peptide R1-P2 inhibits FGF2 induced angiogenesis in vitro. (A) Peptide R1-P2 inhibited tube formation in A549 cells were seeded on matrigel layer and treated with FGF2 in the presence or absence of peptide R1-P2. (B) CAM assay showed less vessels formation in peptide R1-P2 treated group. Bar = 100 μm, the error bars indicate the ± SD from three separate experiments, ###P < 0.001, versus control, **P < 0.01, ***P < 0.001, versus FGF2 alone.
Discussion
Lung cancer is currently the most frequently diagnosed cancer and the most common cause of cancer mortality in worldwide. Lung cancer is classified into two main types, small-cell (SCLC) (15%) and NSCLC (85%) based on their histological features (18). Adenocarcinoma and squamous cell carcinoma (SQC) are the most common histological subtypes in NSCLC cases (19). Targeted therapies, radio sensitizers, internal radiation, and combination treatment regimens have been used in clinic gradually in last decade, but there is still no substantial improvement in lung cancer mortality.
FGFs/FGFRs signaling is involved in a variety of critical cellular function of the development, homeostasis and tumorigenesis of multiple tissues/organs (20-22), such as proliferation, differentiation, apoptosis, drug resistance, and angiogenesis, etc. FGF signaling can also regulate EMT, invasion and metastasis in the tumor development (23,24). FGFR1 plays a critical role in the pathogenesis of lung cancer (5,9,25). Amplification of Fgfr1 has been found in approximately 17% of NSCLC cases and 6% of SCLC cases (26,27). Recently, it has been identified as an independent adverse prognostic marker in early-stage NSCLC (28).
Multiple strategies targeting FGFR1 have been developed, but limited clinical data are currently available. IMC-A1, an isoform-specific monoclonal antibody against FGFR1-IIIc was shown to cause severe anorexia in animal models, and thus was not translated into the clinic application (29). PD173074 and SU5402, two bioavailable FGFR inhibitors, failed to enter phase II clinical trials due to their high toxicities although they have exhibited striking effects in preclinical model experiments (10). Therefore, the exploration of novel FGFR1 inhibitors has attracted extensive attention in recent years.
Peptides can be easily synthesized, and has lower immunogenicity and toxicity, which possess the superior selectivity. Multiple peptide drugs such as α-thymosin have successfully been used in clinic (30). In addition, some peptides have been shown to be effective in phase II/III clinical trials for the treatment of cancer, such as interleukin-2 targeting peptide, or tumor neovasculature homing peptide NGR-hTNF (31,32).
In the present study, we screened a random 12-peptide phage library and identify a novel FGFR1-binding peptide. Furthermore, we demonstrate that this peptide (R1-P2) specifically binds to the human FGFR1 protein with a high affinity, suggesting the specific effect of peptide R1-P2 on FGFR1 activity. We demonstrated that peptide R1-P2 inhibited the phosphorylation levels of FGFR1 and ERK1/2 induced by FGF2 in A549 and NCI-H460 cells, indicating that R1-P2 can effectively inhibit the activities of FGFR1 itself and its downstream signalings.
FGF signaling has been proved to participate the regulation of multiple processes in tumorogenesis, including proliferation, migration, survival and angiogenesis. In in vitro culture model using A549 and NCI-H460 cells, we found that peptide R1-P2 can inhibit the proliferation and induce the apoptosis in both cell lines. We further demonstrated that peptide R1-P2 can effectively inhibit the invasion and migration of cultured A549 and NCI-H460 cells. FGFR1 signaling plays vital roles in vascularization/angiogenesis (23). Next, we confirmed the anti-angiogenic effect of peptide R1-P2 in vitro and in vivo. Tube formation and CAM assay indicates that FGF2 could significantly induce neovascularization, whereas treatment with peptide R1-P2 potently inhibited FGF2-induced neovascularization. Consistently, peptide R1-P2 significantly decreased the tumor volume and weight in xenograft mouse model using A549 cells without grossly observable toxicity. Histological analysis of tumor sections revealed that peptide R1-P2 significantly reduced blood vessel analogue formation.
In summary, we found a novel FGFR1 inhibitory peptide (R1-P2) that can inhibit the in vitro and in vivo growth and neovascularization of cultured A549 and NCI-H460 cells and/or xeno-grafted A549 in nude mice. This novel peptide may be used for the treatment of lung cancer carrying aberrant activated FGFR1.
Supplementary Material
Supplementary figure and tables.
Acknowledgements
This work was supported by Special Funds for Major State Basic Research Program of China (973 program) (No.2014CB942904), National Natural Science Foundation of China (No.81530071, No.81772359).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yang J, Zhao H, Xin Y. et al. MicroRNA-198 inhibits proliferation and induces apoptosis of lung cancer cells via targeting FGFR1. J Cell Biochem. 2014;115:987-95
2. Shtivelman E, Hensing T, Simon G R. et al. Molecular pathways and therapeutic targets in lung cancer. Oncotarget. 2014;5:1392-433
3. Heist R S, Mino-Kenudson M, Sequist L V. et al. FGFR1 amplification in squamous cell carcinoma of the lung. J Thorac Oncol. 2012;7:1775-80
4. Gorringe K L, Jacobs S, Thompson E R. et al. High-resolution single nucleotide polymorphism array analysis of epithelial ovarian cancer reveals numerous microdeletions and amplifications. Clinical Cancer Research. 2007;13:4731-9
5. Preusser M, Berghoff A S, Berger W. et al. High rate of FGFR1 amplifications in brain metastases of squamous and non-squamous lung cancer. Lung Cancer. 2014;83:83-9
6. Edwards J, Krishna S, Witton C J. et al. Gene amplifications associated with the development of hormone-resistant prostate cancer. Clinical Cancer Research. 2003;9:5271-81
7. Lemjabbar-Alaoui H, Hassan O U, Yang Y W. et al. Lung cancer: Biology and treatment options. Biochim Biophys Acta. 2015;1856:189-210
8. Zhang L, Yu H, Badzio A. et al. Fibroblast Growth Factor Receptor 1 and Related Ligands in Small-Cell Lung Cancer. J Thorac Oncol. 2015;10:1083-90
9. Ren M, Hong M, Liu G. et al. Novel FGFR inhibitor ponatinib suppresses the growth of non-small cell lung cancer cells overexpressing FGFR1. Oncol Rep. 2013;29:2181-90
10. Liang G, Chen G, Wei X. et al. Small molecule inhibition of fibroblast growth factor receptors in cancer. Cytokine Growth Factor Rev. 2013;24:467-75
11. Jin M, Yu Y, Qi H. et al. A novel FGFR3-binding peptide inhibits FGFR3 signaling and reverses the lethal phenotype of mice mimicking human thanatophoric dysplasia. Hum Mol Genet. 2012;21:5443-55
12. Liang Y-H, Chang C-C, Chen C-C. et al. Development of an Au/ZnO thin film surface plasmon resonance-based biosensor immunoassay for the detection of carbohydrate antigen 15-3 in human saliva. Clinical Biochemistry. 2012;45:1689-93
13. Yang G W, Jiang J S, Lu W Q. Ferulic Acid Exerts Anti-Angiogenic and Anti-Tumor Activity by Targeting Fibroblast Growth Factor Receptor 1-Mediated Angiogenesis. Int J Mol Sci. 2015;16:24011-31
14. Wu J, Wei T, Tang Q. et al. Discovery and anti-cancer evaluation of two novel non-ATP-competitive FGFR1 inhibitors in non-small-cell lung cancer. BMC Cancer. 2015;15:276
15. Chin Siang Kue K Y T, May Lynn Lam, Hong Boon Lee. Chick embryo chorioallantoic membrane (CAM): an alternative predictive model in acute toxicological studies for anti-cancer drugs. Exp. Anim. 2015:64 129-38
16. Ornitz D M, Marie P J. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes & development. 2002;16:1446-65
17. Yan D, Chen D, Im H J. Fibroblast growth factor-2 promotes catabolism via FGFR1-Ras-Raf-MEK1/2-ERK1/2 axis that coordinates with the PKCdelta pathway in human articular chondrocytes. J Cell Biochem. 2012;113:2856-65
18. Stinchcombe T E, Bogart J, Wigle D A. et al. Annual review of advances in lung cancer clinical research: a report for the year 2009. J Thorac Oncol. 2010;5:935-9
19. Perez-Moreno P, Brambilla E, Thomas R. et al. Squamous cell carcinoma of the lung: molecular subtypes and therapeutic opportunities. Clin Cancer Res. 2012;18:2443-51
20. Su N, Du X, Chen L. FGF signaling: its role in bone development and human skeleton diseases. Front Biosci. 2008;13:2842-65
21. Jin M, Du X, Chen L. Cross-talk between FGF and other cytokine signalling pathways during endochondral bone development. Cell Biol Int. 2012;36:691-6
22. Xie Y, Zhou S, Chen H. et al. Recent research on the growth plate: Advances in fibroblast growth factor signaling in growth plate development and disorders. J Mol Endocrinol. 2014;53:T11-34
23. Babina I S, Turner N C. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer. 2017;17:318-32
24. Yin Y, Ren X, Smith C. et al. Inhibition of fibroblast growth factor receptor 3-dependent lung adenocarcinoma with a human monoclonal antibody. Dis Model Mech. 2016;9:563-71
25. Terai H, Soejima K, Yasuda H. et al. Activation of the FGF2-FGFR1 autocrine pathway: a novel mechanism of acquired resistance to gefitinib in NSCLC. Mol Cancer Res. 2013;11:759-67
26. Peifer M, Fernandez-Cuesta L, Sos M L. et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104-10
27. Yang W, Yao Y W, Zeng J L. et al. Prognostic value of FGFR1 gene copy number in patients with non-small cell lung cancer: a meta-analysis. J Thorac Dis. 2014;6:803-9
28. Cihoric N, Savic S, Schneider S. et al. Prognostic role of FGFR1 amplification in early-stage non-small cell lung cancer. Br J Cancer. 2014;110:2914-22
29. Sun H D, Malabunga M, Tonra J R. et al. Monoclonal antibody antagonists of hypothalamic FGFR1 cause potent but reversible hypophagia and weight loss in rodents and monkeys. Am J Physiol Endocrinol Metab. 2007;292:E964-76
30. Safieh-Garabedian B, Dardenne M, Pleau J M. et al. Potent analgesic and anti-inflammatory actions of a novel thymulin-related peptide in the rat. Br J Pharmacol. 2002;136:947-55
31. Ribas A, Kirkwood J M, Atkins M B. et al. Phase I/II open-label study of the biologic effects of the interleukin-2 immunocytokine EMD 273063 (hu14.18-IL2) in patients with metastatic malignant melanoma. J Transl Med. 2009;7:68
32. Santoro A, Rimassa L, Sobrero A F. et al. Phase II study of NGR-hTNF, a selective vascular targeting agent, in patients with metastatic colorectal cancer after failure of standard therapy. Eur J Cancer. 2010;46:2746-52
Author contact
![]() Corresponding authors: Professor X-L Du or Dr Y-L Xie or Professor L Chen, Department of Rehabilitation Medicine, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, 10# Changjiangzhilu Daping, Yuzhong District, Chongqing 400042, China. E-mail:dxl_xiaolancom or xieyangli841015com or linchen70com
Corresponding authors: Professor X-L Du or Dr Y-L Xie or Professor L Chen, Department of Rehabilitation Medicine, Center of Bone Metabolism and Repair, State Key Laboratory of Trauma, Burns and Combined Injury, Trauma Center, Research Institute of Surgery, Daping Hospital, Third Military Medical University, 10# Changjiangzhilu Daping, Yuzhong District, Chongqing 400042, China. E-mail:dxl_xiaolancom or xieyangli841015com or linchen70com
Received 2018-1-4
Accepted 2018-6-25
Published 2018-7-27