ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2018; 14(13):1859-1872. doi:10.7150/ijbs.27710 This issue Cite
Research Paper
Effect of RIF1 on response of non-small-cell lung cancer patients to platinum-based chemotherapy by regulating MYC signaling pathway
Ying Mei1,2, Yong-Bin Liu1,2, Dong-Li Hu1,2, Hong-Hao Zhou1,2 ![]()
1. Department of Clinical Pharmacology, Xiangya Hospital, Central South University, Changsha 410008, P. R. China;
2. Institute of Clinical Pharmacology, Central South University, Hunan Key Laboratory of Pharmacogenetics, Changsha 410078, P. R. China.
Abstract

Platinum-based chemotherapy is used as first-line therapy for advanced non-small-cell lung cancer (NSCLC). However, there is no effective indicator to predict whether the patient would be chemo-resistant or sensitive to the therapy. In addition, it is urgent to elucidate the mechanisms of cisplatin resistance. RIF1 plays important roles in DNA replication regulation and DNA repair pathway. However, the role of RIF1 in NSCLC progression and chemotherapy response is still unknown. In this study, we found that RIF1 expression was correlated with the response of NSCLC patients to platinum-based chemotherapy (n=89, P=0.002). Among patients who have been treated with platinum chemo-therapy, those with high levels of RIF1 expression had significantly shorter survival than those with low RIF1 expression (P<0.05). RIF1 knockdown increased sensitivity to cisplatin in NSCLC patients both in vitro and in vivo. Moreover, RIF1 knockdown induced G0/G1 phase arrest and increased cisplatin-induced apoptosis and DNA damage. Further investigation showed that RIF1 regulated the expression of MYC and MYC downstream targets, including the cell cycle and double-stranded break (DSB) repair genes which might mediate the effect of RIF1 on cellular response to cisplatin. Overexpression of MYC could reverse the inhibition of MYC targets by RIF1 knockdown. Taken together, these data revealed that RIF1 played an important role in regulating MYC and MYC-activated genes, which in turn contributes to cellular response to cisplatin and NSCLC patients' response to platinum-based chemotherapy. RIF1 might serve as a novel biomarker for predicting platinum-based chemo-sensitivity and the prognosis of NSCLC patients, so as to guide the chemotherapy regimen adjustment for individual patient with NSCLC and improve their clinical outcomes.
Keywords: Lung cancer, RIF1, cisplatin resistance, MYC, DNA damage repair
Introduction
Despite the advances in diagnosis and therapeutic technologies, lung cancer remains the leading cause of cancer mortality both among males and females throughout the world [1]. Non-small-cell lung cancer (NSCLC) represents approximately 85% of all lung cancer cases [2]. A majority of patients are often diagnosed at advanced or metastatic stage which makes the disease difficult to cure [3]. Although cisplatin-based chemotherapy is still used as first-line therapy for advanced lung cancer [4, 5], there is no effective indicator which can predict whether the NSCLC patient would be chemo-resistant or sensitive to the therapy. The truth is that a myriad of patients continue to progress after chemotherapy, thus, finding an indicator to predict the efficacy of chemotherapy in NSCLC is of great significance [6]. Elucidating the underlying mechanism of cisplatin resistance will be beneficial for enhancing the efficiency of chemotherapy as well as for developing novel therapeutic strategies to decrease relapses and to improve long-term clinical outcomes of patients.
The cytotoxicity of cisplatin mainly comes from the formation of cisplatin-containing DNA adducts, which induce DNA double-stranded breaks (DSBs), particularly when DNA is actively replicating [7]. DSBs are repaired by two major pathways: homologous recombination (HR) and non-homologous end joining (NHEJ). HR predominates the DSBs repair process during the S and G2 phases, as it uses sister chromatid to repair the DSBs in a template-guided way. On the contrary, NHEJ predominates the DSBs repair process throughout the whole cell cycle. NHEJ does not require strand homology and is error prone. NHEJ requires the action of KU70, KU80, PARP1, DNA-PKcs, DNA ligase IV, and XRCC4 [8]. If the tumor cells fail to repair their DNA lesions, they may either undergo p53-dependent apoptosis or p53-independent cell death if p53 is mutated [9]. Aberrant activation of DNA repair pathways and subsequent reduction of apoptosis are considered as two major mechanisms of platinum resistance [10, 11].
MYC is a basic helix-loop-helix leucine zipper transcription activator that usually binds to the E-box motifs near the core promoter of target genes to enhance their expression. MYC regulates a considerable number of genes that control the cell proliferation, cell cycle, differentiation, and apoptosis. MYC also controls the expression of DSB repair genes including PARP1 and LIG4 (DNA ligase IV), hence, MYC target genes may be alternative therapeutic target for anticancer therapy to sensitize cancer cells to chemotherapy [12, 13].
RIF1 (replication timing regulatory factor 1) was first identified in budding yeast with a pivotal role in the regulation of telomere length in yeasts [14]. During the past decades, RIF1 has been reported to play significant roles in the replication timing regulation [15-17]. However, the structure and function of RIF1 are different between different species. Most notably, in human cells, RIF1 was reported to have been recruited to DNA double-strand breaks (DSBs), expediting non-homologous end joining (NHEJ) repair [18, 19]. In human cancer cells, RIF1 was reported to be over-expressed in breast cancer tissues and we previously found that RIF1 knockdown increased the cisplatin sensitivity of cervical cancer and ovarian cancer cells [20-22]. However, the role and underlying mechanisms of RIF1 in chemo-sensitivity in NSCLC remain largely unknown.
In this study, we showed that RIF1 regulated the expression of MYC and MYC target cell cycle and DSB repair genes which might mediate the effect of RIF1 on cellular response to cisplatin in NSCLC.
Material and Methods
Patients and samples
A total of 89 patients with advanced NSCLC were enrolled at Xiangya Hospital of Central South University (Changsha, Hunan, China) from 2012 to 2015. Among these patients, 50.6% were squamous cell carcinoma (SCC) and 49.4% were adenocarcinoma. Our study was approved by the Ethics Committee of Xiangya School of Medicine, Central South University (Registration number: CTXY-110008-2). Written informed consent was obtained from all NSCLC patients. The eligible patients for this chemo-sensitivity study had to meet the following inclusion criteria: (1) Primary tumor in the lung with histologically or cytologically confirmed NSCLC; (2) Patients received more than 2 cycles of platinum-based chemotherapy, and had no radiotherapy or surgery before. (3) Chemotherapy response was evaluated by using Response Evaluation Criteria in Solid Tumors (RECIST) [23]. Patients with complete response (CR) or partial response (PR) were identified as responders. Patients with stable disease (SD) or progressive disease (PD) were considered as nonresponders. Exclusion criteria include (1) Lactation or pregnancy, (2) Leptomeningeal or symptomatic brain metastases, (3) Active infection and (4) Previous or concomitant other malignancies. All paraffin-embedded tissue specimens were collected via biopsy of bronchofiberscope and for immunohistochemistry (IHC) analysis in the Pathology Department of Xiangya Hospital.
Immunohistochemistry
Immunohistochemistry (IHC) using paraffin-embedded sections from NSCLC patients and the scoring of the RIF1 staining intensity were performed in the Pathology Department of Xiangya Hospital as previously described [24]. All NSCLC tissue sections were reviewed independently by two pathologists and the staining score of RIF1 was also independently calculated by two pathologists at Xiangya Hospital. The staining intensity was scored according to the following standards: negative = 0, weak = 1, moderate = 2, and strong = 3. RIF1-positive cell proportion was scored as follows: ≤10% = 0, >10% to 25% = 1, >25% to 50% = 2, >50% to 75% = 3 and >75% = 4. A final score was calculated based on the product of the two scores. If the final score was ≤ 5, the tumor was regarded as having low RIF1 expression; and if the score was > 5, then it suggested high RIF1 expression. The cutoff value was set to 5.0 according to receiver operating characteristic curves [25].
Cell culture and cell lines
Two human NSCLC cell lines, A549 and H1299, were bought from the cell banks of the Shanghai Institutes of Biological Sciences. All cell lines were tested and authenticated before application. The cells were cultured in RPMI-1640 medium (Corning) containing 10% fetal bovine serum (FBS).
Plasmids and generation of stable cell lines
The shRNA sequence targeting human RIF1 cDNA was purchased by GenePharma: RIF1 KD: 5'-GCCTTTGAGTTCCATCCAT-3'. The sequence of the scrambled control shRNA is 5'-TTCTCCGAACGTGTCACGT-3'. A549 and H1299 cells were transfected with scrambled control shRNA (SCR) or RIF1 knockdown shRNA (RIF1 KD) using ViaFect Transfection Reagent (Promega) according to the manufacturer's protocol. Lentiviral vectors pGLV3/ H1/GFP were bought from GenePharma. Viral infection was done following the instructions provided by GenePharma. RIF1 stable knockdown NSCLC cell lines were selected with 2 μg/ml puromycin over 2 weeks. Western blot analysis was performed to verify the stable knockdown of RIF1 in NSCLC cell lines. Human MYC expression vectors with C terminal Flag and His tag and the control vectors were purchased from ViGene Biosciences.
RNA isolation and Real-time quantitative PCR
Total RNA was isolated from NSCLC cells or collected tissue samples by TRIzol reagent (Invitrogen) PrimeScript RT reagent Kit (TaKaRa) was used for reverse transcription, and the quantitative RT-PCR was performed by using SYBR Premix DimerEraser kit (TaKaRa) on the Roche LightCycler480 (Roche). -2ΔΔct method was used to analyze the data and the mRNA expression of β-actin was used as normalization control. Sequences of primers are listed in Table 1.
Sequences of primers for quantitative real-time PCR
| Gene name | Forward primer sequence (5′ → 3′) | Reverse primer sequence (5′ → 3′) |
|---|---|---|
| β-actin | CATGTACGTTGCTATCCAGGC | CTCCTTAATGTCACGCACGAT |
| RIF1 | TGGCAGATGACATTGATAGA | TAGATTGTGTAGTAGGAGAAGTT |
| MYC | TTCGGGTAGTGGAAAACCAG | CAGCAGCTCGAATTTCTTCC |
| CDK2 | CCAGGAGTTACTTCTATGCCTGA | TTCATCCAGGGGAGGTACAAC |
| CDK4 | ATGGCTACCTCTCGATATGAGC | CATTGGGGACTCTCACACTCT |
| CCND1 | CAATGACCCCGCACGATTTC | CATGGAGGGCGGATTGGAA |
| PARP1 | CGGAGTCTTCGGATAAGCTCT | TTTCCATCAAACATGGGCGAC |
| LIG4 | AGCAAAAGTGGCTTATACGGATG | TGAGTCCTACAGAAGGATCATGC |
Western blotting
Western blotting was performed as described previously [26]. Briefly, proteins were extracted by using RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 μg/ml leupeptin ,1 mM Na2EDTA) added with phosphatase inhibitors and protease inhibitors (Biotool). The lysate was then centrifuged at 13000 rpm for 15 min at 4°C. The supernatants were collected and protein concentrations were measured using the BCA method. Antibodies against RIF1 were obtained from Santa Cruz Biotechnology and Bethyl Laboratories. Antibodies to BAX, BCL2, Casepase-3, PARP1, LIG4, Cyclin-D1, and c-Myc were purchased from Cell Signaling Technology. Antibodies against CDK2 and CDK4 were obtained from Santa Cruz. Antibody to β-actin was purchased from Sigma-Aldrich and used as a loading control.
Cell viability analysis
5 × 103 RIF1 knockdown and scrambled control cells were seeded in 96-well plates (Corning) and incubated with cisplatin (Sigma) for 48 hours. Cell viability was detected by MTS approach according to the protocol for Cell Titer 96 Aqueous-One-Solution Cell Proliferation Assay kit (MTS) (Promega). 10 µl of MTS diluted in 90 µl of medium was added into each well, then the plate was incubated for 30 minutes at 37°C. Absorbance at 490nm of each well was examined using a spectrophotometer (Bio-Rad Laboratories, Inc.). The IC50 was calculated from the dose-response curves using GraphPad Prism 5.0 program (GraphPad Software, Inc.)
Colony formation assay
The transfected H1299 and A549 cells were seeded in 6-well plates at 500 cells per well with or without cisplatin treatment and cultured for 8 days. The colonies were fixed with 4% paraformaldehyde for 30 minutes and then stained with 1% crystal violet (Beyotime) for 15 minutes. Then, the plates were washed mildly with phosphate-buffered saline (PBS). After the plates were air-dried, the stained colonies were photographed by a high-resolution camera.
Flow cytometric analysis
For the cell apoptosis analysis, H1299 and A549 cells were seeded in 6 well plates. Then the cells were transiently transfected with the scrambled control shRNA or shRNA-RIF1 and 24 hours later, the cells were treated with 0 or 10 μM cisplatin for 48 hours followed by resuspension in complete medium. Next, the NSCLC cells were washed twice with cold PBS and suspended at a concentration of 1 x 106 cells/ml in 1X Binding Buffer. Then, the apoptosis rates were detected using FITC Annexin V Apoptosis Detection Kit I (BD Biosciences) following the protocol.
For cell cycle analysis, transfected A549 and H1299 cells were plated in six-well plates. After 48 hours, the cells were harvested by trypsinization, and fixed with cold 70% ethanol at -20 °C overnight. Cell cycle was measured using the Cell Cycle and Apoptosis Analysis Kit (Beyotime) according to the protocol. 2×104 NSCLC cells were analyzed per sample by FC500 flow cytometry instrument (Beckman Coulter). The proportions of G0/G1 cells were calculated and compared with the ModFit LT software.
Immunostaining
Immunofluorescence experiments were performed according to the recommended instructions of the Fast ImmunoFluorescence Staining Kit (Protein Biotechnologies). Briefly, cells were transiently transfected with scrambled control shRNA (SCR) or RIF1 knockdown shRNA (RIF1 KD) followed by treated with cisplatin (10 μM) and immunostained with anti-RIF1 (red) and anti-γ-H2AX (green) antibodies. Nucleus was stained with 4'6-diamidino-2-phenylindole (DAPI). The primary antibodies were listed as follows: anti-RIF1 (Bethyl Laboratories and Santa Cruz) and anti-γ-H2AX (Bethyl Laboratories). The secondary antibodies used were Alexa Fluor 488 and 594 conjugated second antibodies (Jackson ImmunoResearch). DAPI Fluoromount-G® (SouthernBiotech) was applied to detect the nucleus. The pictures were taken using DeltaVision Elite Imaging System (GE Healthcare Life Sciences). The relative fluorescence intensity was analyzed with Image J.
In vivo end-Joining assay
The plasmid end-joining assay was performed as described previously [27, 28]. In brief. Plasmid pGL2 was completely linearized using HindIII, as confirmed by agarose gel electrophoresis. RIF1 knockdown cells were transfected with the linearized DNA and pRL-TK (Promega) using FuGENE HD Transfection Reagent (Promega). Forty-eight hours after transfection, the transfectants were assayed for luciferase activity following Dual-Luciferase® Reporter Assay Protocol. The relative end-joining efficiency was calculated by comparing luciferase activity expressed in NSCLC cells transfected with HindIII digested DNA with that of the uncut plasmid.
Xenograft tumor model
Female BALB/c nude mice (4-5 w, 18-20 g) were bought from Shanghai SIPPR-B&K Laboratory Animal Corp. A total of 2×106 RIF1 stable knockdown and scrambled control H1299 cells were inoculated subcutaneously into the left and right flanks of the BALB/c nude mice. When the tumors were measurable, cisplatin was administered at a dose of 5 mg/kg every 4 days. The minimum (W) and maximum (L) length of the tumor were examined using a caliper every 4 days. The tumor volumes were calculated as LW2/2. The mice were euthanized 24 days after inoculation and the tumors were embedded in paraffin for IHC and H&E staining. The experimental procedures were approved by the Institutional Review Board of Central South University.
Statistical analysis
All data were presented as mean ±standard deviation (SD). Two-tailed Student's t test was applied for continuous variables between two groups. To compare the differences among more than two groups, one-way ANOVAs was used. χ2 test or Fisher's exact test was used for qualitative variables. Kaplan-Meier method and the log-rank test were used for survival analysis. The correlation between the expressions of indicated genes was analyzed using Pearson's correlation coefficient. Statistical analysis was conducted using SPSS 18.0. P < 0.05 was considered statistically significant.
Results
Correlation of RIF1 expression with chemotherapy response
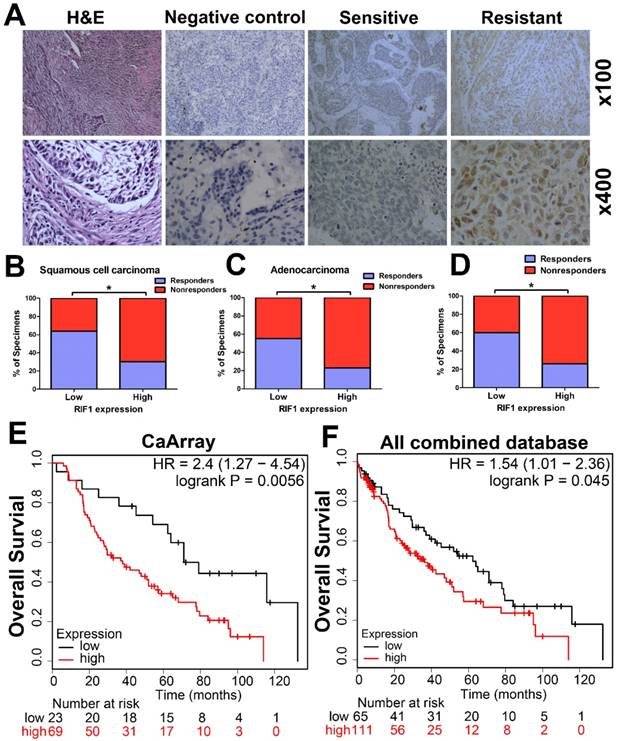
The RIF1 expression level in cancer tissues of 89 patients with advanced NSCLC was evaluated by IHC. Representative images of IHC staining of RIF1 in specimens of primary NSCLC were shown in Fig. 1A. RIF1 expression had significant correlation with chemotherapy responses of NSCLC patients (n=89, P=0.002), squamous cell carcinoma (SCC) patients (n=45, P=0.023) and adenocarcinoma patients (n=44, P=0.03), respectively (Table 2). Generally, chemotherapy responsive patients had lower RIF1 expression in their lung cancer tissues. When taken separately to analysis the disease type, among the chemotherapy responders, about 64.0% of the SCC patients and 54.5% of the adenocarcinoma patients had low RIF1 level. As for the nonresponders, these numbers went down to 36.0% and 45.5%, respectively. While taken together, similar significant correlation was also found in NSCLC patients, of the NSCLC patients having low RIF1 expression, the proportion of responders and nonresponders were 59.6% and 40.4%, respectively (Fig. 1B-D). Moreover, Kaplan-Meier analysis and log-rank test were performed to evaluate the data from Kaplan-Meier plotter database (http://kmplot.com/analysis/). The results showed that overexpression of RIF1 was correlated with poorer overall survival of lung cancer patients who received chemotherapy both in CaArray database (RIF1 low vs high: hazard ratio [HR] of survival = 2.4, 95% confidence interval [CI] = 1.27 - 4.54, P =0.0056) (Fig. 1E) and in 10 combined databases (RIF1 low vs high: hazard ratio [HR] of survival = 1.54, 95% confidence interval [CI] = 1.01-2.36, P =0.045) (Fig. 1F). Therefore, there is a significant correlation between the level of RIF1 expression and the chemotherapy responses of NSCLC patients, and the increased RIF1 expression may induce chemo-resistance.
Chemotherapy response and RIF1 expression in NSCLC
| Chemotherapy response | RIF1 level | P | ||
|---|---|---|---|---|
| Low (%) | High (%) | Ratio (High/Low) | ||
| Squamous cell carcinoma | ||||
| Responders | 16 (64.0) | 6 (30.0) | 0.38 | 0.023 |
| Nonresponders | 9 (36.0) | 14 (70.0) | 1.56 | |
| Adenocarcinoma | ||||
| Responders | 12 (54.5) | 5 (22.7) | 0.42 | 0.03 |
| Nonresponders | 10 (45.5) | 17 (77.3) | 1.70 | |
| Total | ||||
| Responders | 28 (59.6) | 11 (26.2) | 0.39 | 0.002 |
| Nonresponders | 19 (40.4) | 31 (73.8) | 1.63 | |
Clinical significance of RIF1 in human lung cancer patients. (A) Representative images of IHC staining of RIF1 in specimens of primary NSCLC. H&E: representative hematoxylin and eosin staining in NSCLC tissues. Sensitive: representative specimens of RIF1 staining of the patient sensitive to platinum. Resistant: representative specimens of RIF1 staining of the patient resistant to platinum. (B-D) Percentage of NSCLC specimens showing low or high RIF1 expression relative to responders or nonresponders in squamous cell carcinoma (n=45) (B), adenocarcinoma (n=44) (C) and all NSCLC patients (n=89) (D). (E and F) Patients with chemotherapy treatment were stratified by expression of RIF1 and analyzed for overall survival by Kaplan-Meier analysis in CaArray database (E) and 10 combined database (F). CaArray database and 10 combined database were obtained from Kaplan-Meier plotter database (http://kmplot.com/analysis/). (CARRAY: n = 462, GSE14814: n = 90, GSE19188: n = 156, GSE29013: n = 55, GSE31210: n = 246, GSE3141: n = 110, GSE37745: n = 196, GSE4573: n = 130, GSE8894: n = 138, and TCGA: n = 133). HR = hazard ratio. * P<0.05.
Effect of RIF1 knockdown on NSCLC cellular response to cisplatin
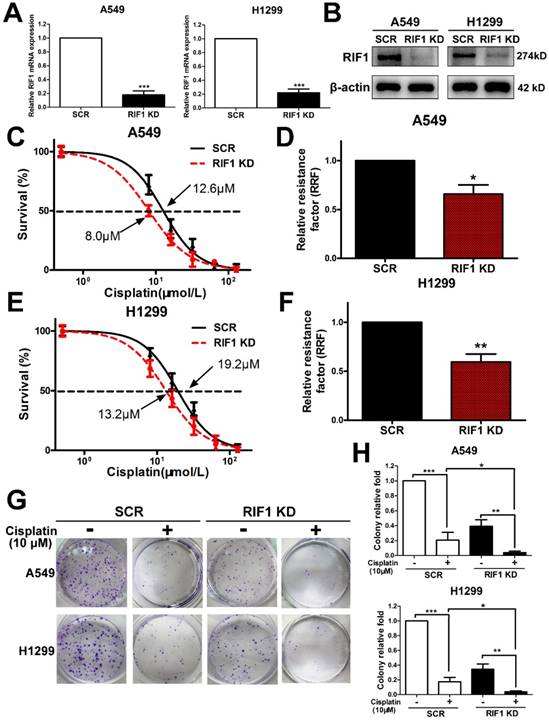
Since all enrolled NSCLC patients received platinum-based chemotherapy, we then used the NSCLC cell lines to explore whether RIF1 expression affected cisplatin sensitivity or not. We knocked down RIF1 expression in A549 and H1299 cells by shRNA, which reduced RIF1 expression effectively compared with the scrambled control. The silencing efficiency was determined using real time RT-PCR and Western blot analyses (Fig. 2A, B). Then A549 and H1299 cells were subjected to MTS assay following treatments with different concentrations of cisplatin. RIF1-knockdown A549 and H1299 cells became more sensitive to cisplatin with about 40%-50% decrease in relative resistance factor (Fig. 2C-F). These results were confirmed by colony formation assays using RIF1-silenced A549 and H1299 cells (Fig. 2G, H).
Effect of RIF1 knockdown on cellular response to cisplatin in A549 and H1299 cells. (A and B) A549 and H1299 cells were transiently transfected with scrambled control shRNA (SCR) or RIF1 knockdown shRNA (RIF1 KD) followed by determination of RIF1 expression by RT-qPCR and Western blot and. β-actin was used as a loading control for Western blot. (C-F) A549 and H1299 cells transiently transfected with scrambled control shRNA (SCR) or RIF1 knockdown shRNA (RIF1 KD) were subjected to treatment with different concentrations of cisplatin. MTS assays were performed to investigate Concentration-dependent growth inhibition of cisplatin in A549 and H1299 cells. Graphpad 5.0 software was used to calculate half maximal inhibitory concentration (IC50). Relative resistance factor (RRF) of cisplatin was calculated by dividing the IC50 of the control cells and by that of the RIF1-knockdown NSCLC cells. (G) Colony formation efficiency of A549 and H1299 cells with or without RIF1 knockdown treated with cisplatin (10 μM). (H) Quantification of relative colony formation efficiency of A549 and H1299 cells. Data were presented as means ± SD of three independent experiments. * P<0.05, ** P<0.01, *** P<0.001.
Knockdown of RIF1 induced G0/G1 phase arrest and enhanced cisplatin induced apoptosis
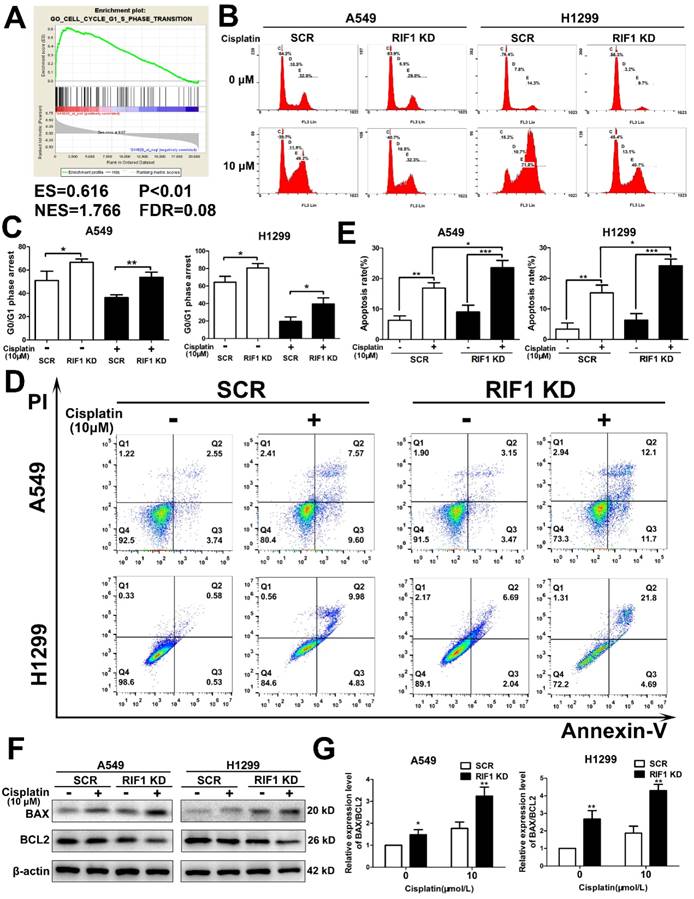
Then we explored whether RIF1 was related to cell cycle progression in NSCLC cells. Gene set enrichment analysis (GSEA) revealed that RIF1 expression was closely correlated with the expression of a set of G1/S phase transition genes in the publicly available NSCLC patient expression profiles (GSE10245; Fig. 3A). To confirm the cell-cycle distribution, flow cytometry was performed in RIF1-knockdown and control cells after cisplatin treatment. The RIF1-knockdown cells displayed a dramatically higher number of cells in G0/G1 phase compared with the control cells (Fig. 3B, C), suggesting that RIF1 regulates G0/G1 phase arrest in response to cisplatin.
It is widely agreed that one of the major mechanisms of cisplatin is that it leads to DNA damage and then induces cell apoptosis [29]. Thus we used flow cytometry to explore whether RIF1 affected cisplatin sensitivity by regulating cisplatin-induced cell apoptosis. The apoptosis rate was increased in RIF1-knockdown A549 and H1299 cells after cisplatin treatment for 48 hours (Fig. 3D, E). We then tested the expression of BAX and BCL2. We observed upregulation of BAX and downregulation of BCL2 (Fig. 3F). We further calculated the ratio of BAX/BCL2 for each treatment (0 and 10 µM), the ratio of BAX/BCL2 for each treatment of RIF1-knockdown A549 and H1299 cells was higher than the scrambled control cells (Fig. 3G), indicating that silencing of RIF1 might enhance cisplatin-induced apoptosis signaling pathway in A549 and H1299 cells. These results indicated that RIF1 contributed to cellular response to cisplatin partly via regulating cell apoptosis.
Effect of RIF1 knockdown on cell cycle progression and cisplatin-induced apoptosis. (A) GSEA plot showing that RIF1 expression was positively correlated with cell cycle G1/S transition genes in the GEO data set (NCBI/GEO/GSE 10245; n = 58). (B and C) Knockdown of RIF1 increased the G0/G1 phase fraction in A549 and H1299 cells. Before detection, the cells were incubated with cisplatin (10 μM) for 24 hours. (D and E) Annexin V/PI staining showed that RIF1 knockdown in A549 and H1299 cells enhanced cellar apoptosis rate compared with scrambled control with cisplatin treatment (10 μM) for 48 hours. (F) RIF1-knockdown or scrambled control A549 and H1299 cells were treated with cisplatin (0 and 10 μM) for 24 h, cellular protein was harvested, and Western blot analysis was performed to investigate the levels of leaved BAX and BCL2. The β-actin was detected as a loading control. (G) The ratio of BAX/BCL2 for each treatment (0 and 10 µM) in RIF1-knockdown or scrambled control A549 and H1299 cells. Date were shown as mean ± SD of three independent experiments. * P<0.05, ** P<0.01, *** P<0.001.
Effect of RIF1 knockdown on DNA Repair
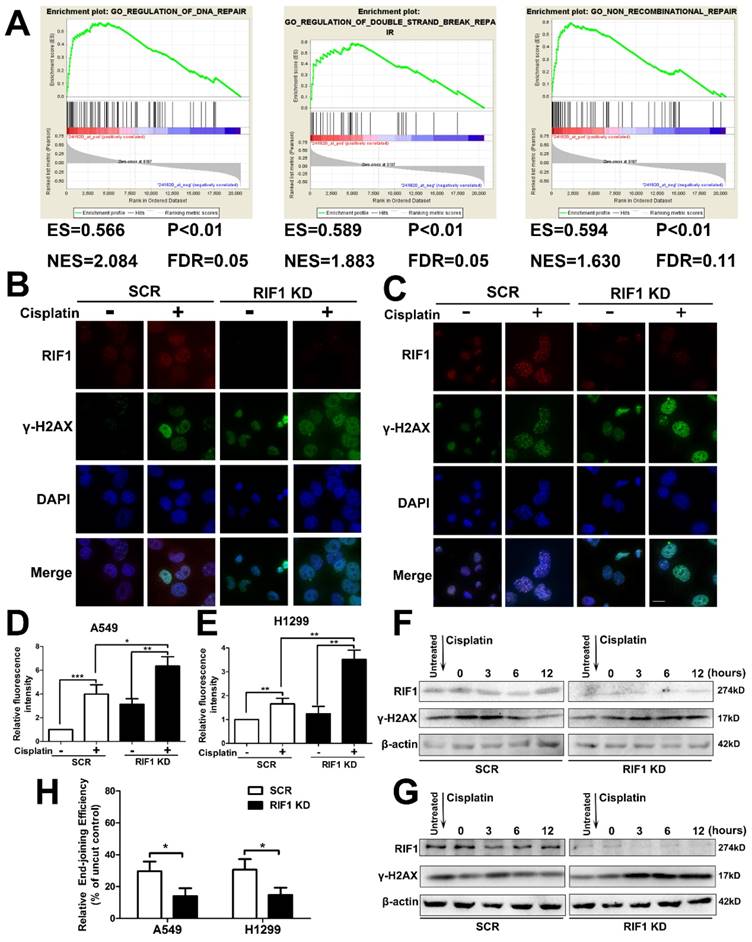
It is established that increased DNA repair capacity is one of the mechanisms of cisplatin resistance and RIF1 has been demonstrated to promote NHEJ repair [19]. Gene set enrichment analysis (GSEA) revealed that RIF1 expression closely correlated with the expression of DSB repair genes and non-recombination repair genes in the publicly available NSCLC patient expression profiles (GSE10245; Fig. 4A). Thus we studied whether knockdown of RIF1 affected NSCLC cellular DNA repair ability upon cisplatin treatment. We did an immunofluorescence assay for γ-H2AX foci (a marker of DSB severity) and RIF1 repair foci after cisplatin treatment, and the results demonstrated that RIF1 foci and γ-H2AX foci colocalized in the nucleus after cisplatin treatment. In addition, knockdown of RIF1 increased γ-H2AX foci compared with scrambled control in A549 and H1299 cells (Fig. 4B-E). Then the time course of γ-H2AX foci formation after cisplatin treatment was also analyzed by immunoblotting analysis. RIF1-knockdown and scrambled control A549 and H1299 cells were treated with cisplatin for 1 hour. Then the cells were washed to make them free of cisplatin. The expression of γ-H2AX increased at 3 hours after cisplatin treatment in both RIF1-knockdown NSCLC and control cells. The difference was that γ-H2AX level significantly decreased along time in control NSCLC cells. However, in RIF1-knockdown cells, γ-H2AX levels were sustained at significantly higher levels till 12 hours after cisplatin removal (Fig. 4F, G). Persistence of high γ-H2AX levels in RIF1-knockdown cells suggested that RIF1 was pivotal for efficient DNA repair upon cisplatin treatment.
Since RIF1 has been demonstrated to promote NHEJ repair, we then measured whether the role of RIF1 in affecting NHEJ capacity was significant in A549 and H1299 cells using a previously reported in vivo end-joining assay [27, 28]. The end-joining capacity tested using pGL2 digested with HindIII reflected overall end-joining, because HindIII cleaved at the site between the promoter and the coding sequences, and any end-joining activity, even that led to small insertions or deletions, would not influence luciferase expression. We examined end-joining capacities in RIF1-knockdown A549 and H1299 cells and found that RIF1-knockdown A549 and H1299 cells had significant lower overall end-joining capacity compared with the corresponding control cells. (Fig. 4H).These results demonstrated that RIF1 promoted overall NHEJ capacity in NSCLC cells.
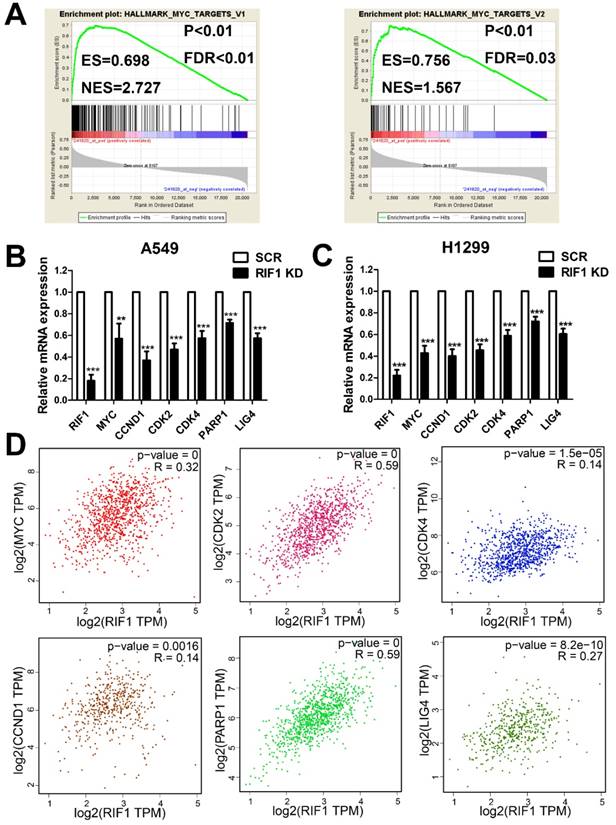
RIF1 correlates with the MYC signaling pathway
To investigate the molecular mechanism underlying the oncogenic role of RIF1 in NSCLC and cisplatin sensitivity, we performed GESA analysis and found that RIF1 expression was positively correlated with MYC target genes expression (Fig. 5A), suggesting RIF1 might be a regulator of MYC, and could activate MYC pathway. Then we performed quantitative real-time PCR to analyze the expression of MYC and MYC targeting genes including CDK2, CDK4 and CCND1 which are involved in G1/S transition and PARP1 and LIG4 which participate in DSB repair. RIF1 knockdown was found to significantly inhibit the expression of these genes in A549 and H1299 cells (Fig. 5B, C). We further confirmed the clinical relationship of RIF1 and MYC pathway in NSCLC tissues using GEPIA database and found that the expression level of RIF1 positively correlated with that of MYC (r = 0.32, P < 0.001), CDK2 (r = 0.59, P < 0.001), CDK4 (r = 0.14, P < 0.001), CCND1 (Cyclin D1) (r = 0.14, P = 0.0016), PARP1 (r = 0.59, P < 0.001) and LIG4 (r = 0.27, P < 0.001) in NSCLC tissues (Fig. 5D). These findings suggested RIF1 was associated with MYC and MYC targeting genes which were involved in cell cycle and DSB repair pathways in NSCLC.
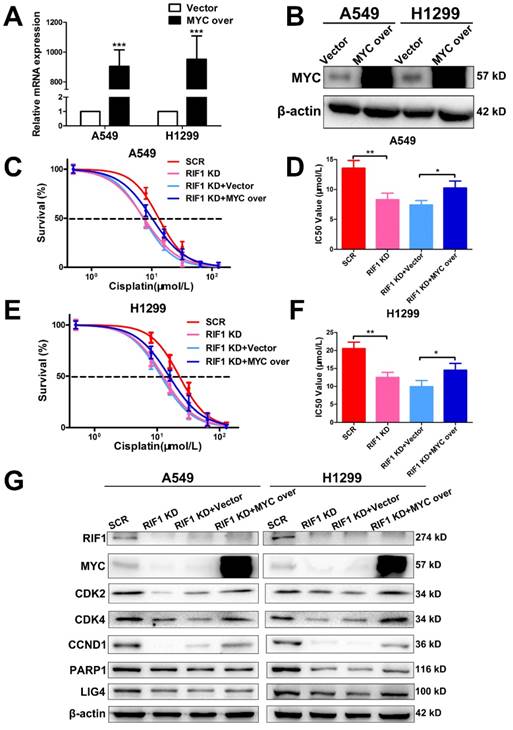
Knockdown of RIF1 increases cisplatin sensitivity by down-regulating MYC
To further investigate whether the effect of RIF1 knockdown on cisplatin sensitivity is mediated by down-regulating MYC pathway or not. We explored whether MYC overexpression could rescue the effect of RIF1 knockdown. The efficiency of MYC overexpression in A549 and H1299 cells was detected by RT-qPCR and western blot (Fig. 6A, B). Through MTS assay, we found that RIF1 knockdown could remarkably increase sensitivity of cells to cisplatin and decrease the IC50 value whereas MYC overexpression could abolish the effect of RIF1 knockdown and increase the IC50 value in RIF1-knockdown A549 and H1299 cells (Fig. 6C-F). Mechanically, western blot assay was performed to determine that RIF1 knockdown can downregulate the expression of MYC and MYC target genes while MYC overexpression could reverse the inhibition of MYC target genes by RIF1 knockdown in A549 and H1299 cells (Fig. 6G). On the one hand, RIF1 knockdown could decrease the expression of CDK2, CDK4 and CCND1 to inhibit G1/S transition. On the other hand, RIF1 knockdown could regulate the expression of PARP1 and LIG4 to inhibit DSB repair. These results illustrated that knockdown of RIF1 increased cisplatin sensitivity by down-regulating MYC pathway.
Knockdown of RIF1 led to defects in DNA repair upon cisplatin treatment. (A) GSEA plot showing that RIF1 expression was positively correlated with a set of genes relating to DNA repair, DSB repair and non-recombinational repair in the GEO data set (NCBI/GEO/GSE 10245; n = 58). (B and C) A549 (B) and H1299 (C) cells were transiently transfected with scrambled control shRNA (SCR) or RIF1 knockdown shRNA (RIF1 KD) followed by treated with cisplatin (10 μM) and immunostained with anti-RIF1 (red) and anti-γ-H2AX (green) antibodies. Nucleus was stained with 4'6-diamidino-2-phenylindole (DAPI). Scale bar 10μM. (D and E) Quantification of relative fluorescence intensity of γ-H2AX in A549 (D) and H1299 cells (E). (F and G) A549 (F) and H1299 (G) were transiently transfected with scrambled control shRNA (SCR) or RIF1 knockdown shRNA (RIF1 KD) followed by incubated with cisplatin (10 μM) for 1 h, then washed free of cisplatin (0 time point) and thenceforth harvested at different time points. The NSCLC cell lysate was immunoblotted for γ-H2AX (a marker for damaged DNA). (H) Knockdown of RIF1 decreased the overall end-joining activity. PGL2 plasmid containing the reporter gene linearize by HindIII was transfected into A549 and H1299 cells and used to detect the overall end-joining activity as mentioned in “Materials and Methods.” Data were presented as means ± SD of three independent experiments. * P<0.05, ** P<0.01, *** P<0.001.
RIF1 correlated with the MYC pathway. (A) GSEA plot showing that RIF1 expression was positively correlated with MYC-activated target genes expression in the GEO data set (NCBI/GEO/GSE 10245; n = 58). (B and C) RT-qPCR analysis of the expression of MYC and MYC targeting genes including CDK2, CDK4, CCND1, PARP1 and LIG4 in RIF1-knockdown and scrambled control A549 and H1299 cells.(D) Bioinformatics analysis based on GEPIA database showed the mRNA expression levels of RIF1 positively correlated with MYC (r = 0.32, P < 0.001), CDK2 (r = 0.59, P < 0.001), CDK4 (r = 0.14, P < 0.001), CCND1 (Cyclin D1) (r = 0.14, P = 0.0016), PARP1 (r = 0.59, P < 0.001) and LIG4 (r = 0.27, P < 0.001) in NSCLC tissues. Data were presented as means ± SD of three independent experiments. ** P<0.01, *** P<0.001.
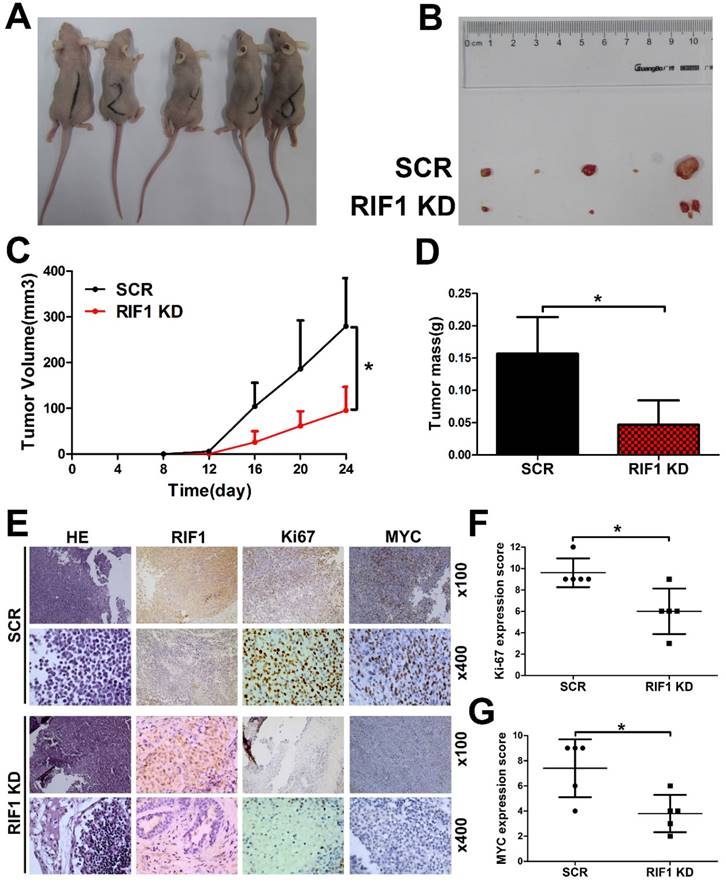
Knockdown of RIF1 increases cisplatin sensitivity in vivo
To further investigate whether knockdown of RIF1 affects cisplatin sensitivity in vivo, RIF1 stable knockdown or control H1299 cells were injected subcutaneously into BALB/c nude mice. Mice were treated intraperitoneally with 5 mg/kg cisplatin every 4 days when the tumors were measurable. RIF1 stable-knockdown cells showed slower growth, forming significantly smaller tumors than control cells with cisplatin treatment. In other words, the tumors derived from the RIF1 stable knockdown cells were significantly more sensitive to cisplatin compared with the control tumors (Fig. 7A-D). The tumors were resected and analyzed by H&E and IHC staining. Tumor tissues derived from RIF1 knockdown cells showed decreased positivity for Ki-67 and MYC compared with the control groups (Fig. 7E-G), indicating that RIF1 knockdown inhibited MYC expression and tumor growth in the mouse model.
Knockdown of RIF1 increased cisplatin sensitivity by down-regulating MYC. (A and B) A549 and H1299 cells transiently transfected with MYC overexpression (MYC over) or vector control (Vector) were subjected to RT-qPCR (A) and western blot (B), β-actin was used as a loading control for Western blot. (C-F) RIF1-kockdown A549 and H1299 cells were subjected to treatment with cisplatin at various concentrations with or without MYC overexpression. Cell viability was determined by absorbance at the 490 nm wavelength and normalized to the absorbance value of the control group by MTS assay. Half maximal inhibitory concentration (IC50) was calculated by Graphpad 5.0 software from 3 independent experiments. (G) Overexpression of MYC reversed the inhibition of MYC targeting genes by RIF1 knockdown in A549 and H1299 cells. Date were shown as mean ± SD of three independent experiments. * P<0.05, ** P<0.01, *** P<0.001.
Discussion
RIF1 has recently been demonstrated to play a pivotal role in DNA repair pathway, especially in NHEJ repair [24]. On the one hand, compared with error-free HR repair pathway, the NHEJ repair is error-prone, thus leading to mutation accumulation and cancer occurrence. On the other hand, it is reasonable to presume that high expression of RIF1 in cancer cells may result in increased DSB repair and confer chemo-resistance, leading to poor prognosis of patients. Actually until now, little is known about the role of RIF1 in chemotherapy response in NSCLC.
Analysis of clinical samples in our study demonstrated that RIF1 was highly expressed in nonresponders compared with the responders of NSCLC patients to platinum-based chemotherapy. Moreover, RIF1 overexpression was associated with worse overall survival of lung cancer patients who received chemotherapy. These results indicate the potentiality of RIF1 as a marker for predicting NSCLC patients' response to cisplatin. We further studied the role of RIF1 in cell cycle progression and cisplatin-induced apoptosis. We found that RIF1 knockdown induced G0/G1 phase arrest and enhanced cisplatin-induced apoptosis. Furthermore, knockdown of RIF1 inhibited DSB repair capacity and cell survival upon cisplatin treatment. These results were consistent with our previous findings in cervical cancer cells [21].
Then we explored how RIF1 regulated these seemingly unrelated pathways in NSCLC cells. GSEA analysis suggested there was a positive correlation between the expression of RIF1 and MYC-activating genes. RIF1 knockdown downregulated the expression of MYC and MYC downstream targets. Clinical association of RIF1 and MYC signaling pathway in human NSCLC tissues was verified using TCGA database. MYC is a famous oncogene, and overexpresses in various cancers. MYC regulates a considerable number of genes controlling tumorigenesis, cell cycle and DSB repair [30]. Hence MYC target genes may be an alternative therapeutic target for anticancer therapy to sensitize cancer cells to chemotherapy [31]. Thus we speculate that MYC signaling might contribute to the effect of RIF1 on cellular response to cisplatin.
Knockdown of RIF1 increased NSCLC cellular sensitivity to cisplatin in vivo. (A and B) 2×106 scrambled control (SCR) and RIF1 stable knockdown (RIF1 KD) H1299 cells were inoculated subcutaneously into the left and right flanks of nude mice. When the tumors were palpable (~50 mm3), the mice were treated with cisplatin (5 mg/kg/4days). Representative images of xenografts are shown in A and B. (C and D) Tumor volume and mass of xenografts were evaluated. (E) Representative images of H&E and IHC staining of the resected tumor. (F and G) IHC analysis showed that RIF1-knockdown decreased the proliferation index Ki-67, and reduced the expression of MYC. Data were presented as means ± SD. * P<0.05.
The MYC target genes include CDK2, CDK4 and CCND1 controlling G1/S phase transition as well as DSB repair genes PARP1 and LIG4 [12, 13, 32]. PARP1 recognizes DSBs, and its activity is involved in the recruitment of factors to promote DSB repair. PARP1 deficiency or inhibition results in delayed activation of DNA damage response proteins such as phosphorylated histone H2AX [33]. LIG4 (Ligase IV), in complex with XLF and XRCC4, and is indispensable to NHEJ repair and absence of either of these factors results in an impaired ability to repair DSBs [34]. The regulation of RIF1 on these MYC target genes may partly explain how RIF1 regulated G1/S phase arrest and DSB repair.
The rescue experiments further confirmed that the effect of RIF1 knockdown on cisplatin sensitivity is mediated by down-regulating MYC pathway. In a word, the role of RIF1 in cellular response to cisplatin is likely to work out via its regulatory effects on the expression of MYC and MYC targeting genes such as CDK2, CDK4 and CCND1 which in turn, affects G1/S phase transition as well as DSB repair gens such as PARP1 and LIG4, hence, affects DNA repair activities and cellular response to cisplatin. However, except for regulating cell cycle and DSB repair, MYC also plays important roles in other biological processes, including the regulation of cell proliferation, pluripotency and cancer stem cells [35, 36]. Thus the concrete regulation mechanism of RIF1-MYC axis await to more profound research in the future. Understanding the underlying mechanisms may allow stratification of treatment selection based on RIF1 expression levels, as well as developing molecular targeting to improve the efficacy of chemotherapy.
In summary, we showed that RIF1 expression correlated with responses of NSCLC patients to platinum-based chemotherapy. We also showed that RIF1 may regulate the expression of MYC signaling pathway which, in turn, affected cellular DSB repair activity and response to cisplatin. A complete understanding of the precise role of RIF1 in NSCLC may allow for the use of RIF1 as a biomarker for predicting patients' response to platinum-based chemotherapy and further assisting in the development of new therapeutic strategies.
Abbreviations
NSCLC: non-small cell lung cancer; RIF1: replication timing regulatory factor 1; CR: complete response; PR: partial response SD: stable disease; PD: progressive disease; IHC: Immunohistochemistry; DSB: double-stranded break, HR: homologous recombination; NHEJ: non-homologous end joining; PBS: phosphate buffered solution; FITC: fluorescein isothiocyanate; PI: propidium iodide; Gene set enrichment analysis; BAX: BCL-2 associated X protein; BCL2: B cell lymphoma-2.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81503073; L1322020).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7-30
2. Chen Z, Fillmore CM, Hammerman PS, Kim CF, Wong KK. Non-small-cell lung cancers: a heterogeneous set of diseases. Nat Rev Cancer. 2014;14:535-46
3. McCloskey P, Balduyck B, Van Schil PE, Faivre-Finn C, O'Brien M. Radical treatment of non-small cell lung cancer during the last 5 years. Eur J Cancer. 2013;49:1555-64
4. Lennon FE, Cianci GC, Cipriani NA, Hensing TA, Zhang HJ, Chen CT. et al. Lung cancer-a fractal viewpoint. Nat Rev Clin Oncol. 2015;12:664-75
5. Cataldo VD, Gibbons DL, Perez-Soler R, Quintas-Cardama A. Treatment of non-small-cell lung cancer with erlotinib or gefitinib. N Engl J Med. 2011;364:947-55
6. Yin JY, Li X, Zhou HH, Liu ZQ. Pharmacogenomics of platinum-based chemotherapy sensitivity in NSCLC: toward precision medicine. Pharmacogenomics. 2016;17:1365-78
7. Xiong Y, Huang BY, Yin JY. Pharmacogenomics of platinum-based chemotherapy in non-small cell lung cancer: focusing on DNA repair systems. Med Oncol. 2017;34:48
8. Bristow RG, Ozcelik H, Jalali F, Chan N, Vesprini D. Homologous recombination and prostate cancer: a model for novel DNA repair targets and therapies. Radiother Oncol. 2007;83:220-30
9. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573-84
10. Stewart DJ. Mechanisms of resistance to cisplatin and carboplatin. Crit Rev Oncol Hematol. 2007;63:12-31
11. Yu WK, Wang Z, Fong CC, Liu D, Yip TC, Au SK. et al. Chemoresistant lung cancer stem cells display high DNA repair capability to remove cisplatin-DNA damage. Br J Pharmacol. 2016
12. Pyndiah S, Tanida S, Ahmed KM, Cassimere EK, Choe C, Sakamuro D. c-MYC suppresses BIN1 to release poly(ADP-ribose) polymerase 1: a mechanism by which cancer cells acquire cisplatin resistance. Sci Signal. 2011;4:a19
13. Luoto KR, Meng AX, Wasylishen AR, Zhao H, Coackley CL, Penn LZ. et al. Tumor cell kill by c-MYC depletion: role of MYC-regulated genes that control DNA double-strand break repair. Cancer Res. 2010;70:8748-59
14. Hardy CF, Sussel L, Shore D. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 1992;6:801-14
15. Hayano M, Kanoh Y, Matsumoto S, Renard-Guillet C, Shirahige K, Masai H. Rif1 is a global regulator of timing of replication origin firing in fission yeast. Genes Dev. 2012;26:137-50
16. Yamazaki S, Ishii A, Kanoh Y, Oda M, Nishito Y, Masai H. Rif1 regulates the replication timing domains on the human genome. EMBO J. 2012;31:3667-77
17. Mattarocci S, Hafner L, Lezaja A, Shyian M, Shore D. Rif1: A Conserved Regulator of DNA Replication and Repair Hijacked by Telomeres in Yeasts. Front Genet. 2016;7:45
18. Silverman J, Takai H, Buonomo SB, Eisenhaber F, de Lange T. Human Rif1, ortholog of a yeast telomeric protein, is regulated by ATM and 53BP1 and functions in the S-phase checkpoint. Genes Dev. 2004;18:2108-19
19. Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5' end resection. Science. 2013;339:700-04
20. Wang H, Zhao A, Chen L, Zhong X, Liao J, Gao M. et al. Human RIF1 encodes an anti-apoptotic factor required for DNA repair. Carcinogenesis. 2009;30:1314-19
21. Mei Y, Peng C, Liu YB, Wang J, Zhou HH. Silencing RIF1 decreases cell growth, migration and increases cisplatin sensitivity of human cervical cancer cells. Oncotarget. 2017;8:107044-51
22. Liu YB, Mei Y, Tian ZW, Long J, Luo CH, Zhou HH. Downregulation of RIF1 Enhances Sensitivity to Platinum-Based Chemotherapy in Epithelial Ovarian Cancer (EOC) by Regulating Nucleotide Excision Repair (NER) Pathway. Cell Physiol Biochem. 2018;46:1971-84
23. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228-47
24. Yin JY, Shen J, Dong ZZ, Huang Q, Zhong MZ, Feng DY. et al. Effect of eIF3a on response of lung cancer patients to platinum-based chemotherapy by regulating DNA repair. Clin Cancer Res. 2011;17:4600-09
25. Zeng ZL, Luo HY, Yang J, Wu WJ, Chen DL, Huang P. et al. Overexpression of the circadian clock gene Bmal1 increases sensitivity to oxaliplatin in colorectal cancer. Clin Cancer Res. 2014;20:1042-52
26. Samaeekia R, Adorno-Cruz V, Bockhorn J, Chang YF, Huang S, Prat A. et al. miR-206 Inhibits Stemness and Metastasis of Breast Cancer by Targeting MKL1/IL11 Pathway. Clin Cancer Res. 2017;23:1091-103
27. Bau DT, Fu YP, Chen ST, Cheng TC, Yu JC, Wu PE. et al. Breast cancer risk and the DNA double-strand break end-joining capacity of nonhomologous end-joining genes are affected by BRCA1. Cancer Res. 2004;64:5013-19
28. Zhong Q, Chen CF, Chen PL, Lee WH. BRCA1 facilitates microhomology-mediated end joining of DNA double strand breaks. J Biol Chem. 2002;277:28641-47
29. Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307-20
30. Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5
31. Chen Y, Xu J, Borowicz S, Collins C, Huo D, Olopade OI. c-Myc activates BRCA1 gene expression through distal promoter elements in breast cancer cells. BMC Cancer. 2011;11:246
32. Muvarak N, Kelley S, Robert C, Baer MR, Perrotti D, Gambacorti-Passerini C. et al. c-MYC Generates Repair Errors via Increased Transcription of Alternative-NHEJ Factors, LIG3 and PARP1, in Tyrosine Kinase-Activated Leukemias. Mol Cancer Res. 2015;13:699-712
33. Ray CA, Nussenzweig A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat Rev Mol Cell Biol. 2017;18:610-21
34. Francis DB, Kozlov M, Chavez J, Chu J, Malu S, Hanna M. et al. DNA Ligase IV regulates XRCC4 nuclear localization. DNA Repair (Amst). 2014;21:36-42
35. Zhang C, Xu B, Lu S, Zhao Y, Liu P. HN1 contributes to migration, invasion, and tumorigenesis of breast cancer by enhancing MYC activity. Mol Cancer. 2017;16:90
36. Wang WJ, Wu SP, Liu JB, Shi YS, Huang X, Zhang QB. et al. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res. 2013;73:1219-31
Author contact
![]() Corresponding author: Hong-Hao Zhou, Department of Clinical Pharmacology, Xiangya Hospital, Central South University, 110 Xiang Ya Road, Changsha, Hunan 410078, China; Phone: +86-73184805380; Fax: +86-73182354476; E-mail: hhzhou2004com; ybliu2014edu.cn
Corresponding author: Hong-Hao Zhou, Department of Clinical Pharmacology, Xiangya Hospital, Central South University, 110 Xiang Ya Road, Changsha, Hunan 410078, China; Phone: +86-73184805380; Fax: +86-73182354476; E-mail: hhzhou2004com; ybliu2014edu.cn
Received 2018-6-7
Accepted 2018-9-5
Published 2018-10-20