Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and methods
3. Results
4. Discussion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(1):239-252. doi:10.7150/ijbs.29063 This issue Cite
Research Paper
Clopidogrel Reduces Fibronectin Accumulation and Improves Diabetes-Induced Renal Fibrosis
Zongyu Zheng1,2, Tianjiao Ma2,3, Xin Lian1, Jialin Gao1, Weigang Wang1, Wenya Weng2,4, Xuemian Lu4, Weixia Sun6, Yanli Cheng6, Yaowen Fu1 ![]() , Madhavi J. Rane7, Evelyne Gozal2,5
, Madhavi J. Rane7, Evelyne Gozal2,5 ![]() , Lu Cai2,5
, Lu Cai2,5
1. Department of Urology, The First Hospital of Jilin University, Changchun 130021, China;
2. Pediatric Research Institute, Department of Pediatrics, University of Louisville, Louisville, KY 40202, USA;
3. Department of Rheumatology and Immunology, China-Japan Union Hospital of the Jilin University, Changchun 130033, China;
4. The Ruian Center of Chinese-American Research Institute for Diabetic Complications, The Third Affiliated Hospital of the Wenzhou Medical University, Ruian 325200, China.
5. Departments of Pharmacology and Toxicology, University of Louisville, Louisville, KY 40202, USA.
6. Department of Nephrology, The First Hospital of Jilin University, Changchun 130021, China.
7. Division of Nephrology, Department of Medicine, University of Louisville, Louisville, KY 40202, USA.
Received 2018-8-7; Accepted 2018-11-11; Published 2019-1-1
Abstract

Hyperglycemia-induced renal fibrosis causes end-stage renal disease. Clopidogrel, a platelet inhibitor, is often administered to decrease cardiovascular events in diabetic patients. We investigated whether clopidogrel can reduce diabetes-induced renal fibrosis in a streptozotocin-induced type 1 diabetes murine model and fibronectin involvement in this protective response. Diabetic and age-matched controls were sacrificed three months after the onset of diabetes, and additional controls and diabetic animals were further treated with clopidogrel or vehicle for three months. Diabetes induced renal morphological changes and fibrosis after three months. Clopidogrel, administered during the last three months, significantly decreased blood glucose, collagen and fibronectin expression compared to vehicle-treated diabetic mice. Diabetes increased TGF-β expression, inducing fibrosis via Smad-independent pathways, MAP kinases, and Akt activation at three months but returned to baseline at six months, whereas the expression of fibronectin and collagen remained elevated. Our results suggest that activation of TGF-β, CTGF, and MAP kinases are early profibrotic signaling events, resulting in significant fibronectin accumulation at the early time point and returning to baseline at a later time point. Akt activation at the three-month time point may serve as an adaptive response in T1D. Mechanisms of clopidogrel therapeutic effect on the diabetic kidney remain to be investigated as this clinically approved compound could provide novel approaches to prevent diabetes-induced renal disease, therefore improving patients' survival.
Keywords: Diabetes, clopidogrel, fibronectin (FN), renal fibrosis
1. Introduction
Diabetes is a chronic, progressive disease. Recent estimates indicated that 30.3 million people in the United States had diabetes in 2017, accounting for 9.4% of the total U.S. population (National Diabetes Statistics Report, 2017) [1]. The prevalence of chronic kidney disease (CKD) among U.S. adults with diagnosed diabetes was approximately 36.5% during 2011-2012 [2].
Diabetic nephropathy (DN) is a diabetic complication caused by glomerular hypertrophy, proteinuria, glomerular filtration, and renal fibrosis. In diabetic patients, hyperglycemia-induced renal fibrosis causes end-stage renal changes such as renal failure, which are the leading causes of death from diabetic complications. A characteristic pathological change in DN is progressive mesangial expansion caused by accumulation of extracellular matrix (ECM) [3].
In general, the ECM consists mainly of fibronectin (FN), collagens (including types I and IV), laminin, and proteoglycans [4]. Elevations of glucose levels lead to a significant increase in FN matrix assembly; meanwhile, collagen IV deposition is dependent on FN assembly [5]. The imbalance between the synthesis and breakdown of the ECM induces glomerulosclerosis and tubulointerstitial fibrosis. Liver-specific FN conditional knockout mice had no circulating FN and these mice were protected from STD-induced T1D, renal fibrosis and albuminuria, suggesting a critical role of circulating FN in these events [6]. The improvement of renal fibrosis in a hyperglycemic state is the most concerned aspect of the current treatment. The mechanism of FN overexpression is not yet completely understood although, several previous studies implicated the upregulation of the transforming growth factor-beta (TGF-β) and the platelet-derived growth factor [7]. The stimulation of rat mesangial cells with TGF-β in another study caused an increase of collagen I and FN expression [8]. However, adverse outcomes may result from anti-TGF-β therapy due to the multiplicity of TGF-β functions. Thus, effective preventive approaches have been actively explored for the reduction of renal complications such as renal fibrosis in diabetic patients.
Significant alterations of platelet function and their hyper activation in diabetic patients [9] have led to current medical guidelines recommendations of prophylactic treatment, such as once-daily administration of low-dose aspirin (acetylsalicylic acid) to decrease platelet activation and for primary and secondary prevention of cardiovascular (CV) events in high-risk diabetic patients [10]. However, emerging clinical evidence indicates that aspirin does not prevent CV events in adult diabetic patients better than placebo [11]. A recent study reported that administration of clopidogrel as an alternative to aspirin, decreased stroke recurrence and CV outcomes in patients with type 2 diabetes (T2D) [12]. However, clopidogrel combined with aspirin increased the total mortality compared to aspirin alone [13], suggesting that it would be better to use clopidogrel alone.
Clopidogrel belongs structurally to the thienopyridine group of antiplatelet medications and specifically inhibits the ADP-dependent platelet aggregation and adhesion via inhibition of the purinergic P2Y12 receptor. A series of studies demonstrated the anti-fibrotic properties of clopidogrel exerted through the TGF-β pathway. Clopidogrel attenuated the renal cortical fibrin deposition of rats which underwent five-sixths nephrectomy and effectively prevented the early progression of renal injury [14]. Clopidogrel decreased TGF-β mRNA expression levels in mice which had received orthotopic tracheal transplantation [15].
Nevertheless, the possible protective effect of clopidogrel on renal fibrosis in diabetes has not yet been studied, which prompted us to conduct our present investigation. Accordingly, in this study, we aimed to elucidate whether treatment with clopidogrel can reduce diabetes-induced renal fibrosis in a murine model and, if so, whether inhibition of FN is involved in this protective response.
2. Materials and methods
2.1 Animals
13-week-old C57BL/6J male mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). The mice were housed at the University of Louisville Research Resources Center and maintained at a temperature of 22 °C with a 12-hour light/dark cycle for free access to food and water. All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Louisville, which is certified by the American Association for Accreditation of Laboratory Animal Care. The experiments were conducted in compliance with the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health (NIH publication, revised 2011).
2.2 Establishment of a T1D mouse model and treatment with clopidogrel
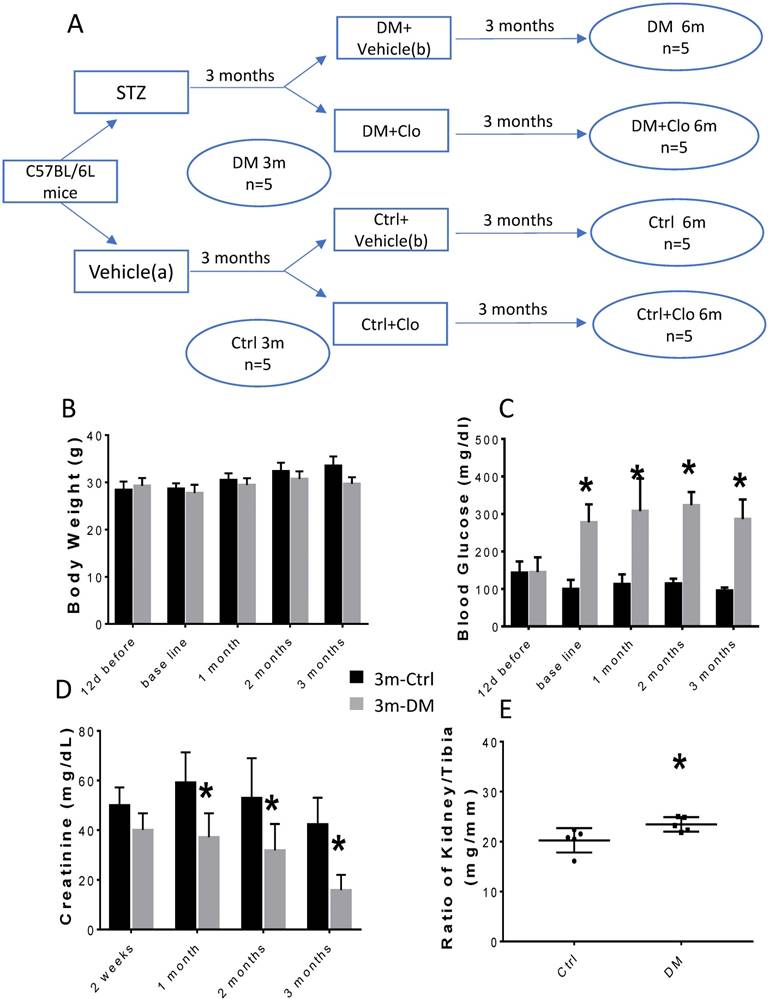
As outlined in Fig. 1A, after 1 week of acclimation, 15 mice were injected I.P. with streptozotocin (STZ) (Sigma-Aldrich, St. Louis, MO, USA) that was freshly dissolved in 0.1 mol/L of citrate acid buffer, pH 4.3-4.5 at a dosage of 55 mg/kg body weight daily for five consecutive days. Additional 15 mice as controls (Ctrl) were injected with a vehicle solution (0.1 mol/L citrate acid buffer, pH 4.3-4.5). Seven days after the last STZ administration, hyperglycemic mice (3-hour fasting blood glucose ≥250 mg/dL) were considered T1D (DM). This time point was defined as a baseline. The mice were raised with free access to food and water all along the protocol. Three months after diabetes induction, five diabetic and five control mice were sacrificed and blood and kidneys harvested. The remaining animals were divided in four groups (Fig. 1A): Normal control with vehicle (Ctrl), Normal control with clopidogrel (Ctrl+ Clo), T1D (DM) with vehicle, and DM with clopidogrel treatment (DM+ Clo) and were treated with 20 mg/kg b.w./day clopidogrel (MedChemExpress, Monmount Junction, NJ, USA) or with vehicle administered in their drinking water for three additional months. At the end of experiment, mice were intraperitoneally anesthetized with Avertin (tribromoethanol, 350 mg/kg) and sacrificed to collect blood and kidneys for mRNA, protein, and histological analyses.
Experimental design and characteristic evaluation of the T1D mouse model at three months. (A) Seven days after STZ injection, mice with hyperglycemia (fasting blood glucose levels ≥ 250 mg/dL) were defined as diabetic (DM). This time point was defined as the baseline. Five DM and five age-matched vehicle injected controls were sacrificed at three months. The remaining DM or Ctrl mice were treated with the vehicle (b) or clopidogrel (Clo) for three additional months. (B) Body weight, (C) fasting blood glucose, (D) urine creatinine, (E) ratio kidney weight/tibia length were determined at three months diabetes. (n = 5 in each group, * p < 0.05, Ctrl vs DM).
2.3 Urine creatinine
Mouse spot urine was collected at each time point using bladder palpation and stored at -80 °C until analysis. Creatinine concentration was measured by specific quantitative colorimetric assay, according to the manufacturer's protocol (BioAssay Systems, Hayward, CA, USA).
2.4 Blood Clotting Time
Blood clotting time was measured by mouse tail vein bleeding assay. In brief, using a surgical blade, the mouse tail was cut 1-2 mm away from the tip; the tail diameter was approximately 1 mm, the tail was immediately dipped in a 50-mL falcon tube filled with 37 °C saline and the bleeding time was recorded for a maximum of 5 min (300 s).
2.5 Histology and immunohistochemical staining
The collected kidneys were fixed in 10% formalin, dehydrated in graded alcohol series, cleared with xylene, embedded in paraffin, and sectioned at a thickness of 5 μm. Kidney sections were stained with hematoxylin and eosin (H&E). Sirius red staining was used to examine collagen deposition for kidney fibrosis as previously described [16]. The proportion of fibrosis (collagen) was quantitated using a Nikon Eclipse E600 microscopy system (Tokyo, Japan).
Standard immunohistochemical (IHC) staining was performed as previously described [17]. For the IHC staining, kidney tissue sections were stained with TGF-β, collagen I, collagen IV, laminin, and FN. The sections were developed with a DAB (3,3-diaminobenzidine) developing system (Vector Laboratories, Inc., Burlingame, CA, USA).
2.6 Western blot analysis
Western blots were performed according to the protocols previously described in detail [16]. The concentrations of primary antibodies varied from 1:1,000 to 1:3,000. The following primary antibodies were used: against the connective tissue growth factor (CTGF), β-Actin (Santa Cruz Biotechnology, Dallas, TX, USA); phosphor-extracellular signal-regulated kinases (p-ERK), total-ERK (t-ERK), phospho-phosphatidylinositide 3-kinases (p-PI3K), total-PI3K (t-PI3K), phospho-AKT (p-AKT), total-AKT (t-AKT), phosphor- P38 mitogen-activated protein kinases (p-p38), total-p38 (t-p38), phosphor-c-Jun N-terminal kinase (p-JNK), total-JNK (t-JNK), p-Smad2, Smad4 (Cell Signaling Technology, Beverly, MA, USA); and FN, TGF-β, collagen I, collagen IV, laminin, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Abcam, Cambridge, MA, USA), Smad2/3 (BD Bioscience, San Jose, CA, USA). The expression levels of the proteins were normalized to that of β-actin or GAPDH, which were used as internal controls.
2.7 RNA isolation and real-time PCR
RNA isolation and real-time PCR were performed according to protocols previously described in detail [16]. In brief, total mRNA from kidney tissue was extracted using the TRIzol reagent (RNA STAT 60 Tel-Test; Ambion, Austin, TX, USA). cDNA was used in the specific assays by Real-Time PCR system (Life Technologies Corporation, Carlsbad, CA, USA) on demand target mixes (Applied Biosystems, Foster City, CA, USA). The expression levels of mRNA encoding for TGF-β, FN, collagen I, and collagen IV in the kidney tissue were determined and normalized to the level of GAPDH (Applied Biosystems). The cycle times (Ct) were then compared to determine the fold differences between samples.
2.8 Statistical analysis
Data were presented as mean ± standard deviation (SD) values (n = 5 per group). Multiple comparisons of data obtained for the different groups were performed by one-way analysis of variance (ANOVA), followed by Tukey's test in pairwise repetitive comparisons. All statistical analyses and graphing were conducted using the software GraphPad Prism 7 (GraphPad Software Inc., San Diego, CA, USA). A p value< 0.05 was considered statistically significant.
3. Results
3.1 Characteristics of the experimental model
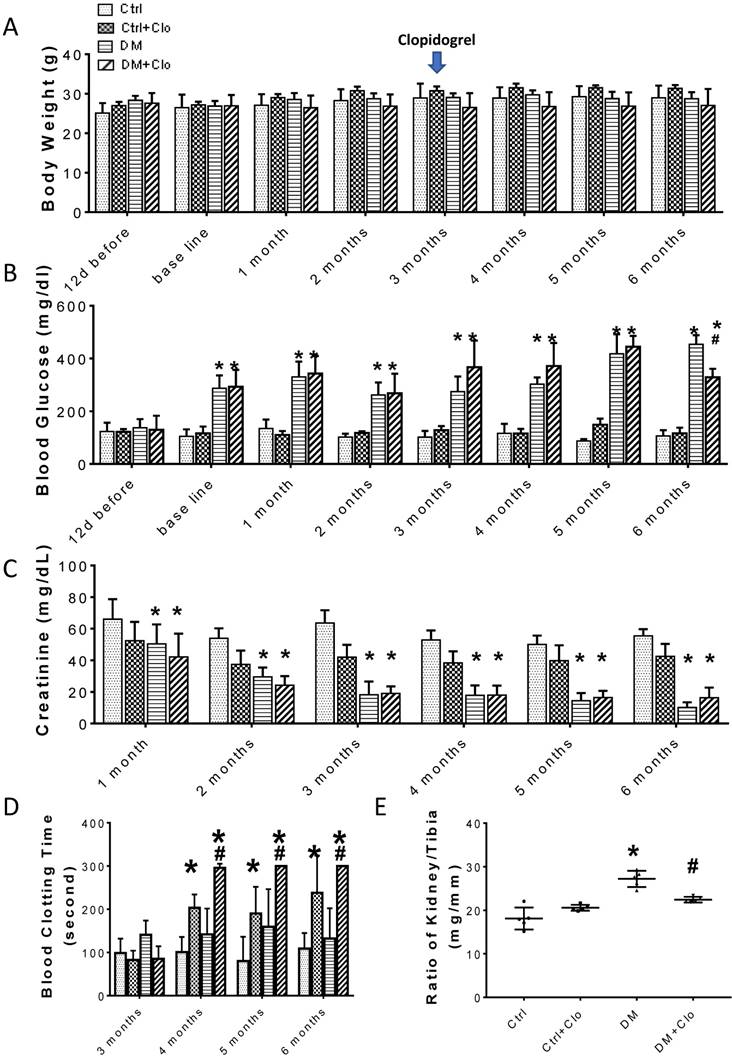
The experimental model is depicted in Figure 1A. The body weight of the experimental animals remained unchanged (Figs. 1B; 2A), whereas, the urine creatinine levels significantly decreased as the disease progressed (Figs. 1D; 2C). It is noteworthy that in DM, the blood glucose levels (Figs. 1C; 2B) and the kidney weight/tibia length ratio (Figs. 1E; 2E) were higher than those of the Ctrl group for the three-six-month course of the experiments.
3.2 Kidney fibrosis in T1D mice
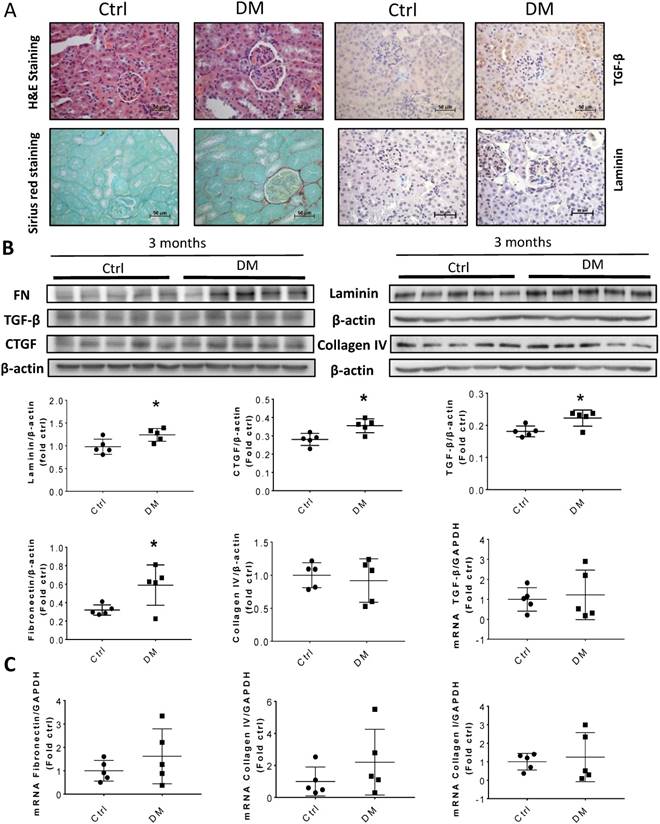
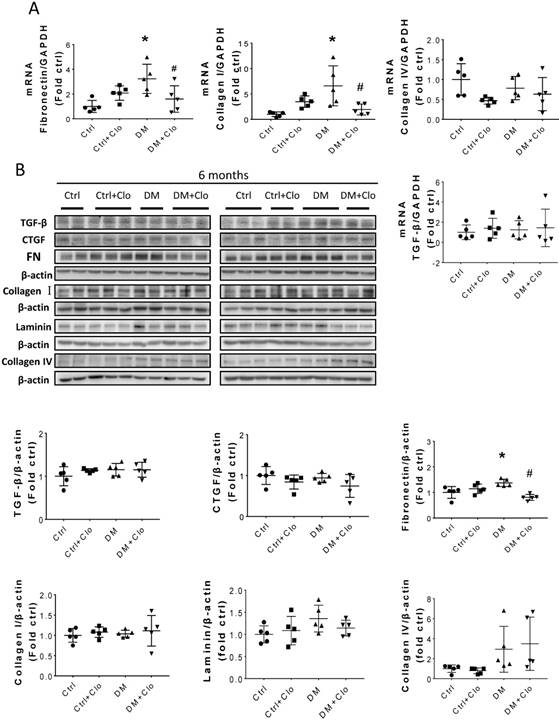
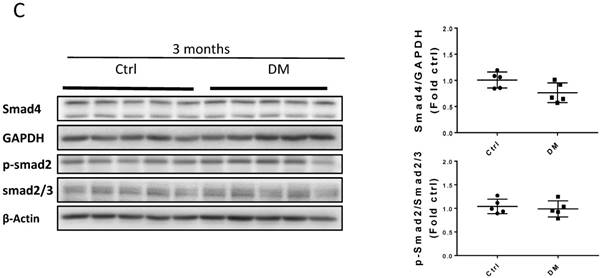
Sirius Red staining and glomerular diameter, as quantified by H&E staining, significantly increased in DM mice compared to those of the controls at three months (Fig. 3A) and higher at six months (Fig. 4A). Immunohistochemistry and Western blotting results at three months revealed that the renal expression of TGF-β in T1D mice and laminin (Fig. 3A) increased concomitantly with the elevation of CTGF and FN levels (Fig. 3B). However, the expression of FN mRNA, as well as the levels of Collagen I and TGF-β mRNA remained unchanged at three months, as determined by determined by real-time PCR (RT-PCR) (Fig. 3C). Collagen IV did not change significantly at both the mRNA level (Fig. 3C) and protein level (Fig. 3B) at three months. In contrast, the levels of FN protein and mRNA in T1D kidneys significantly increased at six months, as established by Western blotting and RT-PCR (Figs. 5A &B). laminin, another important component of ECM, has an increasing trend in the DM group (Fig. 5B). Collagen I mRNA expression in T1D kidneys also increased at six months DM, whereas the expression of CTGF at that point did not differ from that of the control (Fig. 5A). However, TGF-β and collagen IV mRNA (Fig. 5A) and protein (Fig. 5B) levels did not change significantly at six months.
Kidney fibrosis in T1D mice at three months. (A) Representative H&E (top right two panels), Sirius red staining (bottom right two panels) and representative immunohistochemical staining for TGF-β (top left two panels) and laminin (bottom left two panels) (original magnification, ×40); (B) The protein levels of FN, TGF-β, collagen IV, laminin and CTGF were measured by Western blot and quantified; (C) mRNA levels of fibrosis markers, including TGF-β, FN, collagen I, and collagen IV were examined by RT-PCR, and quantified. Data are represented as mean ± SD (n = 5 in each group, * p < 0.05, Ctrl vs DM).
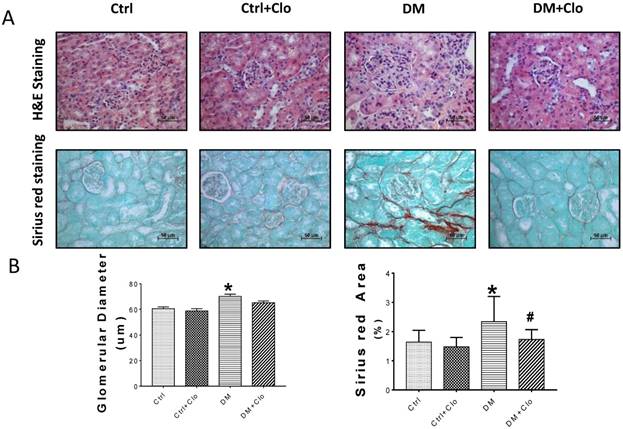
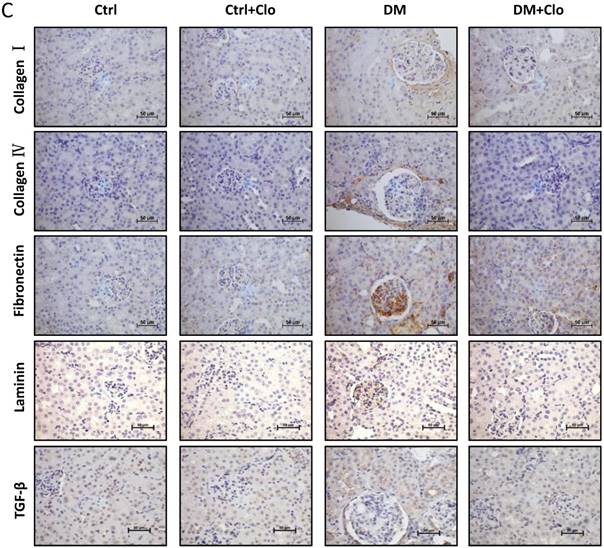
Effect of clopidogrel on T1D-induced kidney fibrosis at six months. (A) Representative images of kidney tissue H&E (upper panel) and Sirius red staining (lower panel) of control (Ctl) and diabetic mice (DM) with or without clopidogrel treatment (Ctl + Clo & DM + Clo), (B) Semi-quantification of the percentage of Sirius red positive area, and glomerular diameter; (C) Representative immunohistochemical staining for FN, collagen I collagen IV and laminin (original magnification, ×20). (n = 5 in each group, *p < 0.05, vs Ctrl; #p < 0.05, vs DM).
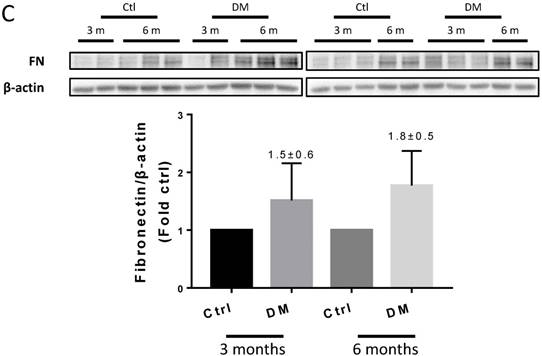
Hyperglycemia induced renal fibrosis in T1D mice at six months. (A) Fibrosis markers mRNA, including TGF-β, FN, collagen I, and collagen IV were examined by RT-PCR and quantified; (B) Protein levels of TGF-β, CTGF, collagen I, collagen IV, laminin and FN were measured by Western blot and quantified. (C) FN expression of Ctrl and T1D mice measured by Western blot and quantification analysis at three and six months. Data are represented as mean ± SD (n = 5 in each group, * p < 0.05, vs Ctrl; #p < 0.05, vs DM).
3.3 Clopidogrel ameliorated hyperglycemia-induced renal remodeling
The clopidogrel treatment started three months after the onset of diabetes, as illustrated in Fig. 1A, when T1D-induced kidney injury was established in DM mice. Treatment efficiency was confirmed by a bleeding test (Fig. 2D), which showed an increase in the bleeding time (>300) in the clopidogrel-treated animals. The clopidogrel treatment did not affect the body weight (Fig. 2A) or urine creatinine levels (Fig. 2C); however, it significantly decreased the blood glucose (Fig. 2B) and kidney tibia ratio (Fig. 2E).
Time-course of biochemical and physical characteristics of the four groups. (A) Body weight, (B) blood Glucose, (C) urine creatinine, (D) blood clotting time, (E) ratio kidney weight/tibia length, were determined six months diabetes. (n = 5 in each group, * p< 0.05, vs Ctrl; #p < 0.05, vs DM).
3.4 Clopidogrel markedly ameliorated hyperglycemia-induced renal fibrosis,
T1D significantly altered the renal function and morphology, decreasing the urinary creatinine and increasing the glomerular diameter and collagen interstitial deposition (Figs. 3 A & 4A, B). Immunohistochemical staining for laminin, collagen I, collagen IV, and FN revealed noticeable increases in the expression of these proteins in T1D at six months, which were attenuated by the clopidogrel treatment during the last three months of the disease (Fig. 4C). RT-PCR analysis of the T1D kidney tissue showed significantly elevated mRNA levels of ECM molecules, such as FN and collagen I, which were reduced by the clopidogrel treatment (Fig. 5A). Western blot analysis confirmed that the T1D-induced increase in the FN expression at six months was decreased by the clopidogrel treatment (Fig 5B), in agreement with our immunochemistry and RT-PCR results (Fig. 4C & 5A). Fibronectin levels increased progressively over time with disease progression, as established by Western blotting analysis quantitation. There was a 1.5-fold increase at three months in T1D and a 1.8-fold increase at six months vs. their respective controls (Fig. 5C). However, despite elevated fibrotic markers, there were no notable differences in the mRNA levels of TGF-β, collagen IV and protein levels of TGF-β, CTGF, laminin, collagen IV and collagen I among all experimental groups at six months (Fig. 5A, B).
3.5 Signaling pathways in the diabetic kidney that may mediate increased FN expression and fibrosis
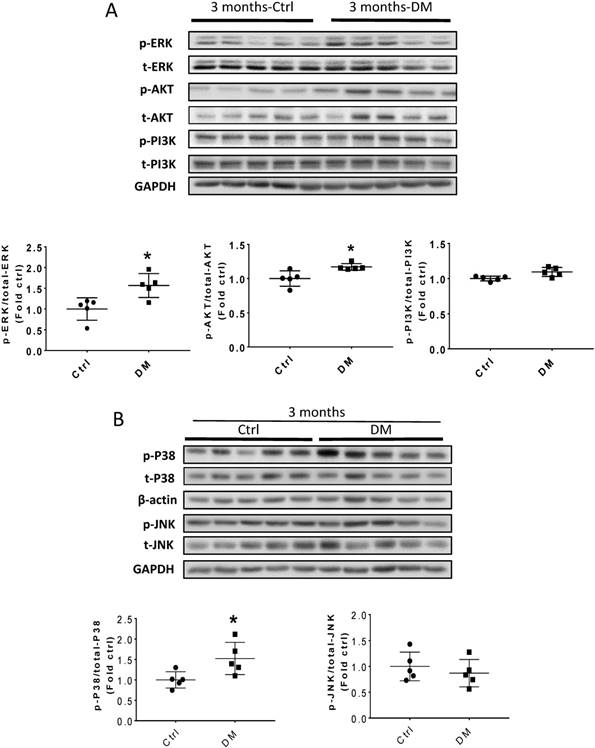
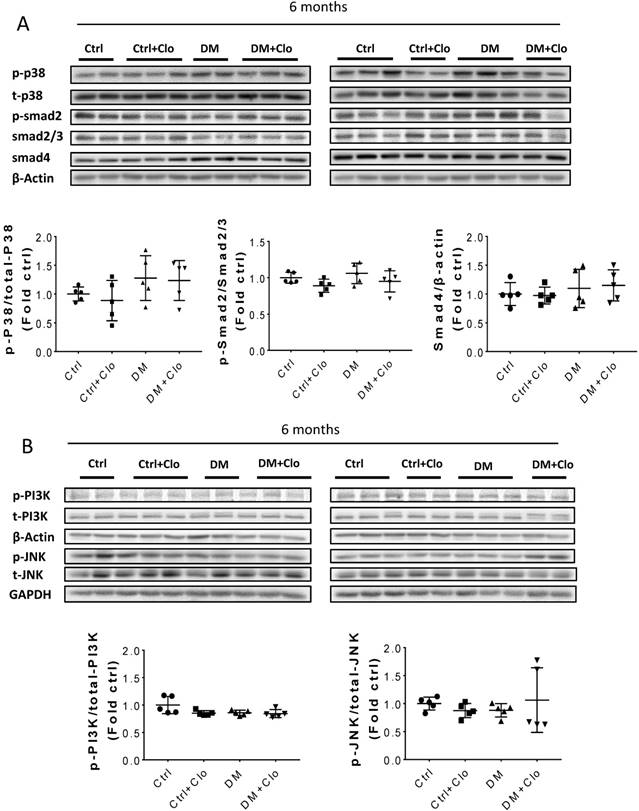
Erk, Akt, and p38 but not JNK signaling pathways were activated at three months T1D (Fig 6A, B). As profibrotic TGF-β expression increased, we investigated whether its downstream pathways, mediated by Smad proteins, were activated to promote FN expression and fibrosis. Western blotting analysis showed that Smad 2 and 4 were not activated (Fig 6C), suggesting that TGF-β fibrogenic activity in the T1D kidney is mediated by Smad-independent pathways. Interestingly, early activation of Erk, Akt, and p38 signaling, as well as pre-fibrotic CTGF and TGF-β levels seen at three months (Fig. 6A, B), returned to the control levels at six months (Fig. 7A, B). JNK and Smad proteins remained inactivated despite the further increase in FN and collagen expression (Fig. 7A, B).
TGF-β-induced pathways and downstream target protein were activated in the kidney after three months T1D. Western blot analysis and quantification of (A) the ratios p-ERK/t-ERK, p-AKT/t-AKT, and p-PI3K/t-PI3K; (B) the ratios p-p38/t-p38 and p-JNK/t-JNK; (C) and the ratios p-Smad2/Smad2/3, and Smad4. Data represented as mean ± SD (n = 5 in each groups, *p < 0.05, vs Ctrl).
TGF-β downstream pathways at six months T1D and effect of clopidogrel. (A) Western blot analysis and quantification of the ratio of p-p38 to t-p38, of p-Smad2 to Smad2/3, and Smad4; (B) Western blot analysis and quantification of the ratio of p-PI3K to t-PI3K and the ratio of p-JNK to t-JNK. Data are presented as mean ± SD (n = 5 in each group), *p < 0.05, vs Ctrl; #p < 0.05, vs DM.
4. Discussion
To date, increasing evidence indicates that FN acts as a marker for the development of renal fibrosis. Here, we provide consistent evidence of T1D-induced renal fibrosis in a time-dependent manner from three to six months after diabetes onset. Platelet inhibition by clopidogrel treatment for the last three months was effective in reducing renal FN accumulation and fibrosis. The novel findings described in our murine T1D model may uncover novel therapeutic strategies that can decrease diabetic renal complications. In addition, this study advanced our knowledge in the following areas and aspects: (1) We describe the morphological and biochemical changes in the kidney of long-term diabetic mice. While most studies reported findings obtained for the period of up to 13-15 weeks after disease onset, our study examines the changes after a period of six months after diabetes onset; (2) We show that the induction of the profibrotic proteins CTGF and TGF-β and their upstream signaling occurs at three-month T1D as also shown in other studies, however being early markers of fibrosis their expression does not persist at the six month time point. However, the fibrogenic alterations in the kidneys continues to progress; (3) We are the first to uncover a role for clopidogrel in the reduction of blood glucose levels and diabetes-induced renal fibrosis in a T1D mouse model; (4) Clopidogrel may decrease FN expression and kidney fibrosis by lowering blood sugar levels or by an independent mechanism.
Renal hypertrophy has been considered a typical feature of renal early damage induced by hyperglycemia in STZ-induced T1D, which may result from glomerular hypertrophy and mesangial matrix overproduction [18]. Kidney hypertrophy, shortly after the onset of diabetes, has been confirmed by a number of other studies [19] and was associated with a poor renal prognosis in patients affected by T1D [20]. Our results concerning diabetes-induced renal morphological changes are in agreement with the evidence mentioned above. Glomerular hypertrophy (represented by the increased glomerular diameter; Fig. 3A and Fig. 4A&B) and kidney hypertrophy (evidenced by increased kidney weight/ tibia length; Fig. 1E and Fig. 2E) progressed from three months to six months after T1D onset and was significantly attenuated by clopidogrel treatment for the last three months of the six-month T1D model.
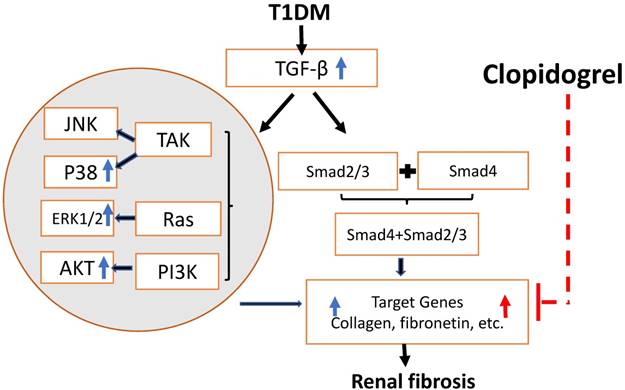
The progressive accumulation of extracellular matrix (ECM) components led to kidney fibrosis. Previously, we found that kidney fibrosis developed in various diabetes mouse models such as both T1D mouse models (STZ-induced [21] and the genetic model in OVE mice) [22], and even in a T2D model of C57BL/6J mice fed with high-fat diet inducing insulin resistance followed by STZ injection inducing mild hyperglycemia [23]. Considerable evidence indicates that collagen accumulation and FN deposition increase in the ECM; FN and collagen increased significantly in diabetes-induced renal fibrosis [4], [24]. Here, we confirmed that the increase in FN accumulation was accompanied with renal fibrosis and progressed in a time-dependent manner and showed that FN at the six-month's time point was significantly higher than that at the three-month's time point (Fig. 5C). The inhibition of Collagen and FN expression reportedly alleviated kidney fibrosis [25]. Our study found that clopidogrel treatment during the last three months significantly inhibited T1D-induced FN expression along with T1D-induced kidney fibrosis, as outlined in Fig. 8.
Schematic representation of the mechanisms of T1D-induced renal fibrosis three and six months after the onset of T1D and the effect of clopidogrel. T1D-increased expression of the TGF-β can induce renal fibrosis via Smad-dependent and independent pathways. In the present studies, we demonstrated that T1D increases TGF-β along with increases in phospho-ERK, phospho-AKT and phospho-p38 expressions at three months diabetes only, TGF-β and renal fibrosis (collagens and FN) at both three months (blue arrowa) and six-months (red arrows). Clopidogrel treatment of DM mice at three months and on, markedly reduced renal fibrosis levels by down-regulating FN and collagen at six months.
Platelets exert pathogenic effects in various CV events, including diabetic CV events [26]. Hyperglycemia has been established to activate platelets in DM patients [27-29]. Therefore, low-dose aspirin has been recommended for primary and secondary prevention of CV events in diabetic patients [26]. However, no effective prevention of CV events by the administration of low-dose aspirin has been shown earlier, which might have been caused by the existence of resistance [11]. Therefore, owing to its effectiveness in the prevention of CV events and improvement of the survival of diabetic patients, clopidogrel may be able to successfully replace aspirin as an attractive therapeutic option in this respect [11]. Evidence revealed that clopidogrel treatment contributed to a greater improvement in the renal function and the inflammatory burden than that with aspirin [30, 31]. However, no report from a clinical study has so far elucidated whether clopidogrel also attenuates renal pathogenesis in T1D either preventively or therapeutically. Using a T1D mouse model, we provide here the first preclinical evidence to confirm that the three-month clopidogrel treatment at a relatively late stage of diabetes in mice with mild renal dysfunction resulted in a significant amelioration of renal fibrosis and reduced FN accumulation as assumed in Fig. 8.
As previously reported, clopidogrel is likely to attenuate renal pathogenesis from different etiologies through inhibition of the TGF-β-mediated fibrotic signaling pathways [32]. Cell cultures, animal models, and clinical studies, provided evidence that TGF-β is a primary mediator of the hyperglycemia-induced fibrosis in diabetic renal injuries [33]. Reportedly, TGF-β mediates fibrotic pathogenesis via Smad-dependent and -independent pathways. TGF-β Smad-independent fibrotic signaling may be mediated by its activation of the MEK/Erk, Rho-like GTPases, and p38/mitogen-activated protein kinase (MAPK) in different tissues under different pathogenic conditions [34]. The activation of extracellular-regulated kinases (ERK) and p38 MAPK was also required for the collagen synthesis and accumulation under certain conditions [36, 37]. However, until now, studies on the TGF pathway have been conducted mainly in vitro, and its underlying mechanism in animals has not yet been completely understood [38]. In our T1D model, we observed an increase in the expression of TGF-β, and ERK, and P-38 MAPK phosphorylation, that may contribute to the development of renal fibrosis through the Smad-independent pathway, and is consistent with the findings reported in the above-mentioned studies [35, 36]. Our results suggest that the Smad-independent signaling activated by TGF-β at the early stage of T1D mice may play an important role in the development of progressive renal fibrosis, determined by increased FN and collagen levels. Nevertheless, this TGF-β -mediated signaling pathway did not subside at six months despite the further progression of the disease, as proposed in Fig. 8. Therefore, additional mechanisms may mediate long-term progression of the disease. Although most studies implicate TGF-β in T1D-induced renal fibrosis, in agreement with our three months findings, longer term data are not available and our study uniquely examined the disease at six months. While the three months clopidogrel treatment clearly attenuates the progression of diabetes-induced renal fibrosis in diabetic mice. , the mechanisms underlying this beneficial effect will require further investigation in our future studies. The effect of clopidogrel treatment starting at the onset of diabetes rather than three months later, and its impact on TGF-β-mediated Smad-independent fibrotic signaling will be investigated in future. Based on previous reports of clopidogrel anti-fibrotic activity via TGF-β [13, 14], administering the treatment at an earlier stage of the disease might have prevented the induction of TGF-β and FN expression and may have further decreased the negative renal consequences of T1D in our model. Clopidogrel may act by preventing platelet-induced vascular injury and oxidative stress, as hyper-reactive platelets in diabetic patients contribute to the pathology by not only triggering thrombus formation and microcapillary embolization, but also releasing constrictive, oxidative, inflammatory, and mitogenic substances [39]. In addition, clopidogrel may reduce fibrosis by systemically improving diabetic conditions. For instance, the present study showed a reduction in the glucose level at the last month of clopidogrel treatment (Fig. 3B). All these assumed hypotheses are summarized in Fig. 8, but remain to be examined in our future work.
In conclusion, we report novel findings specifically implicating clopidogrel-induced platelet inhibition in the improvement of diabetes-induced renal fibrosis. Our current results indicate that diabetes-induced renal fibrosis, accompanied by a significant increase in FN accumulation, can be substantially reduced by clopidogrel treatment for the last three months of the six-month of our T1D model. Although the mechanisms involved in the therapeutic role of clopidogrel in T1D-induced renal fibrotic pathogenesis remain to be elucidated, the potential for clinical application of this commonly used medicine is significant (Fig. 8). Our findings also raise the question whether administering the treatment earlier, closer to the time of diagnosis of diabetes, may prevent the onset of renal complications in diabetic patients.
Abbreviations
Clo: Clopidogrel; Col I: collagen I; Col IV: collagen IV; CTGF: connective tissue growth factor; CV: cardiovascular; ECM: extracellular matrix; FN: fibronectin; H&E: hematoxylin and eosin; p-AKT: phospho-AKT; t-AKT: total-AKT; p-ERK: phosphor-extracellular signal-regulated kinases; t-ERK: total-ERK; p-JNK: phosphor-c-Jun N-terminal kinase; t-JNK: total-JNK; p-p38: phosphor- P38 mitogen-activated protein kinases; t-p38: total-p38; p-PI3K: phospho-phosphatidylinositide 3-kinases; t-PI3K: total-PI3K; RT-PCR: real-time PCR; T1D: type 1 diabetes; T2D: type 2 diabetes; TGF-β: transforming growth factor-beta.
Acknowledgements
This work was supported in part by grants from the National Science Foundation of China (81400725 to W.S.; 81700635 to Y.C.), and the University of Louisville School of Medicine (Collaborative Matching Grant, E.G.).
Author contributions
Z.Y.Z., Y.W.F., E.G. and L.C. originally designed the project. Z.Y.Z. and T.J.M. performed experiments, analyzed data, and wrote the manuscript draft. X.L., W.Y.W, X.M.L, W.X.S., M.R. and Y.L.C. performed partial experiments and data collection. L.C. was responsible for scientific review and manuscript editing. Y.W.F. and E.G. monitored the project progression, modified the experimental designs and manuscript revision. All authors approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Control CfD, Prevention. National diabetes statistics report, 2017. Atlanta, GA: Centers for Disease Control and Prevention, US Dept of Health and Human Services. 2017
2. Murphy D, McCulloch CE, Lin F, Banerjee T, Bragg-Gresham JL, Eberhardt MS. et al. Trends in Prevalence of Chronic Kidney Disease in the United States. Ann Intern Med. 2016;165:473-81
3. Kato M, Park JT, Natarajan R. MicroRNAs and the glomerulus. Exp Cell Res. 2012;318:993-1000
4. Kolset SO, Reinholt FP, Jenssen T. Diabetic nephropathy and extracellular matrix. J Histochem Cytochem. 2012;60:976-86
5. Miller CG, Pozzi A, Zent R, Schwarzbauer JE. Effects of high glucose on integrin activity and fibronectin matrix assembly by mesangial cells. Molecular biology of the cell. 2014;25:2342-50
6. Klemis V, Ghura H, Federico G, Wurfel C, Bentmann A, Gretz N. et al. Circulating fibronectin contributes to mesangial expansion in a murine model of type 1 diabetes. Kidney Int. 2017;91:1374-85
7. Fogo AB. Progression and potential regression of glomerulosclerosis. Kidney Int. 2001;59:804-19
8. Kagami S, Kuhara T, Yasutomo K, Okada K, Löster K, Reutter W. et al. Transforming growth factor-β (TGF-β) stimulates the expression of β1 integrins and adhesion by rat mesangial cells. Experimental cell research. 1996;229:1-6
9. Hess K. The vulnerable blood. Hämostaseologie. 2014;35:25-33
10. American Diabetes A. Standards of medical care in diabetes-2014. Diabetes Care. 2014;37(Suppl 1):S14-80
11. Bell DS. Aspirin in the prevention of cardiovascular events in patients with diabetes. Postgrad Med. 2016;128:180-90
12. Milionis H, Ntaios G, Papavasileiou V, Spengos K, Manios E, Elisaf M. et al. Aspirin Versus Clopidogrel for Type 2 Diabetic Patients with First-Ever Noncardioembolic Acute Ischemic Stroke: Ten-Year Survival Data from the Athens Stroke Outcome Project. J Stroke Cerebrovasc Dis. 2017;26:2769-77
13. Dasgupta A, Steinhubl SR, Bhatt DL, Berger PB, Shao M, Mak KH. et al. Clinical outcomes of patients with diabetic nephropathy randomized to clopidogrel plus aspirin versus aspirin alone (a post hoc analysis of the clopidogrel for high atherothrombotic risk and ischemic stabilization, management, and avoidance [CHARISMA] trial). Am J Cardiol. 2009;103:1359-63
14. Tu X, Chen X, Xie Y, Shi S, Wang J, Chen Y. et al. Anti-inflammatory Renoprotective Effect of Clopidogrel and Irbesartan in Chronic Renal Injury. Journal of the American Society of Nephrology: JASN. 2008;19:77-83
15. Preidl RH, Eckl S, Ramsperger-Gleixner M, Koch N, Spriewald BM, Weyand M. et al. Clopidogrel reduces post-transplant obliterative bronchiolitis. Transpl Int. 2013;26:1038-48
16. Cheng Y, Zhang J, Guo W, Li F, Sun W, Chen J. et al. Up-regulation of Nrf2 is involved in FGF21-mediated fenofibrate protection against type 1 diabetic nephropathy. Free Radic Biol Med. 2016;93:94-109
17. Luo M, Luo P, Zhang Z, Payne K, Watson S, Wu H. et al. Zinc delays the progression of obesity-related glomerulopathy in mice via down-regulating P38 MAPK-mediated inflammation. Obesity (Silver Spring). 2016;24:1244-56
18. Thibodeau JF, Holterman CE, Burger D, Read NC, Reudelhuber TL, Kennedy CR. A novel mouse model of advanced diabetic kidney disease. PLoS One. 2014;9:e113459
19. Lawson ML, Sochett EB, Chait PG, Balfe JW, Daneman D. Effect of puberty on markers of glomerular hypertrophy and hypertension in IDDM. Diabetes. 1996;45:51-5
20. Rigalleau V, Garcia M, Lasseur C, Laurent F, Montaudon M, Raffaitin C. et al. Large kidneys predict poor renal outcome in subjects with diabetes and chronic kidney disease. BMC Nephrol. 2010;11:3
21. Sun W, Wang Y, Miao X, Wang Y, Zhang L, Xin Y. et al. Renal improvement by zinc in diabetic mice is associated with glucose metabolism signaling mediated by metallothionein and Akt, but not Akt2. Free Radic Biol Med. 2014;68:22-34
22. Zheng S, Coventry S, Cai L, Powell DW, Jala VR, Haribabu B. et al. Renal Protection by Genetic Deletion of the Atypical Chemokine Receptor ACKR2 in Diabetic OVE Mice. J Diabetes Res. 2016;2016:5362506
23. Wu H, Kong L, Cheng Y, Zhang Z, Wang Y, Luo M. et al. Metallothionein plays a prominent role in the prevention of diabetic nephropathy by sulforaphane via up-regulation of Nrf2. Free Radic Biol Med. 2015;89:431-42
24. Ma Y, Chen F, Yang S, Chen B, Shi J. Protocatechuic acid ameliorates high glucose-induced extracellular matrix accumulation in diabetic nephropathy. Biomed Pharmacother. 2018;98:18-22
25. Xue HY, Yuan L, Cao YJ, Fan YP, Chen XL, Huang XZ. Resveratrol ameliorates renal injury in spontaneously hypertensive rats by inhibiting renal micro-inflammation. Biosci Rep. 2016:36
26. Ferreiro JL, Angiolillo DJ. Diabetes and antiplatelet therapy in acute coronary syndrome. Circulation. 2011;123:798-813
27. Schneider DJ. Factors contributing to increased platelet reactivity in people with diabetes. Diabetes care. 2009;32:525-7
28. Vaidyula VR, Boden G, Rao AK. Platelet and monocyte activation by hyperglycemia and hyperinsulinemia in healthy subjects. Platelets. 2006;17:577-85
29. Gasparyan AY, Watson T, Lip GY. The role of aspirin in cardiovascular prevention: implications of aspirin resistance. J Am Coll Cardiol. 2008;51:1829-43
30. Bhatt DL, Marso SP. et al. Amplified Benefit of Clopidogrel Versus Aspirin in Patients With Diabetes Mellitus. The American Journal of Cardiology. 2002;90(6):625-8
31. Dash A, Maiti R, Bandakkanavar TK, Bhaskar A, Prakash J, Pandey BL. Prophylactic Add-on Antiplatelet Therapy in Chronic Kidney Disease With Type 2 Diabetes Mellitus: Comparison Between Clopidogrel and Low-dose Aspirin. Int J Prev Med. 2013;4:902-10
32. Jia LX, Qi GM, Liu O, Li TT, Yang M, Cui W. et al. Inhibition of platelet activation by clopidogrel prevents hypertension-induced cardiac inflammation and fibrosis. Cardiovasc Drugs Ther. 2013;27:521-30
33. Hoffman BB, Sharma K, Zhu Y, Ziyadeh FN. Transcriptional activation of transforming growth factor-beta1 in mesangial cell culture by high glucose concentration. Kidney Int. 1998;54:1107-16
34. Papageorgis P, Stylianopoulos T. Role of TGFbeta in regulation of the tumor microenvironment and drug delivery (review). Int J Oncol. 2015;46:933-43
35. Kattla JJ, Carew RM, Heljic M, Godson C, Brazil DP. Protein kinase B/Akt activity is involved in renal TGF-beta1-driven epithelial-mesenchymal transition in vitro and in vivo. Am J Physiol Renal Physiol. 2008;295:F215-25
36. Caraci F, Gili E, Calafiore M, Failla M, La Rosa C, Crimi N. et al. TGF-beta1 targets the GSK-3beta/beta-catenin pathway via ERK activation in the transition of human lung fibroblasts into myofibroblasts. Pharmacol Res. 2008;57:274-82
37. Rodrıguez-Barbero A, Obreo J, Yuste L, Montero J, Rodrı́guez-Peña A, Pandiella A. et al. Transforming growth factor-β1 induces collagen synthesis and accumulation via p38 mitogen-activated protein kinase (MAPK) pathway in cultured L6E9 myoblasts. FEBS letters. 2002;513:282-8
38. Kim SI, Choi ME. TGF-beta-activated kinase-1: New insights into the mechanism of TGF-beta signaling and kidney disease. Kidney Res Clin Pract. 2012;31:94-105
39. Kakouros N, Rade JJ, Kourliouros A, Resar JR. Platelet function in patients with diabetes mellitus: from a theoretical to a practical perspective. Int J Endocrinol. 2011;2011:742719
Author contact
![]() Corresponding author: Dr. Yaowen Fu, the Department of Urology, The First Hospital of Jilin University, No.71 Xinmin Street, Changchun 130021, China, Phone: +86 13756661667. Email: fuyaowencom.cn Or Dr. Evelyne Gozal, Pediatric Research Institute, University of Louisville, 570 South Preston Street, Baxter I, Suite 321C, Louisville, KY 40202, USA, Phone: +1 502-852-2213, Fax: +1 502-852-5634. Email: evelyne.gozaledu
Corresponding author: Dr. Yaowen Fu, the Department of Urology, The First Hospital of Jilin University, No.71 Xinmin Street, Changchun 130021, China, Phone: +86 13756661667. Email: fuyaowencom.cn Or Dr. Evelyne Gozal, Pediatric Research Institute, University of Louisville, 570 South Preston Street, Baxter I, Suite 321C, Louisville, KY 40202, USA, Phone: +1 502-852-2213, Fax: +1 502-852-5634. Email: evelyne.gozaledu