Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Upstream molecules in The...
Downstream effectors
PI3K/AKT pathway in skeletal...
PI3K/AKT pathway in adipose...
PI3K/AKT pathway in the liver
PI3K/AKT pathway in the brain
PI3K/AKT pathway in the pancreas
Therapeutic targets
Conclusion
Acknowledgements
Abbreviations
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(11):1483-1496. doi:10.7150/ijbs.27173 This issue Cite
Review
The PI3K/AKT pathway in obesity and type 2 diabetes
Xingjun Huang1,2, Guihua Liu3, Jiao Guo2 ![]() , Zhengquan Su1,2
, Zhengquan Su1,2 ![]()
1. Guangdong Engineering Research Center of Natural Products and New Drugs, Guangdong Provincial University Engineering Technology Research Center of Natural Products and Drugs, Guangdong Pharmaceutical University, Guangzhou (510006), China.
2. Guangdong Metabolic Diseases Research Center of Integrated Chinese and Western Medicine, Guangdong Pharmaceutical University, Guangzhou (510006), China
3. Shenzhen Center for Disease Control and Prevention, 8 Longyuan Road, Nanshan District, Shenzhen (518055), China.
Received 2018-5-9; Accepted 2018-7-10; Published 2018-8-6
Abstract

Obesity and type 2 diabetes mellitus are complicated metabolic diseases that affect multiple organs and are characterized by hyperglycaemia. Currently, stable and effective treatments for obesity and type 2 diabetes mellitus are not available. Therefore, the mechanisms leading to obesity and diabetes and more effective ways to treat obesity and diabetes should be identified. Based on accumulated evidences, the PI3K/AKT signalling pathway is required for normal metabolism due to its characteristics, and its imbalance leads to the development of obesity and type 2 diabetes mellitus. This review focuses on the role of PI3K/AKT signalling in the skeletal muscle, adipose tissue, liver, brain and pancreas, and discusses how this signalling pathway affects the development of the aforementioned diseases. We also summarize evidences for recently identified therapeutic targets of the PI3K/AKT pathway as treatments for obesity and type 2 diabetes mellitus. PI3K/AKT pathway damaged in various tissues of the body leads to obesity and type 2 diabetes as the result of insulin resistance, and in turn, insulin resistance exacerbates the PI3K/AKT pathway, forming a vicious circle.
Introduction
The prevalence of obesity has grown at an alarming rate although specific public health policies and treatment efforts have been developed to resist the obesity epidemic [1], potentially leading to increased numbers of patients affected by complications of obesity, such as the most devastating type 2 diabetes mellitus (T2DM) [2]. T2DM is also a metabolic disease characterized by chronic hyperglycaemia, along with various comorbidities, such as cardiovascular disease, obesity, microangiopathy and renal failure [3]. Diabetic hyperglycaemia is caused by a decrease in insulin sensitivity, resulting in excessive insulin production, and current therapies either increase insulin sensitivity or introduce exogenous insulin. Unfortunately, these treatments currently cannot ensure long-term glycaemic control or reverse the progress [4].
In the chronically obesity, the prevalence of diabetes significantly increased, which is four times higher than the general population [5]. 80% of people with T2DM are obese or overweight, chronically obese patients, have a greater chance of developing diabetes. Both obesity and T2DM are associated with insulin resistance [6]. However, current therapies are not well designed for the effective treatment of obesity and diabetes. Thus, further researches related to the mechanism of obesity and diabetes need to be identified.
Phosphoinositide 3-kinase (PI3K) was discovered in 1985 and identified as a previously unknown phosphoinositide kinase [7, 8]. After decades of researches, the PI3K/AKT pathway is still worth studying due to its multiple functions. PI3K/AKT signalling plays a central role in cellular physiology by mediating growth factor signals during organismal growth and critical cellular processes, such as glucose homeostasis, lipid metabolism, protein synthesis and cell proliferationand survival[9]. This review mainly focuses on the mechanisms by PI3K/AKT signalling regulates metabolism in normal physiology and morbid conditions, such as obesity and T2D.
Upstream molecules in The PI3K/AKT pathway
PI3Ks are a family of lipid kinases that phosphorylate phosphatidylinositol, which is a component of eukaryotic cell membranes [10]. Based on differences in sequence homology and lipid substrate preference, PI3Ks are divided into three classes (classes I, II, and III). Among these classes, PI3K class I is the most thoroughly researched due to its various activities [8]. Class I PI3K is a heterodimer and is divided into class IA and class IB, according to differences in the molecular structure [11]. The ligands, including growth factors, cytokines and hormones, activate receptor tyrosine kinases (RTKs) and G-protein-coupled receptors (GPCR), activate PI3K. RTKs recruit class I PI3Ks to the plasma membrane, which relieves the inactivation function of p85 and p110 to activate the protein [12, 13]. GPCRs directly interact with PI3Ks through Gα or Gβγ subunits. Meanwhile, RTKs and GPCRs also activates Ras to subsequently activates PI3K [14]. Activated Class I PI3K phosphorylates the substrate phosphatidylinositol 4,5-biphosphate (PIP2) to form phosphatidylinositol 3,4,5-triphosphate (PIP3) on intracellular membranes, subsequently recruiting signalling proteins, including AKT[15]. PIP2 is synthesized by class II PI3Ks using PIP as substrate [16]. Phosphatase and tensin homologue (PTEN), a main negative regulator of the PI3K, dephosphorylates PIP3 to generate PIP2 [17].
AKT contains three domains: pleckstrin homology (PH), middle kinase and regulatory carboxy-terminal domain, of which PH domain regulates the membrane AKT translocation [9]. According to differences in serine/threonine residues, AKTs are divided into three isoforms (AKT1, AKT2 and AKT3). AKT1 expresses ubiquitously, AKT2 mainly expresses in insulin-sensitive tissues, such as skeletal muscle, adipose tissues and liver, and AKT3 expresses in the testes and brain [9, 18]. AKT is activated through two pivotal phosphorylation processes. First, phosphorylation of the threonine 308 (AKT1) in the kinase domain by phosphoinositide-dependent protein kinase 1 (PDK1) initiates the activation process [19], subsequent phosphorylation at serine 473 (AKT1) in the carboxy-terminal regulatory domain through mTOR complex 2 (mTORC2)[17, 20], which is activated by a PI3K-dependent mechanism, completely activates AKT [21]. Similar phosphorylation events are observed at corresponding residues in AKT2 (T309 and S474) and AKT3 (T305 and S472) [22]. Phosphorylation of both residues is necessary for maximum activation of AKT. Protein phosphatase 2A (PP2A) [23] and PH domain leucine-rich repeat protein phosphatases (PHLPP1 and PHLPP2) [24] dephosphorylate AKT T308 and S473, respectively, leading to AKT inactivation. Recently, endomembranes that contain PIP3 and PIP2 have also been shown to directly contribute to AKT activation [25, 26].
Although several studies have reported an absolute requirement for PI3K in AKT activation, AKT activation has also been suggested to be mediated by a PI3K-independent mechanism [27]. However, researchers have not firmly established whether functional AKT activation occurs in the absence of productive PI3K signalling and thus this topic requires further study.
Downstream effectors
Downstream effectors, including protein and lipid kinases, transcription factors, regulators of small G proteins and vesicle trafficking, metabolic enzymes, E3 ubiquitin ligases, cell cycle regulators, and many other effectors, are regulated through serine and/or threonine phosphorylation by AKT (Fig. 1) [22]. Those effectors share a common minimal sequence motif, Arg-Xaa-Arg-Yaa-Zaa-Ser-Hyd [28]. Through effecttors, AKT has important roles in many cellular processes, such as apoptosis, cell survival, cell cycle progression and metabolism. The downstream effectors are described in a previous review [22]. Here, we mainly describe downstream effectors involved in metabolism.
PI3K/AKT pathway. PI3K activates AKT, and AKT phosphorylates downstream substrates that are involved in the regulation of diverse cellular functions, including apoptotic, metabolism and cell cycle progression. As illustrated by these targets, a high degree of functional versatility and overlap exists amongst AKT substrates. Red and black line indicates inhibition and activation respectively. AKT, protein kinase B; AS160, Akt Substrate of 160 kDa; FoxO1, Forkhead box O1; GSK3, glycogen synthase kinase 3; GPCR, G-protein-coupled receptors; IRS-1/2,insulin receptor substrate-1/2; IKK, IkB-kinase; MAD1, MAX dimerization protein 1; MDM2, murine double minute 2; MLK3, mixed lineage kinase 3; mTORC1/2, mTOR complex 1/2; PDK1, phosphoinositide-dependent protein kinase 1; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol 4,5-biphosphate; PIP3, phosphatidylinositol 3,4,5-triphosphate; PTEN, Phosphatase and tensin homologue; RTK, receptor tyrosine kinases; SREBP, sterol regulatory element-binding proteins; TSC1/2, tuberous sclerosis complex 1/2.
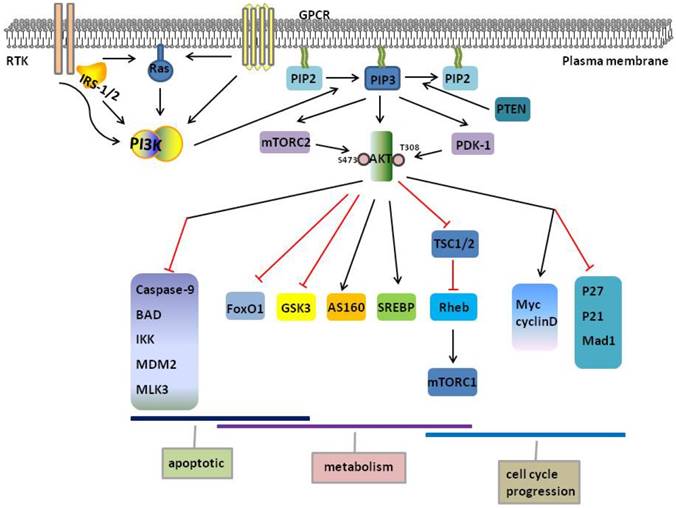
AKT regulates glucose and lipid metabolism. Activated AKT2, which is primarily expressed in insulin-responsive tissues, promotes translation of glucose transporter 4 (GLUT4). And the direct target of AKT is a substrate of 160 kDa (AS160), also known as TBC1D1 [29]. In intracellular compartments, AKT converts glucose to glucose 6-phosphate by stimulating hexokinase. AKT regulates two processes by glycolysis Glucose 6-phosphate and glycogen synthase kinase 3 (GSK3) to produce cellular energy via glycolysis and promotes glycogen production by inhibiting [30]. FoxO proteins, particularly FoxO1, are the main target of AKT and affect energy homeostasis throughout body [31]. FoxO1 and peroxisome proliferator-activated receptor-coactivator 1α (PGC1α) coordinately regulate gene expression to increase gluconeogenesis and fatty acid oxidation [32]. On the other hand, FoxO1 induces the expression of phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase gene (G6PC), subsequently increases gluconeogenesis [33]. AKT directly inhibits FoxO1, reducing glucose levels [31], FoxO1 simultaneously activates AKT to increase energy production and inhibits mTOR complex 1 (mTORC1) to reduce lipid and protein production [34]. Finally, GSK3 inhibits glycogen synthase (GS), which promotes glycogen synthesis. AKT exerts an inhibitory effect on GSK3 by phosphorylation of GSK3 [35]. AKT regulates lipid metabolism through sterol regulatory element-binding proteins (SREBP), which increases cholesterol and fatty acid accumulation, including SREBP-1c, SREBP-1a, and SREBP-2[18, 34]. Therefore, PI3K/AKT regulates glucose metabolism through FoxO1 and GSK-3 and lipid metabolism through mTORC1 and SREBP.
PI3K/AKT pathway in skeletal muscle
Approximately 90% of insulin-stimulated glucose utilization occurs in skeletal muscle and plays an important role in regulating glucose metabolism and energy homeostasis [36]. Insulin regulates skeletal muscle metabolism by promoting glucose transport, glycogen synthesis and protein synthesis through PI3K/AKT signalling pathway (Fig. 2).
PI3K/AKT pathway in muscle tissue when normal state and insulin resistance. In normal sate, insulin mediates PI3K/AKT pathway and then regulates glucose transport through activating AS160, glycogen synthesis through inhibiting GSK3 and protein synthesis through activating S6K1 or inhibiting 4E-BP1. When the excess FFAs enters into skeletal muscle, FFAs forms LCACoAs, and then partitioned to the TAG or toward the mitochondria for oxidation fatty acid, which inhibits PI3K/AKT signalling and leads to skeletal muscle insulin resistance. Red and black line indicates inhibition and activation respectively. See text for detailed descriptions.
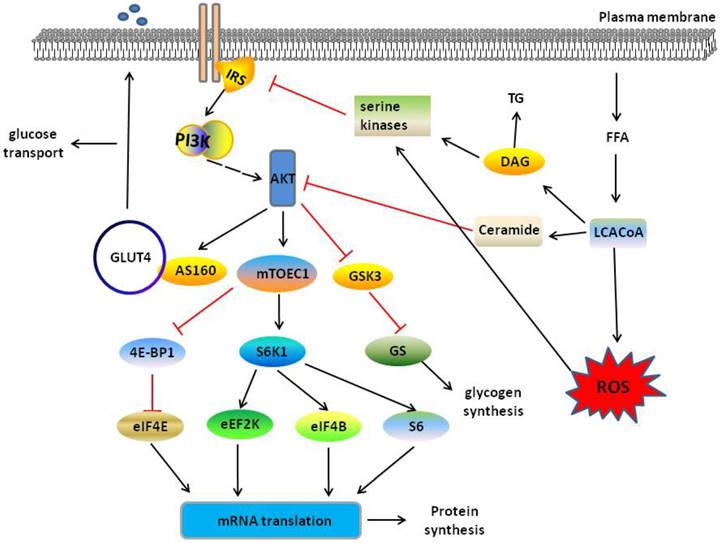
Previous research has found knockout or knockdown of AKT or IRS adaptor proteins obviously reduce insulin-induced glucose uptake, whereas overexpression of AKT increases glucose uptake [37]. AKT directly phosphorylates AS160, inducing GLUT4 translocation, which translocated to the plasma membrane from storage vesicles and transports glucose in skeletal muscle following stimulation of insulin and AKT [38, 39]. GagAKT, constitutively activated form of AKT, promotes glycogen synthesis in L6 myotubes [40]. Following study found activated AKT increases glycogen synthesis in skeletal muscle, activating glycogen synthase (GS) to redirect glucose-6-phosphate to glycogen and inhibiting GSK-3[41]. In skeletal muscle, insulin stimulates protein synthesis and accelerates mRNA translation by regulating the initiation steps of protein translation [42]. AKT-mediated mTORC1 activation regulates a number of downstream effectors proteins through phosphorylation, including the well-characterized effectors p70 ribosomal S6 protein kinase-1 (S6K1) and eukaryotic translation initiation factor-4E (eIF4E)-binding protein-1 (4E-BP1), to increase protein synthesis [43, 44]. Obesity is associated with several processes in skeletal muscle metabolism, leading to insulin resistance. Insulin resistance refers to a blunted response to insulin in organs and as an important marker of T2DM [45]. Therefore, dysfunctional PI3K/AKT-mediated glucose transport and glycogen synthesis play an important role in the development of obesity and T2DM.
Under normal conditions, FFAs enter the skeletal muscle through fatty acid translocase/CD36 and fatty acid-binding protein, and then forms long chain fatty acid CoAs (LCACoAs), which are then partitioned to the synthesis of lipids (TAG) or toward the mitochondria for oxidation [46]. Excessive lipid production and impaired disposal may promote the development of insulin resistance and diabetes in the skeletal muscle [47, 48]. In early studies, insulin resistance was thought to be accompanied by excessive intramyocellular lipid (IMCL) levels [49], and improved insulin sensitivity observed in subjects with T2DM by reduced IMCL levels [50]. Recently, muscle lipid intermediates, such as LCACoAs, ceramides and diacylglycerol (DAG), were shown to be the cause of insulin resistance [46, 48] instead of IMCL [51]. In the presence of insulin resistance, the level of free fatty acids increases in the body resulting in the accumulation of intramyocellular lipids, such as DAG and ceramides [52]. DAG activates serine kinases phosphorylates and inhibits the signal transduction capacity of IRS-1[53]. Ceramides disturb insulin signalling by AKT, leading to reduction of glucose utilization [54]. Mitochondria-dependent mechanisms also cause insulin resistance. Excessive fatty acid oxidation in the mitochondria via the tricarboxylic acid (TCA) cycle and the electron transport chain (ETC) increases the production of fatty acid metabolites and reactive oxygen species (ROS), subsequently activated intracellular stress kinases to inhibit IRS-1 and possibly exerting effects on insulin signalling from GLUT4 translocation to unknown points downstream of AKT and AS160 function [46, 55] (Fig. 2). Excessive lipid intermediate production and the mitochondria disorder, impair the insulin-AKT pathway, leading to insulin resistance in skeletal muscle, and ultimately affect development of obesity and T2DM.
PI3K/AKT pathway in adipose tissue
Adipose tissue has two main functions: energy storage and endocrine function, which plays an important role in maintaining body energy balance, including protecting tissues and organs from cold and hot, thermogenesis and the production of various hormones/cytokines that are collectively referred to as cytokines [56, 57]. Insulin-AKT signalling regulates the metabolism of adipose tissues by promoting glucose utilization, protein synthesis, and lipogenesis. Insulin stimulates the utilization of approximately 10% of glucose in adipose tissue, glucose metabolism in adipose tissue impacts other tissues through its extra-adipose actions [58].
Although most of the lipid stored in adipose tissues is derived from the diet, the adipocytes are fully capable of synthesizing new fatty acids from excessive substrates through de novo lipogenesis (DNL), and fatty acids are then transferred into TGA and eventually exported as very low density lipoproteins (VLDLs) for storage and utilization by peripheral tissues [56]. The two major enzymes of DNL, fatty acid synthase and acetyl CoA carboxylase, are abundantly expressed in adipose tissues under the control of SREBP-1c [56]. In normal physiology, feeding is able to activate synthesis of SREBP-1c via insulin-dependent pathway (mTORC1) or insulin-independent pathway (carbohydrate response element binding protein (ChREBP)) [59, 60]. But insulin pathway is require for expression of SREBP-1c mRNA in obesity or T2DM [60].
PI3K/AKT signalling pathway promotes lipid biosynthesis and inhibits lipolysis. The substrate SREBP, which regulates fatty acid synthase and cholesterol-related genes [18, 61], and FOXO1, which regulates lipolysis by controlling the expression of adipose triglyceride lipase (ATGL) [62], are primary substrates for AKT-mediated lipid metabolism [63]. In the feeding state, SREBP-1c is regulated by four pathways: activated AKT stimulates liver X receptor (LXR), which is require for SREBP-1c transcription [64]; activated AKT stimulates mTORC1 to activate SREBP-1c transcription, and mTORC1 also inhibits Lipin-1, which decreases the half-life of nuclear SREBP-1c [65, 66], and activates S6K1, which promotes SREBP-1c maturation [67, 68]; activated AKT suppresses insulin-induced gene 2A (INSIG2A), which strongly promotes the maturation of SREBP-1c precursors into nuclear forms [69]; and activated AKT inhibits GSK3, which extends the half-life of SREBP-1c [70]. In the fasting state, acute lipolysis is induced by β-adrenergic signalling, leading to cAMP accumulation and subsequent PKA-mediated phosphorylation of hormone sensitive lipase (HSL) and perilipin [71]. In the fed state, PI3K/AKT inhibits protein kinase A (PKA) and thus suppresses lipolysis. AKT-independent, PI3K-dependent pathway also regulates adipocyte lipolysis by directly regulating PKA [72], AKT regulate FoxO1 through three pathway: reducing the expression of the rate-limiting lipolytic enzyme (ATGL), which is responsible for triacylglycerol hydrolase activity (ATGL) [62]; reducing the expression of interferon regulatory factor 4 (IRF4), which promotes lipolysis, at least in part, by inducing the expression of the lipases HSL and ATGL [71]; phosphorylating phosphodiesterase 3b (PDE3B) to reduce intracellular cAMP levels and PKA activity, thus inhibiting lipolysis in adipocytes [73].
Obesity is associated with insulin resistance. Abnormal glucose metabolism caused by insulin resistance in adipose tissues impacts other tissues through its extra-adipose actions. For example, specifically knockdown of GLUT4 in adipose tissues resulted in insulin resistance in skeletal muscle and liver instead of adipose tissues [74]. When the synthesis ability of adipose tissues decreases and fat degradation increases, the release of free fatty acids (FFAs) increases, thus reducing circulating adiponectin levels and lipid oxidation in extra-adipose tissues. However, this process triggers the ectopic accumulation of lipids and subsequently causes lipotoxicity and insulin resistance [75]. How does insulin resistance occur in adipose tissues?
The synthesis and secretion of adiponectin, tumour necrosis factor-α (TNF-α) and interleukin 6 (IL-6), which enhance lipolysis by reducing perilipin and fat-specific protein 27 levels [76]. The increased in subjects with obesity-induced inflammation, aggravating insulin resistance [77]. In the presence of adipose hypertrophy, insulin resistance also occurs because of the expansion of adipose tissues exceeds their storage capacity [45, 78, 79]. Additionally, adipose hypertrophy increases tissue immune cell infiltration, fibrosis, and lipolysis, reduces IRS-1 activation and AKT-induced glucose uptake, and exacerbates systemic insulin resistance and the development of T2DM [79, 80]. Therefore, adipocytokines and adipose hypertrophy cause insulin resistance by blocking PI3K/AKT-mediated inhibition of lipolysis attenuating the capacity of glucose utilization, and weakening the capacity of SREBP to promote lipid synthesis.
PI3K/AKT pathway in the liver
Most of the extracellular glucose is produced in the kidneys and liver, and only the liver acutely responds to insulin by reducing glucose levels [81]. In the fasting sate, glucose is primarily utilized in the liver for gluconeogenesis and glycogenolysis then transported to various tissues while suppressing the synthesis of new fatty acids [82]. In the fed state, the PI3K/AKT signalling pathway reduces hepatic glucose production (HGP) and glycogenolysis, increases glycogen synthesis and the synthesis of fatty acids for storage and subsequent utilization by other tissues [82, 83].
In previous studies, insulin suppressed PEPCK and G6PC expression, which are known to increase hepatic gluconeogenesis by activating the PI3K/AKT pathway [84-86]. However, insulin was recently shown to inhibit gluconeogenic gene expression [87]. Additionally, those mice still maintained the normal postprandial state, even in the absence of canonical liver insulin signalling [85, 87]. Based on these results, AKT may not only be an indispensable intermediate for insulin action, extrahepatic insulin may be stimulated by other pathways to regulate glucose metabolism in the liver. Three pathways may mediate cell-nonautonomous effects on hepatic glucose metabolism [88]. First, insulin action in the hypothalamus through K (ATP) channels decrease expression of G6PC and hepatic glucose production (HGP) in liver [89, 90]. Subsequently, insulin acts in agouti-related peptide (AgRP) neurons through PI3K-PIP3 signalling to control this process [91]. However, further studies should be carried on because the action of insulin in brain may not common in all species [92, 93]. Second, insulin functions through the AKT pathway to inhibit lipolysis and decrease circulating FFA levels in adipose tissues, which suppressing HGP in the liver, even in the absence of hepatic insulin signalling [82, 94]. Third, insulin functions through the AKT pathway suppress α-cell glucagon secretion in the brain, which reducing HGP levels in the liver [92, 95, 96]. All three pathways require the insulin-PI3K pathway regulate to HGP, regardless of whether insulin functions in the brain, liver or adipose tissues (Fig. 3).
PI3K/AKT pathway in liver and other tissue mediate liver metabolism. PI3K/AKT signal increase DNL in four ways: activating S6K1 and SREBP, inhibiting Lipin1 and GSK3. And inhibits HGP through directly inhibiting FoxO1 or indirectly other tissue PI3K/AKT signaling including adipose, brain and pancreas. Red and black line indicates inhibition and activation respectively. See text for detailed descriptions.
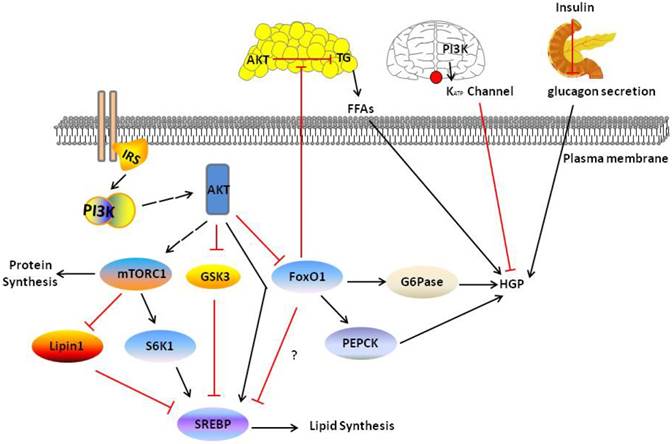
The PI3K/AKT pathway also mediates lipid synthesis. Overexpression of SREBP-1c in the liver selectively induces the expression of lipogenic, but has no effect on cholesterol synthesis genes. AKT has been reported to modulate the mTORC1-S6K1 pathway to regulate SREBP-1c expression in the liver [97]. AKT suppresses the expression of INSIG2A, a liver-specific transcript encoding the SREBP-1c inhibitor INSIG2, through an mTORC1-independent pathway [98]. However, in the absence of AKT2, the activation of mTORC1 and SREBP-1c is not sufficient to drive postprandial lipid generation [99]. Thus AKT-mediated mTORC1-dependent and -independent pathways are required for lipogenesis. The role of FoxO1 in liver lipid metabolism is less well studied; some studies have shown that FoxO1 is directly associated with insulin-promoted expression of fat genes in the liver, whereas others have reported a permissive role for FoxO1 in these processes [100-102]. However, AKT-dependent activation of mTORC1 was recently shown to be required for DNL, but recently inhibition of FoxO1 also is required and sufficient for DNL [82]. Further studies are required to reconcile the relationships between the mTORC1-dependent pathway, INSIG2-dependent pathway and FoxO1 pathway (Fig. 3).
In obesity, the blunted response of adipose tissues leads to decreased FFA uptake and glucose utilization, resulting in ectopic accumulation in other tissues [103]. Ectopic accumulation of lipids in the liver is highly correlated with insulin resistance and non-alcoholic fatty liver disease (NAFLD) [104]. Meanwhile, excessive oxidation of FFAs increases the hepatic acetyl-CoA content and thus activates pyruvate carboxylase, which is required for gluconeogenesis [45], and increases DAG levels, which impair PI3K/AKT pathway and exacerbate insulin resistance [48, 105]. Chronic metabolic inflammation and endoplasmic reticulum (ER) stress also impair hepatic insulin-AKT signalling and subsequently causes insulin resistance [106, 107].
Hepatic glycogen synthesis decreases during hepatic insulin resistance due to decreased insulin-PI3K signalling [48]. However, contradictory findings are observed in insulin-mediated DNL and HGP: why does insulin normally stimulate DNL but inhibit HGP in hepatic insulin resistance. Two possible mechanisms have been reported: in the first, the liver displays selective insulin resistance in which the actions of insulin are suppressed in HGP but are normal in DNL [108]. And recent study finds Irs2-knockout mice develop 'selective insulin resistance', whereas mice lacking in Irs1, or both Irs1 and Irs2, develop 'total insulin resistance', which indicates the IRS 1/2 are the cause of insulin selective resistance [109]; in the second, hepatic insulin signalling remains intact but the cell-nonautonomous pathway may be blocked, which is mentioned on above, resulting in increased HGP during insulin resistance [82]. Others recent study also proved this hypothesis [85, 87]. The first statement can be interpreted as insulin resistance caused by the integrity of the substrate IRS1/2. The two possible mechanisms are, in fact, that insulin signals are intact and both cause increased HGP and DNL when insulin resistance.
PI3K/AKT pathway in the brain
Previously, insulin-sensitive organs included the skeletal muscle, liver and adipose tissues. The brain has recently been reported to be an insulin-sensitive organ, and its function in glucose metabolism and energy regulation has also been recognized.
Leptin is the most important hormone reported to date that regulates food intake and energy balance [110]. In the hypothalamic arcuate nucleus (ARC), anorexigenic proopiomelanocortin (POMC) and orexigenic AgRP coordinately regulate food intake and energy expenditure, as well as peripheral tissue glucose homeostasis and energy partitioning through insulin, leptin and nutrients [111]. The leptin activity-mediated IRS-PI3K pathway was observed in the IRS2-deficient mice [112]. Leptin requires intact PI3K signalling to promote hypothalamic expression of POMC genes and inhibit expression of the NPY and AgRP genes [113, 114]. In a subsequent study, leptin and insulin were shown to coordinately regulate FoxO1 activity, increasing the expression of NPY and AgRP and suppressing POMC expression [115, 116]. Recently, Gpr17, the target of FoxO1, was shown to regulate feeding and sensitivity through insulin and leptin in AgRP neurons [117, 118]. However, leptin and insulin-mediated PI3K signalling was only shown to play a role in POMC expression in the presence of short-term alterations in leptin levels. In the presence of persistent excess energy availability, leptin signalling activates other pathways, such as STAT3 or SH2-containing tyrosine phosphatase, to predominantly regulate food intake in POMC neurons [90, 113, 119]. The PI3K/AKT signalling pathway plays an important role in glucose homeostasis mediated insulin and leptin actions in the hypothalamus and is thought to exert less important effects on food intake [120].
mTOR, a well-known phosphorylation target of AKT, specifically binds to Rheb GTPase with GTPase-activating protein (GAP) activity, which directly activates mTORC1[121]. As shown in a previous study, acute activation of mTOR1 promotes the ability of hypothalamic neural circuits to sense the nutritional milieu and regulate feeding [122, 123]. According to a recent study, mTOR1 is dispensable for the regulation of feeding behaviour and energy metabolism [124], and chronic activation of mTOR1 interferes with the hypothalamic neurons that regulate feeding and energy balance [125]. However, mTOR1 signalling is required for glucose homeostasis in the hypothalamus [126, 127]. One way to regulate HGP is that S6K1, a downstream molecule of mTOR1, regulates HGP, modulates neuronal excitability and peripheral lipid metabolism in POMC neurons, regulates the insulin sensitivity of skeletal muscle via AgRP neurons [128]. Another way to regulate HGP is PI3K-stimulated generation of PIP3 in AgRP neurons, which activates K (ATP) channels, decreasing G6PC expression in the liver [91]. Compared with POMC, AgRP plays a more important role in the long-term regulation of glucose metabolism [91]. The selective inactivation of IR in AgRP neurons in mice results in an inability to suppress HGP whereas only a deficiency in both leptin and IRs in POMC neurons resulted in systemic insulin resistance [45, 129]. Acute activation of AgRP neurons decreases insulin-stimulated glucose uptake and insulin sensitivity by increasing myostatin expression, a muscle-related gene expressed in brown adipose tissue that causes insulin resistance [130]. Recently, Another ligand, glucagon-like peptide (GLP-2), it was recently shown to play a key role in the control of HGP through the PI3K/AKT pathway[131], suggesting that in addition to insulin and leptin, GLP-2 also regulates HGP through PI3K/AKT signalling.
Leptin and insulin-mediate PI3K/AKT signalling is impaired in the brains of obese subjects with insulin resistance [132]. There are two reasons for this: first, the transport of insulin through the blood brain barrier is impaired in obese subjects [45]. second, brain insulin action is attenuated or even diminished in response to both endogenous and exogenous insulin stimulation [133], which impairs hypothalamic AKT activation due to ER stress and the activation of inflammatory pathways [134]. Both factors contribute to the development of insulin resistance in the brain (Fig. 4). Insulin resistance in the brain is followed by disorders of glucose metabolism, such as hepatic glucose production and glucose transport, occurring in other insulin-sensitive organs and may eventually lead to systemic insulin resistance.
PI3K/AKT pathway in normal state and insulin resistance. In normal state, ligand activates PI3K/AKT signalling which inhibits food intake in hypothalamus. And the signalling also inhibits HGP through regulate liver and inhibit lipolysis through regulate adipose tissue. And when the ligand can not pass through BBB, and the ER stress occurs, insulin resistance would occurs in brain, leading to damage PI3K/AKT pathway. Red and black line indicates inhibition and activation respectively. See text for detailed descriptions.
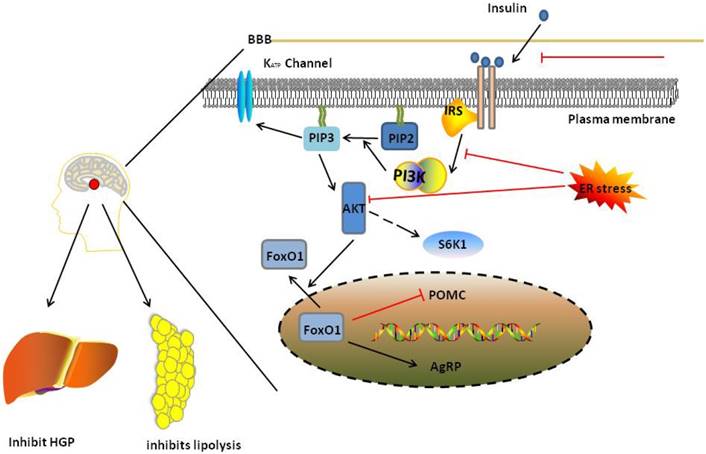
Three functions of the PI3K/AKT pathway are disturbed in insulin resistance in the brain. First, PI3K generates PIP3, which regulates GHP through K (ATP) channels in the hypothalamus. Insulin signalling is blocked during insulin resistance in the brain due to increased protein kinase C (PKC) activity, ER stress and inflammation [134, 135], which blocks the ability of PI3K to generate PIP3. Second, in hypothalamic neurons, FoxO1 upregulates AgRP/NPY expression and downregulates POMC expression under normal conditions, which is inhibited by AKT. In response to insulin resistance in the brain, overexpression of FoxO1 in POMC neurons leads to obesity and hyperphagia [136]. Third, according to studies by Ono et al. [137] and Um, S. H et al. [138], activation of S6K1, a downstream effector of mTORC1, in hypothalamic neurons leads to hepatic insulin resistance because it decreases the stimulation of IRS-1 and AKT.
PI3K/AKT pathway in the pancreas
The pancreas is mainly composed of four types of cells, of which pancreatic β cells are vitally important in maintaining glucose homeostasis by producing and secreting insulin in response to the blood glucose concentration [58]. Insulin regulates β cell function and insulin secretion through the PI3K/AKT pathway.
Insulin was not previously thought to exert an effect on insulin synthesis, differentiation and secretion in pancreatic cells. However, insulin has consistently been shown to play an important role in pancreatic cells [139], mainly through PI3K/AKT signalling. β cell-specific knockout of IRS2 or the IR, which disrupts of insulin signalling in pancreatic β cells, reduces the pancreatic insulin content, β cell mass and glucose-stimulated insulin secretion, followed by the development of a phenotype similar to T2DM [140, 141]. Activation of the PI3K/AKT pathway promotes insulin secretion form pancreatic β cells [142, 143]. Overexpression and constitutive activation of AKT in pancreatic β cells results in an increase in the β cell mass, proliferation and cell size, which are mediated by signalling intermediates downstream of AKT, such as FoxO1, GSK3 and mTOR1, providing further evidence for the role of AKT in pancreatic cells [144]. In contrast, overexpression of a kinase dead mutant in β cells, which reduced AKT activity by 80%, results in a lack of insulin secretion [145]. In addition, the PI3K/AKT pathway prevents lipotoxicity in pancreatic β cells by inhibiting FoxO1, which is abundantly expressed in pancreatic β cells and promotes FFA-induced β cell apoptosis [146, 147].
Obesity and T2DM are associated with insulin resistance. In insulin resistance, the activity of β cells are increased, boosting the release of additional insulin to maintain normal glucose tolerance, resulting in the development of hyperinsulinemia [148]. Glucose tolerance is impaired in response to β cell dysfunction in insulin resistance, resulting in the development of T2DM [149]. In obese subjects, excess levels of circulating FFAs, which are caused by a blunted response of adipose tissues, impairs the function of β cells [150]. This finding also explains why obese individuals readily develop to T2DM. In obesity and T2DM, the insulin-mediated PI3K/AKT pathway is also blocked, which reduces insulin secretion and β cell function [58]. This condition further aggravates insulin resistance by affecting other tissues.
Therapeutic targets
The PI3K/AKT signalling pathway regulates cell proliferation, differentiation, metabolism, and cytoskeletal reorganization, leading to apoptosis and cancer cell survival. Therefore, the pathway is associated with various diseases, such as obesity, diabetes and cancer. The table mainly summarizes recent target treatments which related to PI3K/AKT pathway for obesity and type 2 diabetes (Table 1). In normal condition, PI3K/AKT is activated in regulating the function of the body. When excessive energy intake occurs, the PI3K/AKT pathway is suppressed. At this point, activation of the PI3K/AKT pathway alleviates obesity and insulin resistance. However, when the regulation of PI3K is disturbed, such as overexpression and mutation, it can cause many human diseases (obesity, cancer, etc.) [151]. At this point, the pharmacological inhibition of PI3K is an effective and safe anti-obesity and anti-cancer intervention that can reverse the negative effects of human diseases [152].
Summary of targeting PI3K/AKT pathway in the treatment of obesity and T2DM
| Key cell type/tissue | target | Mechanism | Effect | References |
|---|---|---|---|---|
| C57BL/6 mice HepG2 cell | FAM3A | Activates PI3K/AKT signaling | inhibits hepatic gluconeogenesis | [153] |
| db/db mice HFD-induced diabetic mice | Dapper1 | Activates PI3K/Akt in an insulin-independent manner | attenuates hepatic gluconeogenesis and lipogenesis in T2D | [154] |
| Streptozocin-induced diabetic mice db/db mice HepG2 cell | ATP synthase β subunit (ATPSβ) | Activates PI3K/Akt pathway | promotes hyperglycemia | [155] |
| db/db diabetic mice HepG2 cell | Bone morphogenetic protein-7 (BMP-7) | Activates PDK1 and AKT strengthens insulin signaling | improves glucose uptake promotes peripheral insulin resistance | [156] |
| Streptozotocin-induced diabetic db/db mice Pancreatic beta-cell | glial cell line-derived neurotrophic factor (GDNF) | Activates phosphorylation of AKT | improves glucose tolerance and β cell mass | [157, 158] |
| Pancreatic beta-cell | nuclear factor erythroid 2-related factor 2 (Nrf2) | Through PI3K/AKT pathway | Enhances glucose-stimulated insulin secretion in β-cell and insulin sensitivity | [159] |
| Male C57BL/6J mice HepG2 cell | Irisin | Activate PI3K/Akt/FOXO1 and PI3K/Akt/GSK3 pathway | Reduces gluconeogenesis and increase glycogenesis | [160] |
| Streptozotocin-induced diabetic mice | amniotic fluid stem cells (AFSC) | Activates the IR/PI3K/Akt signaling pathway preserving | promoting endogenous β-cell functionality and proliferation. | [161] |
| C57BL/6 mice HepG2 cell | glycerol kinase (Gyk) | Decreases the expression of FoxO1 | reduced expression of G6Pase and PEPCK | [162] |
| C57BL/6 mice | NPC1L1 | Regulates the AKT-FOXO1-PEPCK/G6Pase pathway | reduced expression of G6Pase and PEPCK | [163] |
| MCF7-tet OFF Sesn2F cells | Sestrins (Sesn1/2/3) | through activation of AMPK and mTORC2, which is necessary for activation of AKT | Regulate both glucose and lipid metabolism | [164, 165] |
| Mesenchymal stem cells | Apelin 13 | Activate PI3K/AKT signaling pathways | enhance the efficacy of MSCs in cell therapy of diabetes | [166] |
| C57BL/6 mice HepG2 cell | miR-451 | regulate the AKT-FOXO1-PEPCK/G6Pase pathway | promote gluconeogenesis and promote glucose homeostasis | [162] |
| C57BL/6 mice HepG2 cell | GSK-3 | Global inhibition of GSK-3 | improves insulin sensitivity and hepatic glycogen deposition significantly | [35, 167] |
| C57BL/6 mice HEK293 Cells HepG2 cell | miR-423-5p | Repress the FAM3A-ATP-Akt pathway | Promote gluconeogenesis and hyperglycemia | [168] |
| Agrp-specific FoxO1 knockouts by matingAgrp-ires-Cre-transgenic mice with Foxo1loxP/loxP mice | Gpr17 | Reduce expression of FoxO1 | Improvedglucose homeostasis, and increased sensitivity toinsulin and leptin | [117, 118] |
| PHLPP-1 knockout (KO) C57BL/6 mice | PHLPP | PHLPP inhibits PI3K/AKT signaling | As inhibitor of AKT | [169, 170] |
| PB-Cre4 mice PTENloxP/loxPmice | PTEN | PTRN inhibits PIP3, thus reduces activation of AKT | As inhibitor of PI3K | [152, 171] |
Conclusion
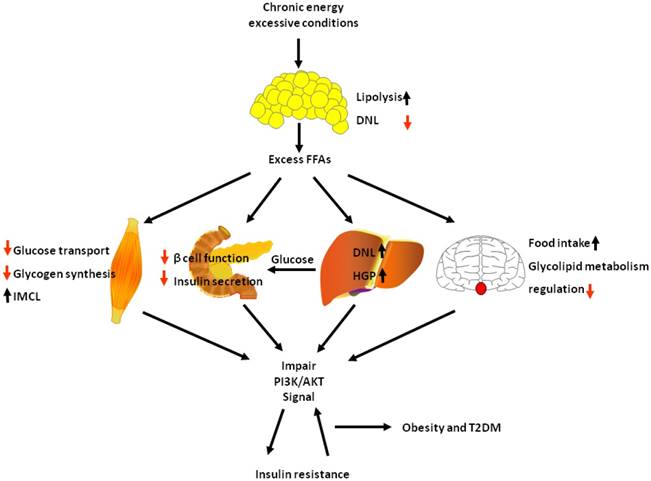
Many ligands such as leptin, insulin, GPL, growth factors act on PI3K/AKT pathway and play a variety of physiological roles. Insulin is the main ligand for PI3K/AKT pathway to regulate metabolism, others only account a small part for metabolism. In obesity and diabetes, insulin guided multiple pathways to regulate lipid and glucose metabolism. Among them, PI3K/AKT is the main pathway of insulin. PI3K/AKT signalling pathway exists various organs in the body and plays an important role in a variety of physiological functions. Here we describe the metabolic functions of PI3K/AKT signalling pathway in the brain, liver, muscle, adipose and pancreas play a role in the metabolism, and the relationship between obesity and diabetes. Under physiological conditions, insulin secreted immediately after a meal activates PI3K/AKT signalling pathway, which increases glucose utilization and reduces gluconeogenesis in liver and muscle, increases body lipid deposition thus reduces FFA circulation in adipose tissue, increases insulin production in the pancreas, regulates lipid and glucose metabolism balance, reduce appetite in the brain. However, in chronic energy excessive conditions as observed in obesity, sustained excess circulating FFAs consistently harm other tissue: lipid accumulation is saturated and results in an increase in lipolysis in adipose tissue, further aggravating content of circulating FFAs; lipid ectopic accumulation in skeletal muscle causes reduce of glucose transport and glycogen synthesis, leading to glucose metabolism unbalance; excess circulating FFAs also destroy β cell function and insulin secretion; and in liver, insulin action is normal, but inhibition of extra-hepatic insulin signalling and lipid ectopic accumulation causes increase of HGP and excess insulin causes increasing DNL. All of those would impair PI3K/AKT signal then causing insulin resistance, and insulin resistance further exacerbates PI3K/AKT signal, forming a vicious circle. Finally, the vicious circle would lead to obesity and T2DM (Fig. 5).
In chronic energy excessive conditions, the causes of insulin resistance. In chronic energy excessive conditions, lipid accumulation is saturated and results in an increase in lipolysis in adipose tissue, causing excess FFAs. Lipid ectopic accumulation in skeletal muscle causes reduce of glucose transport and glycogen synthesis; excess circulating FFAs also destroy β cell function and insulin secretion; and in liver, insulin action is normal, but inhibition of extra-hepatic insulin signalling and lipid ectopic accumulation causes increase of HGP and excess insulin causes increasing DNL; and in brain, excess FFAs causes glucose and lipid metabolism disorder. All those ultimately impair PI3K/AKT signal, causing insulin resistance, and insulin resistance further exacerbates PI3K/AKT signalling, forming a vicious circle. Black arrow indicates activation or augment. Red arrow indicates reduction.
Over the past decade, people have made a surprising understanding of the regulation and function of PI3K/AKT signalling pathway. There is no doubt that PI3K/AKT signalling is closely related to metabolism. And manipulation of PI3K/AKT signalling and its downstream molecule are promising therapeutic target for the treatment of obesity and T2DM. However, the PI3K/AKT pathway is still complex and still needs a lot of energy to study.
Acknowledgements
This work was financially supported by the Industry University Research Collaborative Innovation Major Projects of Guangzhou Science Technology Innovation Commission, China (no.201604020164); the Science and Technology Planning Project of Yunfu, Guangdong, China (no.201702-9) and the National Science Foundation of China (no.81173107).
Abbreviations
PI3K, Phosphoinositide 3-kinase; AKT, protein kinase B; T2DM, type 2 diabetes mellitus; RTKs, receptor tyrosine kinases; GPCR, G-protein-coupled receptors; PIP2, phosphatidylinositol 4,5-biphosphate; PIP3, phosphatidylinositol 3,4,5-triphosphate; PTEN, Phosphatase and tensin homologue; PDK1, phosphoinositide-dependent protein kinase 1; mTORC2, mTOR complex 2; PP2A, Protein phosphatase 2A; PHLPP1, PH domain leucine-rich repeat protein phosphatises 1; GLUT4, glucose transporter 4; GSK3, glycogen synthase kinase 3; FoxO, forkhead box O; PGC1α, peroxisome proliferator-activated receptor-coactivator 1α; PEPCK, phosphoenolpyruvate carboxykinase; G6PC, glucose-6-phosphatase gene; SREBP, sterol regulatory element-binding proteins; IGF-1, insulin-like growth factor 1; KLF14, Krüppel-like factor 14; GS, glycogen synthase; UCP1, uncoupling protein 1; HDL, high density lipoprotein; S6K1, S6 protein kinase-1; 4E-BP1, eukaryotic translation initiation factor-4E binding protein-1; eIF4F, Eukaryotic translation initiation factor 4F; eIF4B, eukaryotic translation initiation factor 4B; eEF2K, eukaryotic elongation factor-2 kinase; eEF2, elongation factor 2; FFAs, free fatty aids; LCACoAs, long chain fatty acid CoAs; IMCL, intramyocellular lipid; DAG, diacylglycerol; TCA, tricarboxylic acid; ETC, electron transport chain; ROS, reactive oxygen species; DNL, de novo lipogenesis; VLDLs, very low density lipoproteins; ChREBP, carbohydrate response element binding protein; ATGL, adipose triglyceride lipase; LXR, liver X receptor; INSIG2A, insulin-induced gene 2A; HSL, hormone sensitive lipase; PKA, protein kinase A; IRF4, interferon regulatory factor 4; PDE3B, phosphodiesterase 3b; IL-6, interleukin 6; NAFLD, non-alcoholic fatty liver disease; ER, endoplasmic reticulum; ARC, arcuate nucleus; POMC, proopiomelanocortin; AgRP, agouti-related peptide; VMH, ventral medial nucleus; TH, tyrosine hydroxylase; JAK2, Janus kinase 2; STAT3, Signal transducer and activator of transcription 3; GAP, GTPase-activating protein; GLP-2, glucagon-like peptide; HGP, hepatic glucose production; PKC, protein kinase C.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Barnett R. Obesity. The Lancet. 2017;389(10069):591
2. Wang YC, McPherson K, Marsh T, Gortmaker SL, Brown M. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet. 2011;378(9793):815-25
3. Rask-Madsen C, King GL. Vascular complications of diabetes: mechanisms of injury and protective factors. Cell metabolism. 2013;17(1):20-33
4. Holman RR. Type 2 diabetes mellitus in 2012: Optimal management of T2DM remains elusive. Nat Rev Endocrinol. 2013;9(2):67-8
5. Bhupathiraju SN, Hu FB. Epidemiology of Obesity and Diabetes and Their Cardiovascular Complications. Circ Res. 2016;118(11):1723-35
6. Scheen AJ, Van Gaal LF. Combating the dual burden: therapeutic targeting of common pathways in obesity and type 2 diabetes. The Lancet Diabetes & Endocrinology. 2014;2(11):911-22
7. Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature. 1985;315(6016):239-42
8. Guo H, German P, Bai S, Barnes S, Guo W, Qi X. et al. The PI3K/AKT Pathway and Renal Cell Carcinoma. Journal of Genetics and Genomics. 2015;42(7):343-53
9. Abeyrathna P, Su Y. The critical role of Akt in cardiovascular function. Vascular pharmacology. 2015;74:38-48
10. David A. Fruman REM, and Lewis C. Cantley. PHOSPHOINOSITIDE KINASES. Annu Rev Biochem. 1998:481-507
11. Graupera M, Potente M. Regulation of angiogenesis by PI3K signaling networks. Experimental cell research. 2013;319(9):1348-55
12. Burke JE, Williams RL. Synergy in activating class I PI3Ks. Trends in biochemical sciences. 2015;40(2):88-100
13. Castillo SD, Vanhaesebroeck B, Sebire NJ. Phosphoinositide 3-kinase: a new kid on the block in vascular anomalies. The Journal of pathology. 2016;240(4):387-96
14. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nature reviews Molecular cell biology. 2010;11(5):329-41
15. Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275(5300):665-8
16. Hawkins PT, Stephens LR. Emerging evidence of signalling roles for PI(3,4)P-2 in Class I and II PI3K-regulated pathways. Biochemical Society Transactions. 2016;44:307-14
17. Zhang J, Yu XH, Yan YG, Wang C, Wang WJ. PI3K/Akt signaling in osteosarcoma. Clinica chimica acta; international journal of clinical chemistry. 2015;444:182-92
18. Krycer JR, Sharpe LJ, Luu W, Brown AJ. The Akt-SREBP nexus: cell signaling meets lipid metabolism. Trends Endocrinol Metab. 2010;21(5):268-76
19. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PRJ, Reese CB. et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase B alpha. Current Biology. 1997;7(4):261-9
20. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307(5712):1098-101
21. Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J. et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer discovery. 2015;5(11):1194-209
22. Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell. 2017;169(3):381-405
23. Andjelkovic M, Jakubowicz T, Cron P, Ming XF, Han JW, Hemmings BA. Activation and phosphorylation of a pleckstrin homology domain containing protein kinase (RAC-PK/PKB) promoted by serum and protein phosphatase inhibitors. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(12):5699-704
24. Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18(1):13-24
25. Jethwa N, Chung GHC, Lete MG, Alonso A, Byrne RD, Calleja V. et al. Endomembrane PtdIns(3,4,5)P-3 activates the PI3K-Akt pathway. Journal Of Cell Science. 2015;128(18):3456-65
26. Braccini L, Ciraolo E, Campa CC, Perino A, Longo DL, Tibolla G. et al. PI3K-C2gamma is a Rab5 effector selectively controlling endosomal Akt2 activation downstream of insulin signalling. Nat Commun. 2015;6:7400
27. Mahajan K, Mahajan NP. PI3K-independent AKT activation in cancers: a treasure trove for novel therapeutics. J Cell Physiol. 2012;227(9):3178-84
28. Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS letters. 1996;399(3):333-8
29. Govers R. Cellular regulation of glucose uptake by glucose transporter GLUT4. Advances in clinical chemistry. 2014;66:173-240
30. Manning BD, Cantley LC. AKT/PKB Signaling: Navigating Downstream. Cell. 2007;129(7):1261-74
31. Kousteni S. FoxO1, the transcriptional chief of staff of energy metabolism. Bone. 2012;50(2):437-43
32. Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447(7147):1012-6
33. Webb AE, Brunet A. FOXO transcription factors: key regulators of cellular quality control. Trends in biochemical sciences. 2014;39(4):159-69
34. Hay N. Interplay between FOXO, TOR, and Akt. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2011;1813(11):1965-70
35. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785-9
36. Kraegen EW, James DE, Jenkins AB, Chisholm DJ. Dose-response curves for in vivo insulin sensitivity in individual tissues in rats. Am J Physiol. 1985;248(3 Pt 1):E353-62
37. DE NYRGLJJ. Rapid activation of Akt2 is sufficient to stimulate GLUT4translocation in 3T3-L1 adipocytes. Cell metabolism. 2008;7(4):348-23
38. Mueckler BTM. Glucose transporters in the 21st Century. American Journal of Physiology: Endocrinology and Metabolism. 2010;298(2):E141-E5
39. Cesar Osorio-Fuentealba, Ariel E. Contreras-Ferrat, Altamirano. F. Electrical Stimuli Release ATP to Increase GLUT4 Translocation and Glucose Uptake via PI3Kg-Akt-AS160 in Skeletal Muscle Cells. Diabetes. 2013;62(5):1519-26
40. Ueki K, Yamamoto-Honda R, Kaburagi Y, Yamauchi T, Tobe K, Burgering BM. et al. Potential role of protein kinase B in insulin-induced glucose transport, glycogen synthesis, and protein synthesis. The Journal of biological chemistry. 1998;273(9):5315-22
41. Wan M, Leavens KF, Hunter RW, Koren S, von Wilamowitz-Moellendorff A, Lu M. et al. A noncanonical, GSK3-independent pathway controls postprandial hepatic glycogen deposition. Cell metabolism. 2013;18(1):99-105
42. Vary TCJ, Leonard S.;Kimball, Scot R. Insulin fails to stimulate muscle protein synthesis in sepsis despite unimpaired signaling to 4E-BP1 and S6K1. American Journal of Physiology: Endocrinology and Metabolism. 2001;44(5):1045-53
43. Liu Y, Vertommen D, Rider MH, Lai YC. Mammalian target of rapamycin-independent S6K1 and 4E-BP1 phosphorylation during contraction in rat skeletal muscle. Cellular signalling. 2013;25(9):1877-86
44. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274-93
45. Chen W, Balland E, Cowley MA. Hypothalamic Insulin Resistance in Obesity: Effects on Glucose Homeostasis. Neuroendocrinology. 2017;104(4):364-81
46. Kraegen EW, Cooney GJ. Free fatty acids and skeletal muscle insulin resistance. Current opinion in lipidology. 2008;19(3):235-41
47. Pagel-Langenickel I, Bao J, Pang L, Sack MN. The role of mitochondria in the pathophysiology of skeletal muscle insulin resistance. Endocr Rev. 2010;31(1):25-51
48. Erion DM, Shulman GI. Diacylglycerol-mediated insulin resistance. Nature medicine. 2010;16(4):400-2
49. Schrauwen P. High-fat diet, muscular lipotoxicity and insulin resistance. Proc Nutr Soc. 2007;66(1):33-41
50. Jazet IM, Schaart G, Gastaldelli A, Ferrannini E, Hesselink MK, Schrauwen P. et al. Loss of 50% of excess weight using a very low energy diet improves insulin-stimulated glucose disposal and skeletal muscle insulin signalling in obese insulin-treated type 2 diabetic patients. Diabetologia. 2008;51(2):309-19
51. Schenk S, Horowitz JF. Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J Clin Invest. 2007;117(6):1690-8
52. Holland WL, Knotts TA, Chavez JA, Wang LP, Hoehn KL, Summers SA. Lipid mediators of insulin resistance. Nutr Rev. 2007;65(6 Pt 2):S39-46
53. Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307(5708):384-7
54. Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, Liu Y. et al. Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell metabolism. 2007;5(3):167-79
55. Hesselink MKC, Schrauwen-Hinderling V, Schrauwen P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nature Reviews Endocrinology. 2016;12(11):633-45
56. Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156(1-2):20-44
57. Kubota T, Kubota N, Kadowaki T. Imbalanced Insulin Actions in Obesity and Type 2 Diabetes: Key Mouse Models of Insulin Signaling Pathway. Cell metabolism. 2017;25(4):797-810
58. Zhang J, Liu F. Tissue-specific insulin signaling in the regulation of metabolism and aging. IUBMB Life. 2014;66(7):485-95
59. Denechaud PD, Bossard P, Lobaccaro JM, Millatt L, Staels B, Girard J. et al. ChREBP, but not LXRs, is required for the induction of glucose-regulated genes in mouse liver. J Clin Invest. 2008;118(3):956-64
60. Haas Joel T, Miao J, Chanda D, Wang Y, Zhao E, Haas Mary E. et al. Hepatic Insulin Signaling Is Required for Obesity-Dependent Expression of SREBP-1c mRNA but Not for Feeding-Dependent Expression. Cell metabolism. 2012;15(6):873-84
61. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL. et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39(2):171-83
62. Chakrabarti P, Kandror KV. FoxO1 controls insulin-dependent adipose triglyceride lipase (ATGL) expression and lipolysis in adipocytes. The Journal of biological chemistry. 2009;284(20):13296-300
63. Porstmann T, Griffiths B, Chung YL, Delpuech O, Griffiths JR, Downward J. et al. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene. 2005;24(43):6465-81
64. Chen W, Chen G, Head DL, Mangelsdorf DJ, Russell DW. Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell metabolism. 2007;5(1):73-9
65. Peterson TR, Sengupta SS, Harris TE, Carmack AE, Kang SA, Balderas E. et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell. 2011;146(3):408-20
66. Li S, Brown MS, Goldstein JL. Bifurcation of insulin signaling pathway in rat liver: mTORC1 required for stimulation of lipogenesis, but not inhibition of gluconeogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(8):3441-6
67. Owen JL, Zhang Y, Bae SH, Farooqi MS, Liang G, Hammer RE. et al. Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(40):16184-9
68. Bae EJ, Xu J, Oh DY, Bandyopadhyay G, Lagakos WS, Keshwani M. et al. Liver-specific p70 S6 kinase depletion protects against hepatic steatosis and systemic insulin resistance. The Journal of biological chemistry. 2012;287(22):18769-80
69. Yecies Jessica L, Zhang Hui H, Menon S, Liu S, Yecies D, Lipovsky Alex I. et al. Akt Stimulates Hepatic SREBP1c and Lipogenesis through Parallel mTORC1-Dependent and Independent Pathways. Cell metabolism. 2011;14(2):280
70. Bengoechea-Alonso MT, Ericsson J. A phosphorylation cascade controls the degradation of active SREBP1. The Journal of biological chemistry. 2009;284(9):5885-95
71. Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J. et al. Transcriptional control of adipose lipid handling by IRF4. Cell metabolism. 2011;13(3):249-59
72. Choi SM, Tucker DF, Gross DN, Easton RM, DiPilato LM, Dean AS. et al. Insulin regulates adipocyte lipolysis via an Akt-independent signaling pathway. Molecular and cellular biology. 2010;30(21):5009-20
73. Ahmad F, Lindh R, Tang Y, Ruishalme I, Ost A, Sahachartsiri B. et al. Differential regulation of adipocyte PDE3B in distinct membrane compartments by insulin and the beta3-adrenergic receptor agonist CL316243: effects of caveolin-1 knockdown on formation/maintenance of macromolecular signalling complexes. The Biochemical journal. 2009;424(3):399-410
74. Vazirani RP VA, Sadacca LA, Buckman MS, Picatoste B, Beg M. Disruption of Adipose Rab10-Dependent Insulin Signaling Causes Hepatic Insulin Resistance. diabetes. 2016;65:1577-89
75. Lionetti L, Mollica MP, Lombardi A, Cavaliere G, Gifuni G, Barletta A. From chronic overnutrition to insulin resistance: the role of fat-storing capacity and inflammation. Nutr Metab Cardiovasc Dis. 2009;19(2):146-52
76. Ranjit S, Boutet E, Gandhi P, Prot M, Tamori Y, Chawla A. et al. Regulation of fat specific protein 27 by isoproterenol and TNF-alpha to control lipolysis in murine adipocytes. J Lipid Res. 2011;52(2):221-36
77. Amrani A, Jafarian-Tehrani M, Mormede P, Durant S, Pleau JM, Haour F. et al. Interleukin-1 effect on glycemia in the non-obese diabetic mouse at the pre-diabetic stage. The Journal of endocrinology. 1996;148(1):139-48
78. Rutkowski JM, Stern JH, Scherer PE. The cell biology of fat expansion. Journal Of Cell Biology. 2015;208(5):501-12
79. Gustafson B, Hedjazifar S, Gogg S, Hammarstedt A, Smith U. Insulin resistance and impaired adipogenesis. Trends In Endocrinology And Metabolism. 2015;26(4):193-200
80. Arner E, Westermark PO, Spalding KL, Britton T, Ryden M, Frisen J. et al. Adipocyte Turnover: Relevance to Human Adipose Tissue Morphology. Diabetes. 2010;59(1):105-9
81. Eileen L. Whiteman, Han Cho, Birnbaum MJ. Role of Akt protein kinase B in metabolism. TRENDS in Endocrinology & Metabolism. 2002;13(10):444-51
82. Titchenell PM, Quinn WJ, Lu M, Chu Q, Lu W, Li C. et al. Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell metabolism. 2016;23(6):1154-66
83. Kubota N, Kubota T, Itoh S, Kumagai H, Kozono H, Takamoto I. et al. Dynamic functional relay between insulin receptor substrate 1 and 2 in hepatic insulin signaling during fasting and feeding. Cell metabolism. 2008;8(1):49-64
84. Schmoll D, Walker KS, Alessi DR, Grempler R, Burchell A, Guo S. et al. Regulation of glucose-6-phosphatase gene expression by protein kinase Balpha and the forkhead transcription factor FKHR. Evidence for insulin response unit-dependent and -independent effects of insulin on promoter activity. The Journal of biological chemistry. 2000;275(46):36324-33
85. Lu MJ, Wan M, Leavens KF, Chu QW, Monks BR, Fernandez S. et al. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nature medicine. 2012;18(3):388-U254
86. Dong XC, Copps KD, Guo S, Li Y, Kollipara R, DePinho RA. et al. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell metabolism. 2008;8(1):65-76
87. Titchenell PM, Chu Q, Monks BR, Birnbaum MJ. Hepatic insulin signalling is dispensable for suppression of glucose output by insulin in vivo. Nature Communications. 2015;6:7078
88. Titchenell PM, Lazar MA, Birnbaum MJ. Unraveling the Regulation of Hepatic Metabolism by Insulin. Trends Endocrinol Metab. 2017;28(7):497-505
89. Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J. et al. Hypothalamic K(ATP) channels control hepatic glucose production. Nature. 2005;434(7036):1026-31
90. Myers MG Jr, Olson DP. Central nervous system control of metabolism. Nature. 2012;491(7424):357-63
91. Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X. et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell metabolism. 2007;5(6):438-49
92. Ramnanan CJ, Edgerton DS, Cherrington AD. Evidence against a physiologic role for acute changes in CNS insulin action in the rapid regulation of hepatic glucose production. Cell metabolism. 2012;15(5):656-64
93. Ramnanan CJ, Saraswathi V, Smith MS, Donahue EP, Farmer B, Farmer TD. et al. Brain insulin action augments hepatic glycogen synthesis without suppressing glucose production or gluconeogenesis in dogs. J Clin Invest. 2011;121(9):3713-23
94. Scherer T, O'Hare J, Diggs-Andrews K, Schweiger M, Cheng B, Lindtner C. et al. Brain Insulin Controls Adipose Tissue Lipolysis and Lipogenesis. Cell metabolism. 2011;13(2):183-94
95. Paranjape SA, Chan O, Zhu W, Horblitt AM, McNay EC, Cresswell JA. et al. Influence of insulin in the ventromedial hypothalamus on pancreatic glucagon secretion in vivo. Diabetes. 2010;59(6):1521-7
96. Lefebvre PJ, Paquot N, Scheen AJ. Inhibiting or antagonizing glucagon: making progress in diabetes care. Diabetes Obes Metab. 2015;17(8):720-5
97. Li S, Ogawa W, Emi A, Hayashi K, Senga Y, Nomura K. et al. Role of S6K1 in regulation of SREBP1c expression in the liver. Biochem Biophys Res Commun. 2011;412(2):197-202
98. Yecies Jessica L, Zhang Hui H, Menon S, Liu S, Yecies D, Lipovsky Alex I. et al. Akt Stimulates Hepatic SREBP1c and Lipogenesis through Parallel mTORC1-Dependent and Independent Pathways. Cell metabolism. 2011;14(1):21-32
99. Wan M, Leavens KF, Saleh D, Easton RM, Guertin DA, Peterson TR. et al. Postprandial hepatic lipid metabolism requires signaling through Akt2 independent of the transcription factors FoxA2, FoxO1, and SREBP1c. Cell metabolism. 2011;14(4):516-27
100. Banks AS, Kim-Muller JY, Mastracci TL, Kofler NM, Qiang L, Haeusler RA. et al. Dissociation of the glucose and lipid regulatory functions of FoxO1 by targeted knockin of acetylation-defective alleles in mice. Cell metabolism. 2011;14(5):587-97
101. Haeusler RA, Hartil K, Vaitheesvaran B, Arrieta-Cruz I, Knight CM, Cook JR. et al. Integrated control of hepatic lipogenesis versus glucose production requires FoxO transcription factors. Nat Commun. 2014;5:5190
102. Deng X, Zhang W, I OS, Williams JB, Dong Q, Park EA. et al. FoxO1 inhibits sterol regulatory element-binding protein-1c (SREBP-1c) gene expression via transcription factors Sp1 and SREBP-1c. The Journal of biological chemistry. 2012;287(24):20132-43
103. Bugianesi E, Gastaldelli A, Vanni E, Gambino R, Cassader M, Baldi S. et al. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: sites and mechanisms. Diabetologia. 2005;48(4):634-42
104. Utzschneider KM, Kahn SE. Review: The role of insulin resistance in nonalcoholic fatty liver disease. Journal Of Clinical Endocrinology & Metabolism. 2006;91(12):4753-61
105. Qu X, Seale JP, Donnelly R. Tissue and isoform-selective activation of protein kinase C in insulin-resistant obese Zucker rats - effects of feeding. Journal Of Endocrinology. 1999;162(2):207-14
106. Hotamisligil GS. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell. 2010;140(6):900-17
107. Yang L, Calay ES, Fan J, Arduini A, Kunz RC, Gygi SP. et al. S-Nitrosylation links obesity-associated inflammation to endoplasmic reticulum dysfunction. Science. 2015;349(6247):500-6
108. Brown MS, Goldstein JL. Selective versus total insulin resistance: A pathogenic paradox. Cell metabolism. 2008;7(2):95-6
109. Kubota N, Kubota T, Kajiwara E, Iwamura T, Kumagai H, Watanabe T. et al. Differential hepatic distribution of insulin receptor substrates causes selective insulin resistance in diabetes and obesity. Nat Commun. 2016;7:12977
110. Kwon O, Kim KW, Kim MS. Leptin signalling pathways in hypothalamic neurons. Cell Mol Life Sci. 2016;73(7):1457-77
111. Varela L, Horvath TL. Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep. 2012;13(12):1079-86
112. Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S. et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391(6670):900-4
113. Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R. et al. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest. 2008;118(5):1796-805
114. Morrison CD, Morton GJ, Niswender KD, Gelling RW, Schwartz MW. Leptin inhibits hypothalamic Npy and Agrp gene expression via a mechanism that requires phosphatidylinositol 3-OH-kinase signaling. Am J Physiol Endocrinol Metab. 2005;289(6):E1051-7
115. Kim KW, Donato J Jr, Berglund ED, Choi YH, Kohno D, Elias CF. et al. FOXO1 in the ventromedial hypothalamus regulates energy balance. J Clin Invest. 2012;122(7):2578-89
116. Kitamura T, Feng Y, Kitamura YI, Chua SC Jr, Xu AW, Barsh GS. et al. Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nature medicine. 2006;12(5):534-40
117. Ren H, Orozco IJ, Su Y, Suyama S, Gutierrez-Juarez R, Horvath TL. et al. FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell. 2012;149(6):1314-26
118. Hongxia Ren JRC, Ning Kon, Domenico Accili. Gpr17 in AgRP Neurons Regulates Feeding and Sensitivity to Insulin and Leptin. Diabetes. 2015;64:3670-9
119. Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E. et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382(6588):250-2
120. Al-Qassab H, Smith MA, Irvine EE, Guillermet-Guibert J, Claret M, Choudhury AI. et al. Dominant role of the p110beta isoform of PI3K over p110alpha in energy homeostasis regulation by POMC and AgRP neurons. Cell metabolism. 2009;10(5):343-54
121. Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E. et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25(6):903-15
122. Mori H, Inoki K, Munzberg H, Opland D, Faouzi M, Villanueva EC. et al. Critical role for hypothalamic mTOR activity in energy balance. Cell metabolism. 2009;9(4):362-74
123. Dalamaga M, Chou SH, Shields K, Papageorgiou P, Polyzos SA, Mantzoros CS. Leptin at the intersection of neuroendocrinology and metabolism: current evidence and therapeutic perspectives. Cell metabolism. 2013;18(1):29-42
124. Albert V, Cornu M, Hall MN. mTORC1 signaling in Agrp neurons mediates circadian expression of Agrp and NPY but is dispensable for regulation of feeding behavior. Biochem Biophys Res Commun. 2015;464(2):480-6
125. Miller AM, Brestoff JR, Phelps CB, Berk EZ, Reynolds THt. Rapamycin does not improve insulin sensitivity despite elevated mammalian target of rapamycin complex 1 activity in muscles of ob/ob mice. Am J Physiol Regul Integr Comp Physiol. 2008;295(5):R1431-8
126. Blouet C, Schwartz GJ. Hypothalamic nutrient sensing in the control of energy homeostasis. Behav Brain Res. 2010;209(1):1-12
127. Haissaguerre M, Saucisse N, Cota D. Influence of mTOR in energy and metabolic homeostasis. Mol Cell Endocrinol. 2014;397(1-2):67-77
128. Smith Mark A, Katsouri L, Irvine Elaine E, Hankir Mohammed K, Pedroni Silvia MA, Voshol Peter J. et al. Ribosomal S6K1 in POMC and AgRP Neurons Regulates Glucose Homeostasis but Not Feeding Behavior in Mice. Cell Reports. 2015;11(3):335-43
129. Hill JW, Elias CF, Fukuda M, Williams KW, Berglund ED, Holland WL. et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell metabolism. 2010;11(4):286-97
130. Steculorum SM, Ruud J, Karakasilioti I, Backes H, Engstrom Ruud L, Timper K. et al. AgRP Neurons Control Systemic Insulin Sensitivity via Myostatin Expression in Brown Adipose Tissue. Cell. 2016;165(1):125-38
131. Shi X, Zhou F, Li X, Chang B, Li D, Wang Y. et al. Central GLP-2 enhances hepatic insulin sensitivity via activating PI3K signaling in POMC neurons. Cell metabolism. 2013;18(1):86-98
132. Carvalheira JB, Ribeiro EB, Araujo EP, Guimaraes RB, Telles MM, Torsoni M. et al. Selective impairment of insulin signalling in the hypothalamus of obese Zucker rats. Diabetologia. 2003;46(12):1629-40
133. Kullmann S, Heni M, Hallschmid M, Fritsche A, Preissl H, Haring HU. Brain Insulin Resistance at the Crossroads of Metabolic and Cognitive Disorders in Humans. Physiol Rev. 2016;96(4):1169-209
134. Yue JT, Lam TK. Lipid sensing and insulin resistance in the brain. Cell metabolism. 2012;15(5):646-55
135. Benoit SC, Kemp CJ, Elias CF, Abplanalp W, Herman JP, Migrenne S. et al. Palmitic acid mediates hypothalamic insulin resistance by altering PKC-theta subcellular localization in rodents. J Clin Invest. 2009;119(9):2577-89
136. Iskandar K, Cao Y, Hayashi Y, Nakata M, Takano E, Yada T. et al. PDK-1/FoxO1 pathway in POMC neurons regulates Pomc expression and food intake. Am J Physiol Endocrinol Metab. 2010;298(4):E787-98
137. Ono H, Pocai A, Wang Y, Sakoda H, Asano T, Backer JM. et al. Activation of hypothalamic S6 kinase mediates diet-induced hepatic insulin resistance in rats. J Clin Invest. 2008;118(8):2959-68
138. Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M. et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431(7005):200-5
139. Kubota N, Tobe K, Terauchi Y, Eto K, Yamauchi T, Suzuki R. et al. Disruption of insulin receptor substrate 2 causes type 2 diabetes because of liver insulin resistance and lack of compensatory beta-cell hyperplasia. Diabetes. 2000;49(11):1880-9
140. Cantley J, Choudhury AI, Asare-Anane H, Selman C, Lingard S, Heffron H. et al. Pancreatic deletion of insulin receptor substrate 2 reduces beta and alpha cell mass and impairs glucose homeostasis in mice. Diabetologia. 2007;50(6):1248-56
141. RN; K, JC; B, JN; W, C; P, MA; M, CR K. Tissue-Specific Knockout of the Insulin Receptor in Pancreatic b Cells Creates an Insulin Secretory Defect Similar to that in Type 2 Diabetes. Cell. 1999;96(3):329-39
142. Yuval Dor JB, Olga I. Martinez & Douglas A. Melton. Adult pancreatic b-cells are formed by self-duplication rather than stem-cell differentiation. nature. 2004;429(6):41-6
143. Georgia S, Bhushan A. β cell replication is the primary mechanism for maintaining postnatal β cell mass. Journal of Clinical Investigation. 2004;114(7):963-8
144. Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest. 2001;108(11):1631-8
145. Bernal-Mizrachi E, Fatrai S, Johnson JD, Ohsugi M, Otani K, Han Z. et al. Defective insulin secretion and increased susceptibility to experimental diabetes are induced by reduced Akt activity in pancreatic islet beta cells. J Clin Invest. 2004;114(7):928-36
146. Wang W, Liu Y, Chen Y, Cao C, Xiang Y, Zhang D. et al. Inhibition of Foxo1 mediates protective effects of ghrelin against lipotoxicity in MIN6 pancreatic beta-cells. Peptides. 2010;31(2):307-14
147. Wang HW, Mizuta M, Saitoh Y, Noma K, Ueno H, Nakazato M. Glucagon-like peptide-1 and candesartan additively improve glucolipotoxicity in pancreatic beta-cells. Metabolism. 2011;60(8):1081-9
148. Kahn SE, Prigeon RL, McCulloch DK, Boyko EJ, Bergman RN, Schwartz MW. et al. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes. 1993;42(11):1663-72
149. Oliveira JM, Rebuffat SA, Gasa R, Gomis R. Targeting type 2 diabetes: lessons from a knockout model of insulin receptor substrate 2. Canadian journal of physiology and pharmacology. 2014;92(8):613-20
150. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444(7121):840-6
151. Dornan GL, Burke JE. Molecular Mechanisms of Human Disease Mediated by Oncogenic and Primary Immunodeficiency Mutations in Class IA Phosphoinositide 3-Kinases. Frontiers in immunology. 2018;9:575
152. Ortega-Molina A, Lopez-Guadamillas E, Mattison JA, Mitchell SJ, Munoz-Martin M, Iglesias G. et al. Pharmacological inhibition of PI3K reduces adiposity and metabolic syndrome in obese mice and rhesus monkeys. Cell metabolism. 2015;21(4):558-70
153. Wang C, Chi Y, Li J, Miao Y, Li S, Su W. et al. FAM3A activates PI3K p110alpha/Akt signaling to ameliorate hepatic gluconeogenesis and lipogenesis. Hepatology. 2014;59(5):1779-90
154. Kuang JR, Zhang ZH, Leng WL, Lei XT, Liang ZW. Dapper1 attenuates hepatic gluconeogenesis and lipogenesis by activating PI3K/Akt signaling. Mol Cell Endocrinol. 2017;447:106-15
155. Wang C, Chen Z, Li S, Zhang Y, Jia S, Li J. et al. Hepatic overexpression of ATP synthase beta subunit activates PI3K/Akt pathway to ameliorate hyperglycemia of diabetic mice. Diabetes. 2014;63(3):947-59
156. Chattopadhyay T, Singh RR, Gupta S, Surolia A. Bone morphogenetic protein-7 (BMP-7) augments insulin sensitivity in mice with type II diabetes mellitus by potentiating PI3K/AKT pathway. BioFactors (Oxford, England). 2017;43(2):195-209
157. Mwangi S, Anitha M, Mallikarjun C, Ding X, Hara M, Parsadanian A. et al. Glial cell line-derived neurotrophic factor increases beta-cell mass and improves glucose tolerance. Gastroenterology. 2008;134(3):727-37
158. Anitha M, Gondha C, Sutliff R, Parsadanian A, Mwangi S, Sitaraman SV. et al. GDNF rescues hyperglycemia-induced diabetic enteric neuropathy through activation of the PI3K/Akt pathway. J Clin Invest. 2006;116(2):344-56
159. Wang X, Hai CX. ROS acts as a double-edged sword in the pathogenesis of type 2 diabetes mellitus: is Nrf2 a potential target for the treatment? Mini reviews in medicinal chemistry. 2011;11(12):1082-92
160. Liu TY, Shi CX, Gao R, Sun HJ, Xiong XQ, Ding L. et al. Irisin inhibits hepatic gluconeogenesis and increases glycogen synthesis via the PI3K/Akt pathway in type 2 diabetic mice and hepatocytes. Clin Sci (Lond). 2015;129(10):839-50
161. Villani V, Milanesi A, Sedrakyan S, Da Sacco S, Angelow S, Conconi MT. et al. Amniotic fluid stem cells prevent beta-cell injury. Cytotherapy. 2014;16(1):41-55
162. Zhuo S, Yang M, Zhao Y, Chen X, Zhang F, Li N. et al. MicroRNA-451 Negatively Regulates Hepatic Glucose Production and Glucose Homeostasis by Targeting Glycerol Kinase-Mediated Gluconeogenesis. Diabetes. 2016;65(11):3276-88
163. Kurano M, Hara M, Satoh H, Tsukamoto K. Hepatic NPC1L1 overexpression ameliorates glucose metabolism in diabetic mice via suppression of gluconeogenesis. Metabolism. 2015;64(5):588-96
164. Lee JH, Budanov AV, Talukdar S, Park EJ, Park HL, Park HW. et al. Maintenance of metabolic homeostasis by Sestrin2 and Sestrin3. Cell metabolism. 2012;16(3):311-21
165. Peng M, Yin N, Li MO. Sestrins function as guanine nucleotide dissociation inhibitors for Rag GTPases to control mTORC1 signaling. Cell. 2014;159(1):122-33
166. Mottaghi S, Larijani B, Sharifi AM. Apelin 13: a novel approach to enhance efficacy of hypoxic preconditioned mesenchymal stem cells for cell therapy of diabetes. Medical hypotheses. 2012;79(6):717-8
167. MacAulay K, Woodgett JR. Targeting glycogen synthase kinase-3 (GSK-3) in the treatment of Type 2 diabetes. Expert opinion on therapeutic targets. 2008;12(10):1265-74
168. Yang W, Wang J, Chen Z, Chen J, Meng Y, Chen L. et al. NFE2 Induces miR-423-5p to Promote Gluconeogenesis and Hyperglycemia by Repressing the Hepatic FAM3A-ATP-Akt Pathway. Diabetes. 2017;66(7):1819-32
169. Mathur A, Pandey VK, Kakkar P. PHLPP: a putative cellular target during insulin resistance and type 2 diabetes. J Endocrinol. 2017;233(3):R185-R98
170. Miyamoto S, Purcell NH, Smith JM, Gao T, Whittaker R, Huang K. et al. PHLPP-1 negatively regulates Akt activity and survival in the heart. Circ Res. 2010;107(4):476-84
171. Malek M, Kielkowska A, Chessa T, Anderson KE, Barneda D, Pir P. et al. PTEN Regulates PI(3,4)P2 Signaling Downstream of Class I PI3K. Mol Cell. 2017;68(3):566-80.e10
Author contact
![]() Corresponding author: E-Mails: suzhqedu.cn (Z.-Q.S.) and gyguoyzcom (J.G.). Tel.: +86-20-3935-2067 and Fax: +86-20-3935-2067.
Corresponding author: E-Mails: suzhqedu.cn (Z.-Q.S.) and gyguoyzcom (J.G.). Tel.: +86-20-3935-2067 and Fax: +86-20-3935-2067.