International Journal of Biological Sciences
ISSN: 1449-2288
10
Impact Factor
ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(13):1883-1891. doi:10.7150/ijbs.27854 This issue Cite
Research Paper
PKM2 Inhibitor Shikonin Overcomes the Cisplatin Resistance in Bladder Cancer by Inducing Necroptosis
Yonggang Wang 1, Fangshi Hao1, Yonghao Nan2, Licheng Qu3, Wanli Na1, Chunshu Jia3, ![]() , Xiaoliang Chen1
, Xiaoliang Chen1 ![]()
1. Department of Urology, China-Japan Union Hospital, Jilin University, Changchun, China
2. Department of Urology, The Frist Affiliated Hospital of Zhengzhou University, Zhengzhou
3. Center for Reproductive Medicine and Center for Prenatal Diagnosis, First Hospital, Jilin University, Changchun, China
Received 2018-6-13; Accepted 2018-9-8; Published 2018-10-20
Citation:
Wang Y, Hao F, Nan Y, Qu L, Na W, Jia C, Chen X. PKM2 Inhibitor Shikonin Overcomes the Cisplatin Resistance in Bladder Cancer by Inducing Necroptosis. Int J Biol Sci 2018; 14(13):1883-1891. doi:10.7150/ijbs.27854. https://www.ijbs.com/v14p1883.htm
Other stylesAbstract

Cisplatin-based chemotherapy often results in the development of chemo-resistance when used to treat bladder cancer (BC), which is difficult to overcome. Recent data indicate that pyruvate kinase M2 (PKM2), a glycolytic enzyme for Warburg effect, is strongly upregulated in BC, and contributes to the cisplatin resistance in BC. However, the underlying mechanisms remain unclear. In this study, we also found that the expression level of PKM2 is also higher in cisplatin resistant BC cells and tumors. Down-regulation of PKM2 by siRNA or inhibition of PKM2 by shikonin re-sensitized the cisplatin resistant T24 cells. Shikonin and cisplatin together exhibit significantly greater killing effects than when used alone. Interestingly, we found shikonin kills the T24 cisplatin resistant cells by inducing necroptosis, as the cell death could not inhibited by apoptosis inhibitor, z-VAD, but compromised by RIP3 inhibitor, GSK872, or RIP3 siRNA. In contrast, shikonin induced apoptosis in T24 parental cells. We further investigate the underlying mechanism, and found that the dysregulation of Bcl-2 family proteins, including Bcl-2, PUMA, Bax, play an important role in deciding that shikonin kills the BC cells by necroptosis or apoptosis. Collectively, our results suggested that inducing necroptosis is an alternative way to overcome the apoptosis resistant in BC therapy, and orchestrating the regulation of Bcl-2, PUMA, and Bax in BC cisplatin resistant cells may improve the therapy effect of cisplatin in BC tumor.
Introduction
Bladder cancer (BC) or urothelial carcinoma of the bladder is one of the leading prevalent cancer in men, and the expense for the therapy is higher [1]. While the diagnosis and treatment of low grade BC is generally favorable [2], advanced BC is one of the most aggressive cancers with high morbidity and mortality [3]. Currently, cisplatin is the major chemotherapy drug for advanced BC, which can be used as neoadjuvant therapy combined with radical cystectomy, or as a single agent or key component for metastatic BC [4, 5]. However, a large amount of BC patients encounter preexisting chemo-resistance, which limit the therapy effect of cisplatin [6]. Those patients with drug resistance usually have initial response to cisplatin treatment, but eventually develop resistance in the final stage, resulting in treatment failure and disease progression [7]. Therefore, there is a pressing need to explore additional avenues to more effectively treat advanced BC as a whole, which is very important to predict treatment outcome and develop effective chemotherapeutic agents.
Recently, several studies reported that pyruvate kinase isozyme M2 (PKM2) is highly expressed in human cancers, including bladder cancer [8-10], and contributes to chemo-resistance [10, 11]. Aerobic glycolysis commonly exist in most of cancer cells, which allow them to produce energy followed by lactic acid fermentation even in the presence of oxygen, known as the “Warburg effect” [12]. Thus, glycolysis, the major pathway for energy generation, is vital for the proliferation and survival of cancer cells [13, 14]. Although it is not completely understood why cancer cells shift energy production from Krebs cycle to glycolysis, it is believed that PKM2 has important roles in this shift [15, 16]. PKM2 has been shown to have an important role in cancer cell metabolism and growth, because inhibition of PKM2 by peptide aptamer inhibited cell growth [16], and PKM2 knockdown by siRNA or displacement of PKM2 with PKM1 significantly reduced the ability of human cancer cell lines to form tumor in nude mice [17]. As PKM2 is necessary for cancer cells' aerobic glycolysis, which is a hallmark of cancer metabolism and the major energy source essential for cancer cell growth and survival, PKM2 is a potential molecular target for disrupting glucose metabolism in cancer cells.
Since PKM2 plays an important role in cancer metabolism, it could potentially serve as a drug target for cancer therapy. Recently, shikonin, a major active chemical component extracted from lithospermum erythrorhizon, was identified as a novel inhibitor of tumor PKM2 and prevent cancer cell glycolysis [18]. In the last four decades, shikonin and its derivatives have been investigated as potential anticancer drugs for various aspects of cancer treatment [15, 18, 19]. Moreover, a clinical study indicated that shikonin was effective in treating later-stage lung cancer patients [20]. Shikonin was reported to inhibit the tumor growth by inducing apoptotic cell death in a variety of cancers [21-23]. In apoptosis resistant cells, it triggered necroptosis that contributed to overcome Bcl-2- and Bcl-XL-mediated apoptotic resistance [24]. It was previous reported that inhibition of PKM2 by shikonin could overcome the cisplatin-resistance in bladder cancer, but without clear mechanism [10]. In current study, we further confirmed that depletion or inhibition of PKM2 by its siRNA or shikonin re-sensitized the cisplatin resistant BC cells. Mechanically, we found that shikonin treatment kills the cisplatin resistant BC cells via necroptosis, which is modulated by Bcl-2 family proteins, including PUMA, Bcl-2, and Bax. Our results provide a further insight into the role of PKM2 in the bladder cancer chemoresistance and the implication of shikonin, which might be developed as new therapy strategy to overcome drug resistance in bladder cancer.
Materials and Methods
Cell Lines, Reagents, and Treatment
The bladder cancer cells, T24 and SW870 cell line, were purchased from American Type Culture Collection (ATCC; Manassas, VA). The cells were cultured in Dulbecco's modified Eagle's medium (Gibco, Carlsbad, CA, USA) supplemented with 10% v/v fetal bovine serum (Gibco) and antibiotics (100 U/ml penicillin and 100 mg/ml streptomycin) in a humidified 5% CO2 atmosphere at 37.8°C (Thermo). The establishment of cisplatin-resistant bladder cancer cell line was developed by cisplatin concentration gradient progressive increase induction method as previous described [7]. For cell culture, the levels of mycoplasma were routinely (once every three months) assessed in these cells using a MycoAlert Mycoplasma Detection Kit purchased from Lonza (Rockland, ME), and the results were negative.
Shikonin, purchased from ChromaDex (ASB-00019210-005), was dissolved in dimethyl sulfoxide (DMSO, Sigma, St. Louis, MO). Cisplatin was from Sigma-Aldrich (479306, Shanghai, China), and diluted in 0.9 % NaCl. The cell-permeant pan caspase inhibitor of apoptosis, Z-VAD (627610), MLKL inhibitor, Necrosulfonamide (NSA, 5025), and RIP3 inhibitor, GSK-872 (480073), were purchased from EMD Millipore company (Bedford, MA, USA), and dissolved in DMSO before used.
Clinical Specimens
Human bladder cancer tissues were collected from 14 patients with bladder cancer in First Hospital of Jilin University (Changchun, China) who signed the written informed consents before therapy and surgery. These patients received cisplatin-based chemotherapy, and had an age range of 50-79 years and a median age of 60 years. The patient were divided into two group (7 in each group) based on their therapy outcomes. The patients received cisplatin treatment with relapse were considered as cisplatin resistant patients. Patient anonymity was also preserved. All specimens had a confirmed pathological diagnosis and were classified according to World Health Organization (WHO) criteria.
Western Blot Analysis
Cells were collected and lysed using a mammalian protein extraction reagent containing a protease inhibitor cocktail. After centrifugation at 17 000 g for 15 min, the protein content in the supernatant was determined using the BCA protein assay kit (Bio-Rad, Shanghai, China). Whole Cell Lysate was used for the assay. Antibodies against pyruvate kinase M2 (PKM2), PUMA (ab9643), RIP3 (ab56164), p-RIP3 (S227, ab209384), p-MLKL (S358, ab187091), Bax (ab32503), Bcl-2 (ab32124), Bid (ab32060), Bcl-XL (ab32370) were purchased from Abcam (Cambridge, MA, USA), antibodies against β-Actin (sc-47778) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
MTS assay and ATP assay
After different concentrations of drugs treatment, 20 μL MTS (Promega) was added to each well. The absorbance at 490 nm was recorded on a Varioskan Flash Multiplate Reader (Thermo Scientific) after incubation for 3 hours at 37°C. The ATP level of cell was determined by CellTiter-Glo Luminescent cell viability assay (Promega). Assays were performed in triplicate and repeated three times.
Figure 1
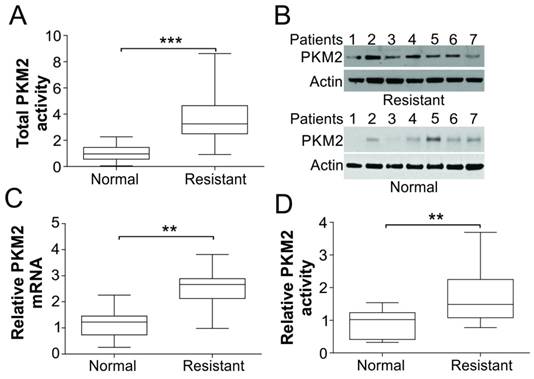
PKM2 was upregulated in tumors of cisplatin resistant BC patients. (A) The total enzymatic activity of PKM2 in tumor tissue specimens from normal (n=7) and cisplatin resistant BC patients (n=7). (B) The protein expression of PKM2 in tumor tissue specimens from normal and cisplatin resistant BC patients. (C) The mRNA expression of PKM2 in tumor tissue specimens from normal and cisplatin resistant BC patients. (D) The relative PKM2 activity in tumor tissue specimens from normal and cisplatin resistant BC patients, which is normalized to its proteins expression level as in (B). **, p<0.01; ***, p<0.001.
Apoptosis Analysis
After treatment, adherent and floating cells were harvested and resuspended with PBS solution containing 3.7 % formaldehyde, 0.5 % Nonidet P-40, and 10 ug/ml Hoechst 33258 (Invitrogen). Apoptosis was assessed through microscopic visualization of condensed chromatin and micronucleation as described [25]. The caspase-3/7 activity assay was determined using Caspase-Glo 3/7 assay system as described by manufacturer (G8090, Promega).
Plasmid and siRNA Transfection
The T24 human BC cell line was chosen for the knockdown of mRNA encoding PKM2 (143814), Bcl-2 (214532), and PUMA (134322) using specific siRNAs from Thermo Fisher Scientific. Non-specific, scrambled siRNA from Sigma was used as a negative control. The pCEP4-HA-Bax (#16587) plasmid was purchase from Addgene. The control plasmid pCEP4 (V04450) was purchase from Thermofisher Scientific. The transfection was conducted by lipofetamin 2000 according to the manufacturer's instruction.
PKM Activity Assay
PKM activities were analyzed using the lactate dehydrogenase (LDH)-coupled assay as described previously [26]. The following reagents in a final volume of 400 μl (10 mmol/L Tris-acetate, pH 7.5; 10 mmol/L MgCl2; 50 mmol/L KCl; 2 mmol/L ADP; 10 mmol/L phosphoenolpyruvate; 4.4 units of LDH; and 0.12 mmmol/L NADH) was used as standard assay mixture. The reaction was started by adding 5 μl of Whole Cell Lysate and 4 μl of 5′-AMP brings PKM2 to its maximal velocity. The baseline was measured without the addition of phosphoenolpyruvate and 5′-AMP.
Statistical Analysis
Two-way ANOVA was used for multi-group comparisons. The PKM2 activity data in human skin tissue were analyzed using the Student's t-test. All of the experiments have been repeated at least three times. Data were reported as mean ± standard error (SEM). P < 0.05 was considered significant.
Results
PKM2 is Upregulated in Human bladder Cancer Tissue Samples with cisplatin resistance
We first detected the activity and expression levels of PKM2 in human bladder cancer frozen tissue samples. Seven patients sample and seven samples from patient resistant to cisplatin therapy have been obtained. As shown in Fig. 1A, the total PKM2 activity was increased by about 3-folds in cisplatin resistance patient tumor samples than normal tissues. And the protein and mRNA expression levels of PKM2 were higher in almost all of the cisplatin resistant tumor samples than normal control (Fig. 1B, C). Furthermore, we normalized the PKM2 activity to its proteins expression level, and found that the relative PKM2 activity is also significantly higher in the cisplatin resistant patients (Fig. 1D). These results suggest that PKM2 could play an important role on human BC resistant to cisplatin treatment.
Figure 2
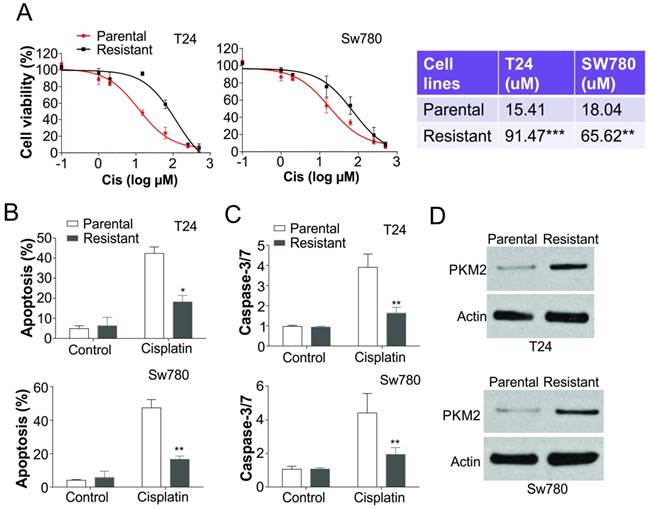
PKM2 was upregulated in cisplatin resistant BC cells. (A) T24 and SW780 parental and resistant BC cells were treated cisplatin (Cis) as indicated dosages for 24 hours, and subsequently analyzed with MTT assay. Left, the survival curve; right, the IC50 values. (B) Apoptosis of T24 and SW780 parental and resistant BC cells treated with 25 µM cisplatin was determined by Hoechst 33258 staining. (C) The caspase-3/7 activity in T24 and SW780 parental and resistant BC cells treated with 25 µM cisplatin. (D) The expression of PKM2 in T24 and SW780 parental and resistant BC cells. *, p<0.05; **, p<0.01; ***, p<0.001.
The expression level of PKM2 is also higher in the bladder cancer cells resistant to cisplatin treatment
To further confirm the regulation of PKM2 in cisplatin resistant bladder tumor, we generated two bladder cancer cell lines with resistance to cisplatin treatment. The T24 and SW780 cisplatin resistant cells were insensitive to the different doses of cisplatin when compared with their parental cells (Fig. 2A). The IC50 values of cisplatin in T24 and SW780 resistant cells are significantly higher than that in parental cells (Fig. 2A). Since apoptosis is an important cell death caused by cisplatin, we also analyzed the apoptosis in T24 and SW780 parental (P) and resistant (R) cells in response to cisplatin treatment. Consistently, cisplatin treatment barely induced apoptosis or caspase-3/7 activation in the T24R or SW780R cells (Fig. 2B, C). We further analyzed the PKM2 expression in the T24 and SW780 parental and resistant cells, and found that the expression level of PKM2 was also much higher than the parental cells (Fig. 2D). Collectively, our results suggested that PKM2 is induced in the cisplatin resistant bladder cancer in vitro.
Depletion of PKM2 re-sensitized the resistant cells to cisplatin treatment
Since PKM2 is highly expressed in human bladder tumor samples, we further studied if shikonin, a specific inhibitor of PKM2, can re-sensitize cisplatin resistant cell to drug treatment. We found that shikonin treatment of T24R cells significant reduced the PKM2 kinase activity (Fig. 3A), but did not change the expression of PKM2 (Fig. 3B), which further confirm that shikonin is the specific inhibitor of PKM2. Inhibition of PKM2 by shikonin alone triggered more cell death in T24R cells, compared with cisplatin treatment alone (Fig. 3C). Furthermore the combination treatment of shikonin and cisplatin induced higher percentage of cell death in T24R cells (Fig. 3C), suggested that inhibition of PKM2 by shikonin enhanced the killing effect of cisplatin. However, shikonin alone or in combined with cisplatin did not significantly or merely increased the apoptotic cell death in T24R cells (Fig. 3D), suggesting that shikonin might re-sensitized the T24R to cisplatin via other form of cell death. The treatment of shikonin also lead to drop of ATP level in T24 resistant cells (Fig. 3E), further suggested the non-apoptotic cell death induced by shikonin. To further confirm the effects of PKM2, We depleted PKM2 by siRNA in cisplatin resistant T24 cells, and found that absence of PKM2 resensitized the resistant cells to cisplatin treatment (Fig. 3F, G). Therefore, our result suggested inhibition of PKM2 re-sensitized the cisplatin resistant cells by non-apoptotic cell death.
Shikonin kills bladder cancer resistant cells by inducing necroptosis
As shikonin treatment alone or in combination with cisplatin actually killed the T24R cells, but did not significantly induced more apoptosis, we further investigated the underlying mechanism. It was reported that shikonin could induce necroptosis in various cancer cells [27, 28], we hypothesized that shikonin triggered necroptosis in T24R cells. As predicted, shikonin treatment alone or in combined with cisplatin led to phosphorylation of RIP3 in T24R cells, but did not change the expression level of RIP3 (Fig. 4A). Furthermore, pretreatment of RIP3 inhibitor (GSK872) or MLKL inhibitor (necrosulfonamide, NSA) suppressed the cell death caused by shikonin treatment or its combination with cisplatin (Fig. 4B). However, pretreatment of z-VAD, the apoptosis inhibitor, did not affected the cell death caused by shikonin or its combination with cisplatin (Fig. 4C). Furthermore, knockdown of RIP3 by siRNA has the similar effect with GSK872, which also suppressed the cell death, and phosphorylation of RIP3 induced by shikonin or its combination with cisplatin (Fig. 4D,E). Similarly, shikonin also sensitized the T24 parental cells to cisplatin treatment by triggering more apoptosis (Fig. 4F, G). Collectively, our results suggested that shikonin resensitized the cisplatin resistant bladder cancer cells by inducing necroptosis instead of apoptosis.
Figure 3
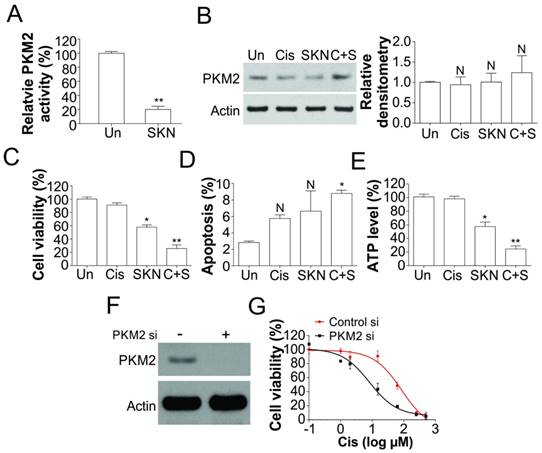
Shikonin inhibited PKM2 activity and re-sensitized T24 resistant BC cells to cisplatin. (A) PKM2 activity in T24 resistant cells treated with 0.4 μM shikonin (SKN). (B) The expression of PKM2 in T24 resistant cells treated with 0.4 μM shikonin and 25 µM cisplatin (Cis) alone or their combination (C+S). Left, the representative western blots; right, the densitometry analysis of western blot (n=3). (C, D, and E) T24 resistant cells were treated with 0.4 μM shikonin and 25 µM cisplatin alone or their combination for 24 h. C, the cell viability was analyzed by MTT assay. D, the apoptosis was analyzed by Hoechst 33258 staining. E, the ATP level was analyzed. (F) The expression of PKM2 in T24 resistant cell transfected with PKM2 siRNA. (G) The T24 resistant cells were transfected with PKM2 siRNA, and subsequently treated with cisplatin as indicated for 24 h. The cell viability was analyzed by MTT assay. N, p>0.05; *, p<0.05; **, p<0.01.
Dysregulation of PUMA contributes to shikonin induced necroptosis in cisplatin resistant cells
It would be interesting to study the reason why shikonin induced necroptosis in cisplatin resistant cells, but apoptosis in parental cells. As Bcl-2 family proteins play an important role in both apoptosis and necroptosis [29-31], we firstly study the regulation of several important Bcl-2 family proteins. We found that PUMA and Bax were induced by cisplatin treatment in T24 parental cells, but blunted in the T24 resistant cells (Fig. 5A). In contrast, the expression of Bcl-2 in T24 parental cells was suppressed by cisplatin treatment, but was increased in T24 resistant cells, and did not significant change upon cisplatin treatment (Fig. 5A). However, shikonin treatment in T24R cells only re-activated the induction of PUMA, but did not change the expression of Bax or Bcl-2 (Fig. 5B). Since PUMA was recently reported to engage into RIP3 dependent necroptosis [29], we therefore test its role in shikonin induced necroptosis. We found that depletion of PUMA by siRNA abrogated the phosphorylation of RIP3 (Fig. 5C), and cell death caused by shikonin or its combination with cisplatin in T24R cells (Fig. 5D, E). However, absence of PUMA did not significantly change the apoptosis in T24R cells treated with cisplatin and/or shikonin (Fig. 5F). Therefore, our results suggested that PUMA mediated the necroptosis caused by shikonin in the T24 resistant cells.
Figure 4
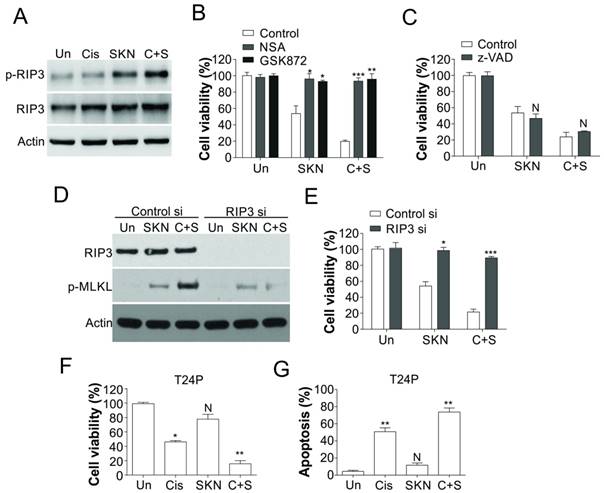
Shikonin induced necroptosis in T24 resistant BC cells. (A) T24 resistant cells were treated with 0.4 μM shikonin (SKN) and 25 µM cisplatin (Cis) alone or their combination (C+S) for 24 h. The expression of p-RIP3 and RIP3 was analyzed by western blot. (B) T24 resistant cells were pretreated with RIP3 inhibitor (GSK872, 2 µM) or MLKL inhibitor (NSA, 2 µM) for 1 h, and treated with 0.4 μM shikonin and 25 µM cisplatin alone or their combination for another 24 h. The cell viability was analyzed by MTT assay. (C) T24 resistant cells were pretreated with caspases inhibitor (z-VAD, 10 µM) for 1 h, and treated with 0.4 μM shikonin and 25 µM cisplatin alone or their combination for another 24 h. The cell viability was analyzed by MTT assay. (D) T24 resistant cells were transfected with RIP3 siRNA, and subsequently treated with 0.4 μM shikonin and 25 µM cisplatin alone or their combination for 24 h. The expression of RIP3 and p-MLKL was analyzed by western blot. (E) The T24 resistant cells were treated as in (D), the cell viability was analyzed by MTT assay. (F and G) T24 parental cells were treated as in (A). F, the cell viability was analyzed by MTT assay. G, the apoptosis was analyzed by Hoechst 33258 staining. N, p>0.05; *, p<0.05; **, p<0.01; ***, p<0.001.
Figure 5
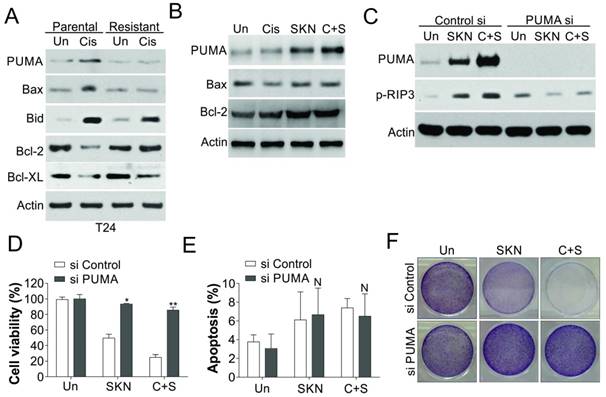
Induction of PUMA contributes to shikonin induced necroptosis. (A) The expression of indicated Bcl-2 family proteins in T24 parental and resistant cells treated with 25 µM cisplatin (Cis). (B) The expression of indicated proteins in T24 resistant cells treated with 0.4 μM shikonin (SKN) and 25 µM cisplatin (Cis) alone or their combination (C+S) for 24 h. (C) T24 resistant cells were transfected with PUMA siRNA, and subsequently treated with 0.4 μM shikonin and 25 µM cisplatin alone or their combination for 24 h. The expression of PUMA and p-MLKL was analyzed by western blot. (D, E, and F) The T24 resistant cells were treated as in (C). D, the cell viability was analyzed by MTT assay. E, the apoptosis was analyzed by Hoechst 33258 staining. F. Crystal violet staining for the cell viability. N, p>0.05; *, p<0.05; **, p<0.01.
Bcl-2 and Bax dysregulation decides PUMA induce apoptosis or necroptosis
Since PUMA induction can also promote apoptosis, we further studied why the induction of PUMA trigger T24R cells necroptosis but not apoptosis. As Bcl-2 and Bax was dysregulation in T24R cell in response to cisplatin treatment, but did not change in response to shikonin treatment alone or in combination with cisplatin (Fig. 5A,B), we hypothesized that Bcl-2 and Bax might also play an important role in shikonin induced necroptosis in T24R cells. As predicted, knockdown of Bcl-2 expression in T24R cells suppressed the phosphorylation of RIP3, but increased the activation of caspase-3 (Fig. 6A).Although absence of Bcl-2 did not sensitize the T24R cells to cisplatin and/or shikonin treatment (Fig. 6B), it increased the apoptosis (Fig. 6C), which compensated the loss of necroptosis induced by cisplatin and/or shikonin. Similarly, enhanced expression of Bax also suppressed the phosphorylation of RIP3, and increased activation of caspase-3 (Fig. 6D). Besides, increased expression of Bax did not enhance the killing effects of shikonin and/or cisplatin in T24R cells (Fig. 6E), but induced more apoptosis in response to drug treatment (Fig. 6F). Therefore, our result suggested that Bcl-2 and Bax mediated PUMA induced necroptosis in response to shikonin treatment in cisplatin resistant bladder cancer cells.
Figure 6
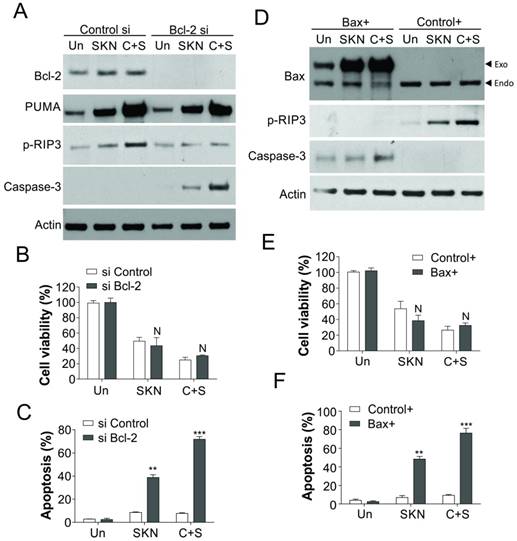
Bcl-2 and Bax modulate the cell death induced by shikonin. (A) T24 resistant cells were transfected with Bcl-2 siRNA, and subsequently treated with 0.4 μM shikonin (SKN) and 25 µM cisplatin (Cis) alone or their combination (C+S) for 24 h. The expression of indicated proteins was analyzed by western blot. (B and C) The T24 resistant cells were treated as in (A). B, the cell viability was analyzed by MTT assay. C, the apoptosis was analyzed by Hoechst 33258 staining. (D) T24 resistant cells were transfected with Bax plasmid, and subsequently treated with 0.4 μM shikonin and 25 µM cisplatin alone or their combination for 24 h. The expression of indicated proteins was analyzed by western blot. (E and F) The T24 resistant cells were treated as in (D). E, the cell viability was analyzed by MTT assay. F, the apoptosis was analyzed by Hoechst 33258 staining. N, p>0.05; **, p<0.01, ***, p<0.001.
Discussion
In summary, we found in this study that PKM2 was upregulated in cisplatin resistant bladder cancer cells and tumors. The induction of PKM2 contributes to the cisplatin resistant in BC. Depletion or inhibition of PKM2 re-sensitized the BC cells to cisplatin treatment. As to the mechanism, inhibition of PKM2 by shikonin re-sensitized the BC resistant cells by inducing necroptosis. Furthermore, we also found the dysregulation of Bcl-2 family proteins decides whether shikonin kill the BC cells via necroptosis or apoptosis. In human bladder cancer tissue samples, our results indicate that PKM2 is upregulated, making PKM2 a potential anti-cancer target. PKM2 can exist in either active protein kinase (dimer) or pyruvate kinase (tetramer) [32], the dual kinase activity can connect metabolism switch and gene regulation during tumor transformation and progression. Upregulation of PKM2 is commonly found in many human cancers, including retinal cancer, ovarian cancer, breast cancer, and lung cancer [33]. It was also reported that transgenic mice expressing a constitutively activated HRAS have marked higher expression of PKM2 during the initiation of low-grade non-invasive bladder tumors, and high expression level of PKM2 contribute to cisplatin resistance in BC [10, 33]. In the current study, the expression of PKM2 was also examined in BC tissues, as well as cisplatin resistant BC cell lines. Consistent with previous studies, our result further suggested that PKM2 expression contributes to the cisplatin resistance in BC, which further confirmed the oncogene role of PKM2.
Besides, we also extended the investigation on the effect of PKM2 inhibition by its specific inhibitor, shikonin. Shikonin, a natural product isolated from the dried root of lithospermum erythrorhizon, is a newly identified specific inhibitor of PKM2 activity [18]. In previous reports, it was that shikonin could circumvent drug-transporter and apoptotic defect-mediated drug resistance by inducing necroptosis [24]. In our study, we also found that the cell death in cisplatin resistant BC cells induced by shikonin was accompanied with a sharp reduction of cellular ATP, and phosphorylation of RIP3, which are important marker for necroptosis [29, 34]. As PKM2 is essential for maintaining the homeostasis of cellular energy for cancer cells [15, 17], its inhibition by shikonin leads to energy shut-off in cancer cells, and results in necroptosis. Necroptosis regulated by RIP3 has been established as a new type of programmed cell death and could be induced in various cancer cells [35, 36]. Consistent with previous reports [24, 37], RIP3 were found to be activated by shikonin in cisplatin resistant BC cells, which indicated that necroptosis is an alternative way to kill apoptosis resistant BC cells.
The clinical cancer drug resistance was usually relevant to dysregulation Bcl-2 family members. In our study, we also found that stabilization of Bcl-2 and suppression of Bax and PUMA contribute to the cisplatin resistant in BC cells. However, shikonin-induced cell death is not inhibited by overexpression of Bcl-2 or suppression of Bax, since shikonin induces a dominant necroptotic death, which is a separate death pathway from apoptosis [24]. Interestingly, the necroptosis induced by shikonin is modulated by PUMA induction. PUMA, also known as Bcl-2-binding component 3 (BBC3), is originally found to be pro-apoptotic protein [38]. Other than apoptosis, more and more studies showed that PUMA is also involved in several necroptosis related diseases, such as inflammation bowl disease, ischemia reperfusion [39, 40]. Recently, PUMA was also reported to modulated RIP3 phosphorylation by targeting mitochondria dysfunction in TNFα induced necroptosis [29]. However, the specific function of PUMA related necroptosis in cancer therapy is still unclear. In our study, we firstly reported that induction of PUMA by shikonin can overcome the cisplatin resistance in BC cells, and kill BC cells by necroptosis. Furthermore, our study also illustrated that the function of PUMA in shikonin induced necroptosis is also modulated by Bcl-2 and Bax, as depletion of Bcl-2 or enhanced expression of Bax in cisplatin resistant cells reverted the cell death from necroptosis into apoptosis.
Therefore, our studies demonstrate that shikonin, as a specific PKM2 inhibitor, suppresses PKM2 activity in cisplatin resistant BC cells. Inhibition of PKM2 reduced the cisplatin resistance in BC cells. Moreover, our study also revealed that necroptosis induced by shikonin is an alternative method to conquer the drug resistance in BC therapy.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Knowles MA, Hurst CD. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015;15:25-41
2. Cancer Genome Atlas Research N. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315-322
3. Chavan S, Bray F, Lortet-Tieulent J, Goodman M, Jemal A. International variations in bladder cancer incidence and mortality. Eur Urol. 2014;66:59-73
4. James ND, Hussain SA, Hall E, Jenkins P, Tremlett J, Rawlings C, Crundwell M, Sizer B, Sreenivasan T, Hendron C, Lewis R, Waters R, Huddart RA, Investigators BC. Radiotherapy with or without chemotherapy in muscle-invasive bladder cancer. N Engl J Med. 2012;366:1477-1488
5. Choi W, Czerniak B, Ochoa A, Su X, Siefker-Radtke A, Dinney C, McConkey DJ. Intrinsic basal and luminal subtypes of muscle-invasive bladder cancer. Nature reviews Urology. 2014;11:400-410
6. Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, Roth B, Cheng T, Tran M, Lee IL, Melquist J, Bondaruk J, Majewski T, Zhang S, Pretzsch S, Baggerly K, Siefker-Radtke A, Czerniak B, Dinney CP, McConkey DJ. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer cell. 2014;25:152-165
7. Fan Y, Shen B, Tan M, Mu X, Qin Y, Zhang F, Liu Y. Long non-coding rna uca1 increases chemoresistance of bladder cancer cells by regulating wnt signaling. FEBS J. 2014;281:1750-1758
8. Benesch C, Schneider C, Voelker HU, Kapp M, Caffier H, Krockenberger M, Dietl J, Kammerer U, Schmidt M. The clinicopathological and prognostic relevance of pyruvate kinase m2 and pakt expression in breast cancer. Anticancer research. 2010;30:1689-1694
9. Shi HS, Li D, Zhang J, Wang YS, Yang L, Zhang HL, Wang XH, Mu B, Wang W, Ma Y, Guo FC, Wei YQ. Silencing of pkm2 increases the efficacy of docetaxel in human lung cancer xenografts in mice. Cancer science. 2010;101:1447-1453
10. Wang X, Zhang F, Wu XR. Inhibition of pyruvate kinase m2 markedly reduces chemoresistance of advanced bladder cancer to cisplatin. Scientific reports. 2017;7:45983
11. Su Q, Tao T, Tang L, Deng J, Darko KO, Zhou S, Peng M, He S, Zeng Q, Chen AF, Yang X. Down-regulation of pkm2 enhances anticancer efficiency of thp on bladder cancer. Journal of cellular and molecular medicine. 2018;22:2774-2790
12. Warburg O. On the origin of cancer cells. Science. 1956;123:309-314
13. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell metabolism. 2008;7:11-20
14. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029-1033
15. Mazurek S, Grimm H, Boschek CB, Vaupel P, Eigenbrodt E. Pyruvate kinase type m2: A crossroad in the tumor metabolome. The British journal of nutrition. 2002;87(Suppl 1):S23-29
16. Spoden GA, Mazurek S, Morandell D, Bacher N, Ausserlechner MJ, Jansen-Durr P, Eigenbrodt E, Zwerschke W. Isotype-specific inhibitors of the glycolytic key regulator pyruvate kinase subtype m2 moderately decelerate tumor cell proliferation. International journal of cancer. 2008;123:312-321
17. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The m2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230-233
18. Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Shikonin and its analogs inhibit cancer cell glycolysis by targeting tumor pyruvate kinase-m2. Oncogene. 2011;30:4297-4306
19. Xuan Y, Hu X. Naturally-occurring shikonin analogues-a class of necroptotic inducers that circumvent cancer drug resistance. Cancer letters. 2009;274:233-242
20. Guo XP, Zhang XY, Zhang SD. [clinical trial on the effects of shikonin mixture on later stage lung cancer]. Zhong xi yi jie he za zhi = Chinese journal of modern developments in traditional medicine. 1991;11:598-599 580
21. Yeh CC, Kuo HM, Li TM, Lin JP, Yu FS, Lu HF, Chung JG, Yang JS. Shikonin-induced apoptosis involves caspase-3 activity in a human bladder cancer cell line (t24). In vivo. 2007;21:1011-1019
22. Yoon Y, Kim YO, Lim NY, Jeon WK, Sung HJ. Shikonin, an ingredient of lithospermum erythrorhizon induced apoptosis in hl60 human premyelocytic leukemia cell line. Planta medica. 1999;65:532-535
23. Shilnikova K, Piao MJ, Kang KA, Ryu YS, Park JE, Hyun YJ, Zhen AX, Jeong YJ, Jung U, Kim IG, Hyun JW. Shikonin induces mitochondria-mediated apoptosis and attenuates epithelial-mesenchymal transition in cisplatin-resistant human ovarian cancer cells. Oncology letters. 2018;15:5417-5424
24. Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo J, Hu X. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Molecular cancer therapeutics. 2007;6:1641-1649
25. Chen D, Ming L, Zou F, Peng Y, Van Houten B, Yu J, Zhang L. Tap73 promotes cell survival upon genotoxic stress by inhibiting p53 activity. Oncotarget. 2014;5:8107-8122
26. Chiappetta G, Tallini G, De Biasio MC, Pentimalli F, de Nigris F, Losito S, Fedele M, Battista S, Verde P, Santoro M, Fusco A. Fra-1 expression in hyperplastic and neoplastic thyroid diseases. Clinical cancer research: an official journal of the American Association for Cancer Research. 2000;6:4300-4306
27. Han W, Xie J, Li L, Liu Z, Hu X. Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis. Apoptosis: an international journal on programmed cell death. 2009;14:674-686
28. Hu X, Han W, Li L. Targeting the weak point of cancer by induction of necroptosis. Autophagy. 2007;3:490-492
29. Chen D, Tong J, Yang L, Wei L, Stolz DB, Yu J, Zhang J, Zhang L. Puma amplifies necroptosis signaling by activating cytosolic DNA sensors. Proceedings of the National Academy of Sciences of the United States of America. 2018;115:3930-3935
30. Sulkshane P, Teni T. Bh3 mimetic obatoclax (gx15-070) mediates mitochondrial stress predominantly via mcl-1 inhibition and induces autophagy-dependent necroptosis in human oral cancer cells. Oncotarget. 2017;8:60060-60079
31. Yan C, Oh JS, Yoo SH, Lee JS, Yoon YG, Oh YJ, Jang MS, Lee SY, Yang J, Lee SH, Kim HY, Yoo YH. The targeted inhibition of mitochondrial hsp90 overcomes the apoptosis resistance conferred by bcl-2 in hep3b cells via necroptosis. Toxicology and applied pharmacology. 2013;266:9-18
32. Gao X, Wang H, Yang JJ, Liu X, Liu ZR. Pyruvate kinase m2 regulates gene transcription by acting as a protein kinase. Molecular cell. 2012;45:598-609
33. Huang C, Huang Z, Bai P, Luo G, Zhao X, Wang X. Expression of pyruvate kinase m2 in human bladder cancer and its correlation with clinical parameters and prognosis. OncoTargets and therapy. 2018;11:2075-2082
34. He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to tnf-alpha. Cell. 2009;137:1100-1111
35. Chen D, Yu J, Zhang L. Necroptosis: An alternative cell death program defending against cancer. Biochimica et biophysica acta. 2016;1865:228-236
36. Meng MB, Wang HH, Cui YL, Wu ZQ, Shi YY, Zaorsky NG, Deng L, Yuan ZY, Lu Y, Wang P. Necroptosis in tumorigenesis, activation of anti-tumor immunity, and cancer therapy. Oncotarget. 2016;7:57391-57413
37. Lu B, Wang Z, Ding Y, Wang X, Lu S, Wang C, He C, Piao M, Chi G, Luo Y, Ge P. Rip1 and rip3 contribute to shikonin-induced glycolysis suppression in glioma cells via increase of intracellular hydrogen peroxide. Cancer letters. 2018;425:31-42
38. Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. Puma induces the rapid apoptosis of colorectal cancer cells. Molecular cell. 2001;7:673-682
39. Qiu W, Wu B, Wang X, Buchanan ME, Regueiro MD, Hartman DJ, Schoen RE, Yu J, Zhang L. Puma-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. The Journal of clinical investigation. 2011;121:1722-1732
40. Wu B, Qiu W, Wang P, Yu H, Cheng T, Zambetti GP, Zhang L, Yu J. P53 independent induction of puma mediates intestinal apoptosis in response to ischaemia-reperfusion. Gut. 2007;56:645-654
Author contact
![]() Corresponding authors: Chunshu Jia, Centre for Reproductive Medicine, Centre for Prenatal Diagnosis, First Hospital, Jilin University, No.71 Xinmin Str Changchun, 130021, China. Email: jiacs08jlu.edu.cn or Xiaoliang Chen, Department of Urology, China-Japan Union Hospital, Jilin University, No. 126 Xiantai Road, Changchun, 130033, China. E-Mail: xlchen15jlu.edu.cn
Corresponding authors: Chunshu Jia, Centre for Reproductive Medicine, Centre for Prenatal Diagnosis, First Hospital, Jilin University, No.71 Xinmin Str Changchun, 130021, China. Email: jiacs08jlu.edu.cn or Xiaoliang Chen, Department of Urology, China-Japan Union Hospital, Jilin University, No. 126 Xiantai Road, Changchun, 130033, China. E-Mail: xlchen15jlu.edu.cn
Citation styles
APA
Wang, Y., Hao, F., Nan, Y., Qu, L., Na, W., Jia, C., Chen, X. (2018). PKM2 Inhibitor Shikonin Overcomes the Cisplatin Resistance in Bladder Cancer by Inducing Necroptosis. International Journal of Biological Sciences, 14(13), 1883-1891. https://doi.org/10.7150/ijbs.27854.
ACS
Wang, Y.; Hao, F.; Nan, Y.; Qu, L.; Na, W.; Jia, C.; Chen, X. PKM2 Inhibitor Shikonin Overcomes the Cisplatin Resistance in Bladder Cancer by Inducing Necroptosis. Int. J. Biol. Sci. 2018, 14 (13), 1883-1891. DOI: 10.7150/ijbs.27854.
NLM
Wang Y, Hao F, Nan Y, Qu L, Na W, Jia C, Chen X. PKM2 Inhibitor Shikonin Overcomes the Cisplatin Resistance in Bladder Cancer by Inducing Necroptosis. Int J Biol Sci 2018; 14(13):1883-1891. doi:10.7150/ijbs.27854. https://www.ijbs.com/v14p1883.htm
CSE
Wang Y, Hao F, Nan Y, Qu L, Na W, Jia C, Chen X. 2018. PKM2 Inhibitor Shikonin Overcomes the Cisplatin Resistance in Bladder Cancer by Inducing Necroptosis. Int J Biol Sci. 14(13):1883-1891.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.