Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results and Discussion
Conclusion
Materials and Methods
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(2):341-350. doi:10.7150/ijbs.28904 This issue Cite
Short Research Communication
UDP-glucose Dehydrogenase: The First-step Oxidation Is an NAD+-dependent Bimolecular Nucleophilic Substitution Reaction (SN2)
Jun Chen, Yang Yu, Jiaojiao Gao, Shulin Yang ![]()
School of Environmental & Biological Engineering, Nanjing University of Science and Technology, Xiaolingwei 200, Nanjing, China, 210094
Received 2018-7-31; Accepted 2018-11-11; Published 2019-1-1
Abstract

UDP-glucose dehydrogenase (UGDH) catalyzes the conversion of UDP-glucose to UDP-glucuronic acid by NAD+-dependent two-fold oxidation. Despite extensive investigation into the catalytic mechanism of UGDH, the previously proposed mechanisms regarding the first-step oxidation are somewhat controversial and inconsistent with some biochemical evidence, which instead supports a mechanism involving an NAD+-dependent bimolecular nucleophilic substitution (SN2) reaction. To verify this speculation, the essential Cys residue of Streptococcus zooepidemicus UGDH (SzUGDH) was changed to an Ala residue, and the resulting Cys260Ala mutant and SzUGDH were then co-expressed in vivo via a single-crossover homologous recombination method. Contrary to the previously proposed mechanisms, which predict the formation of the capsular polysaccharide hyaluronan, the resulting strain instead produced an amide derivative of hyaluronan, as validated via proteinase K digestion, ninhydrin reaction, FT-IR and NMR. This result is compatible with the NAD+-dependent SN2 mechanism.
Keywords: bimolecular nucleophilic substitution reaction (SN2), catalytic mechanism, NAD+-dependent, two-fold oxidation, UDP-glucose dehydrogenase
Introduction
UDP-glucose dehydrogenase (UGDH; EC 1.1.1.22) catalyzes the NAD+-dependent two-fold oxidation of UDP-glucose to UDP-glucuronic acid (UDP-GlcUA), which was originally reported by Strominger and collaborators more than sixty years ago [1]. UGDH is required for the formation of the zebrafish cardiac valve [2], the synthesis of hemicellulose and pectin precursors that are present in newly formed plant cell walls [3,4] and multiple functions in pathogenic bacteria [5]. UDP-GlcUA is a major precursor of many polysaccharides, especially glycosaminoglycans, e.g., hyaluronan (HA), heparin, chondroitin sulfate, and dermatan sulfate. These polysaccharides play important roles in metabolism [6-14] and are promising materials for tissue engineering [15,16]. The significance of UGDH and these polysaccharides necessitates resolving the currently controversial mechanism of the first-step oxidation.
The oxidation of a free alcohol to a free acid generally occurs through an aldehyde intermediate. Nelsestuen and Kirkwood discovered that bovine UGDH catalyzes not only the oxidation of UDP-α-D-gluco-hexodialdose (UDP-Glc-6-CHO, the corresponding aldehyde of UDP-glucose) to UDP-GlcUA but also the reduction of the aldehyde to UDP-glucose [17], resulting in their proposal that UDP-Glc-6-CHO acts as an intermediate throughout the UGDH catalytic pathway.
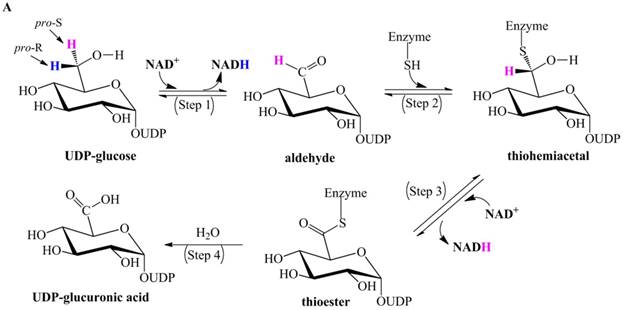
Ridley et al. proposed a classic four-step catalytic mechanism (Figure 1A) [18]: (i) the first step generates the aldehyde UDP-Glc-6-CHO and NADH via transfer of pro-R hydride to NAD+; (ii) UDP-Glc-6-CHO is attacked by the nucleophile Cys to generate a thiohemiacetal intermediate, resulting in covalent binding of the substrate to the enzyme; (iii) the thiohemiacetal is oxidized to a thioester intermediate, coupled to the formation of a second NADH; and (iv) the last step is the irreversible hydrolysis of the thioester, yielding UDP-GlcUA.
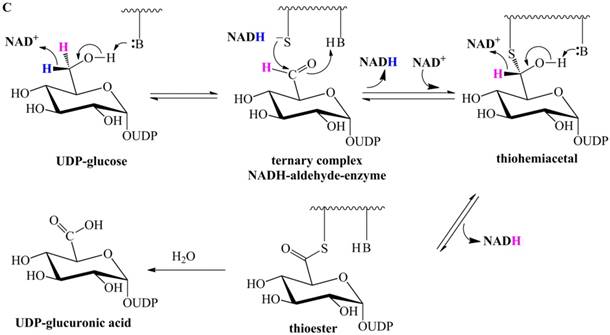
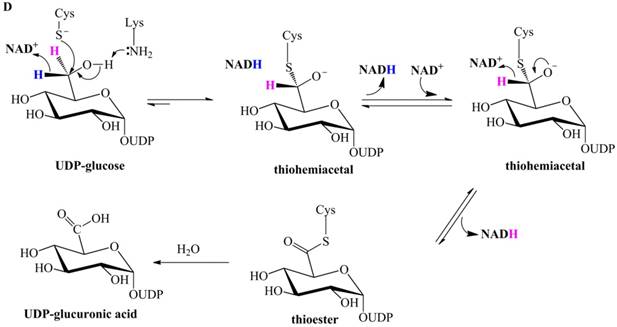
Previously proposed mechanisms of UGDH. (A) A four-step catalytic mechanism of UGDH. First, pro-R H is abstracted by the first NAD+ to produce UDP-Glc-6-CHO and NADH. Second, an essential Cys is added to the aldehyde to produce a thiohemiacetal intermediate, covalently binding the substrate to UGDH. Third, the second NAD+ removes pro-S H, generating a thioester and NADH. These three steps are reversible. The last step is the irreversible hydrolysis of the thioester to yield UDP-GlcUA. (B) Mechanism in which UDP-Glc-6-CHO is not generated. UDP-glucose is attacked by Lys to generate a Schiff base intermediate, which is concomitantly attacked by a nearby Cys residue to generate a thiohemiacetal. The second oxidation and hydrolysis of the thioester are similar to those in the mechanism described by Ridley et al. [18]. (C) An improved version of the mechanism reported by Ridley et al. [18]. Wavy lines indicate peptides containing the necessary active amino acid residues. A general base (-B:) serves as the acceptor of the hydroxyl proton. UDP-glucose is oxidized to UDP-Glc-6-CHO via hydride transfer, but the aldehyde forms a ternary complex with NADH and the enzyme. The nucleophilic addition of Cys-S- destroys this complex to generate a thiohemiacetal, accompanied by the release of the first NADH. The following two steps are similar to those in the mechanism described by Ridley et al. [18]. (D). One mechanism in which the incipient aldehyde is trapped by the Cys thiolate. The first oxidation generates UDP-Glc-6-CHO via hydride transfer, which is kinetically coupled to the addition of the deprotonated Cys276. The subsequent steps are similar to those in the mechanism described by Ge et al. [19].
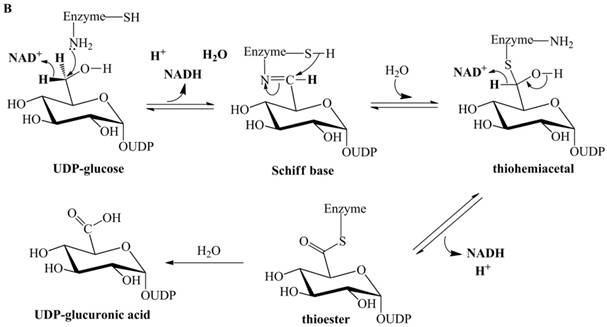
UGDH accepts UDP-Glc-6-CHO and catalyzes its oxidation to UDP-GlcUA [17], but UDP-Glc-6-CHO is neither released into solution nor derivatized by carbonyl-derivatizing agents [1]. A seemingly simple explanation for this finding is that the aldehyde is covalently bound to the enzyme in a thiohemiacetal form (as described in Figure 1A). However, UGDH still does not release UDP-Glc-6-CHO after the essential thiol group is derivatized with cyanide to prevent the formation of the thiohemiacetal [18]. This finding suggests that the aldehyde should not act as an intermediate; therefore, Ordman and Kirkwood proposed a mechanism [20] (Figure 1B) in which UDP-glucose is directly oxidized to a Schiff base intermediate. The Schiff base is hydrolyzed to the thiohemiacetal intermediate through concomitant attack by a nearby Cys. The remaining steps are similar to those in the mechanism proposed by Ridley et al. [18].
Biochemical evidence supports the possibility that Streptococcus pyogenes UGDH (SpUGDH) and bovine UGDH share an identical catalytic pathway [21-24]. Ge et al. performed oxidation of UDP-glucose, catalyzed by SpUGDH in an H218O solvent [19]. Only a single 18O atom was incorporated into UDP-GlcUA. However, the mechanism proposed by Ordman and Kirkwood [20] (Figure 1B) indicates that both oxygens of the carboxyl group in UDP-GlcUA should be isotopically labeled.
To explain the failure to observe UDP-Glc-6-CHO in the absence of the essential Cys, Ge et al. proposed that UDP-Glc-6-CHO forms the ternary complex NADH-aldehyde-Cys260Ala, such that it is slowly released and not readily hydrated [19]. Unless otherwise stated, the numbering is according to the sequence of SpUGDH. Accordingly, Ge et al. [19] proposed a mechanism (Figure 1C) in which the first oxidation gives rise to the ternary complex, whereas the following steps are similar to those of the previously suggested mechanism (Figure 1A). Note that the aldehyde can be reduced by bovine UGDH in the absence of the essential Cys [18], consistent with the fact that UDP-Glc-6-CHO can be readily reduced by Cys260Ala of SpUGDH in the presence of NADH [19]. Therefore, once NADH and UDP-Glc-6-CHO are located at the center of Cys260Ala, UDP-Glc-6-CHO will be immediately reduced to UDP-glucose. i.e., this putative ternary complex [19] does not exist. As a result, this ternary-complex hypothesis cannot explain the failure to observe the aldehyde in the absence of the essential Cys.
Subsequently, Egger et al. investigated human UGDH using multiple methods and proposed that the oxidation of UDP-glucose to the thiohemiacetal occurs via a hydride transfer kinetically coupled to the nucleophilic attack from the Cys-S- (Figure 1D) [25,26]. This mechanism supports the notion that the aldehyde is generated in the first-step oxidation and then immediately trapped by the deprotonated Cys276 (homologous to Cys260 of SpUGDH) to generate the thiohemiacetal, i.e., Egger et al. merged together the classic two steps proposed by Ridley et al. [18]. Likewise, this mechanism cannot explain the failure to detect UDP-Glc-6-CHO in the absence of the essential Cys.
These previously proposed catalytic mechanisms of UGDH (Figure 1) diverge mainly in the first oxidation step, with the subsequent steps being nearly identical and consistent with biochemical evidence. However, the first-step oxidation mechanism remains controversial and ambiguous. Kinetic studies indicate that UGDH from mammals [18,27-29], humans [25,26], bacteria [21,22,24,30] and plants [31,32] catalyzes an identical reaction, i.e., NAD+-dependent two-fold oxidation. It is easier to obtain UGDH from a bacterium (e.g., Streptococcus) than from eukaryotes (e.g., bovine, human, plant); moreover, the corresponding characters of a bacterium are readily observable when UGDH is studied in vivo. Streptococcus zooepidemicus was thus chosen in this study to investigate the mechanism of UGDH.
Results and Discussion
A single-crossover homologous recombination strain is distinct from the starting strain
Hyaluronan (HA) is a naturally occurring, linear polysaccharide that consists of repeating disaccharide units of N-acetyl-D-glucosamine (GlcNAc) and D-glucuronic acid (GlcUA) connected via alternating β(1→3) and β(1→4) glycosidic linkages. HA is synthesized as an extracellular capsule by S. zooepidemicus via the has operon [33]. Briefly, UGDH of S. zooepidemicus (SzUGDH) releases UDP-GlcUA into the cytoplasm, and then HA synthase (EC 2.4.1.212) embedded in the cytomembrane incorporates the activated sugars UDP-GlcUA and UDP-N-acetyl glucosamine (UDP-GlcNAc) to generate HA, which is then released into solution (Figure S1). The product of SzUGDH (i.e., UDP-GlcUA) is a substrate of HA synthase, thereby facilitating in vivo investigation into the mechanism of the first-step oxidation of UDP-glucose.
The essential nucleophilic Cys residue (at the 260th site) of SzUGDH was mutated to Ala via site-directed mutagenesis of the hasB gene (encoding the native SzUGDH) to hasB* (encoding the Cys260Ala mutant) based on an erroneous-matching PCR method [34]. Then, the recombined plasmid pKSV7-hasB* was transformed into S. zooepidemicus (labeled as S-1) via electroporation [35]. The colonies of the resulting strain (labeled as S-2) were distinct from those of S-1 (Figure S2). A single-crossover homologous recombination strain (S-3), in which hasB* is incorporated into the genomic DNA, was screened out (Figure S2) to better purify and characterize the new product released by HA synthase. The difference between S-1 and S-3 is that S-3 harbors the C260A mutant (note that C260A represents the Cys260Ala mutant of SzUGDH in the following text).
The proposed mechanisms (Figure 1A and 1D) and the postulated ternary complex (Figure 1C) support the production of UDP-Glc-6-CHO, which should be halted in the C260A mutant [19], and the mechanism (Figure 1B) indicates that a Schiff base should be covalently bound to C260A. According to these mechanisms (Figure 1), S-3 should secrete HA because C260A can neither release the putative intermediate (the aldehyde or the Schiff base) nor accept any other substrates. However, the colony characteristics among S-1, S-2 and S-3 were strikingly distinct (Figure S2), and the cultivation medium of S-1 was significantly clearer than that of S-2 or S-3 (Figure S3). These differences indicate that the C260A mutant in S-3 releases a derivative of UDP-GlcUA, and this derivative is subsequently incorporated by HA synthase to generate an HA derivative (denoted by HAd). HAd is then released from the inner surface of the cytomembrane into the cultivation medium, resulting in the noteworthy turbidity of S-3. Is it possible that UDP-Glc-6-CHO or the Schiff base, as proposed in the previous mechanisms (Figure 1), reacts with other reagents in the center of C260A to give rise to HAd?
If this is the case, the potential reagent for a -CHO is either H2O molecules or the primary amines from the heart extract powder and peptone in the Todd-Hewitt broth medium, whereas the putative Schiff base is a stable intermediate and therefore does not react with ingredients from the medium. It was assumed that peptone and heart extract are totally composed of amino acids and oligopeptides, which belong to the primary amine family, and their average molecular mass was considered to be 75 g/mol (corresponding to the lowest-molecular-weight amino acid, Gly), so the molar concentration of primary amines was 0.3 M. The nucleophilic form (:NH2R) was less than 1/10 that of the total forms (:RNH2 and RNH3+) when pH was 8, due to the protonated amino pKa (approx. 9~10). Therefore, the H2O concentration (55.56 M) was 1800-fold greater than the concentration of the nucleophile (:NH2R), i.e., UDP-Glc-6-CHO (if present) should be virtually completely surrounded by H2O molecules and, thus, inaccessible to primary amines. Moreover, UDP-Glc-6-CHO is intrinsically readily hydrated [23] and the hydrated form is rapidly oxidized to UDP-GlcUA [22], whereas the addition of a primary amine to an aldehyde is slow and requires slightly acidic catalysis conditions (pH 4~5). If UDP-Glc-6-CHO were to be generated in C260A, it would be hydrated and then oxidized to UDP-GlcUA, i.e., the product of S-3 should still be HA. This observation is compatible with the failed trapping of UDP-Glc-6-CHO [36] because H2O molecules essentially act as carbonyl-derivatizing agents. The resulting HAd cannot be explained by these previous mechanisms (Figure 1), suggesting a new mechanism for UGDH.
A new mechanism for UGDH is suggested based on existing experimental evidence
(i) The first oxidation should involve the nucleophilic addition of the essential Cys
In the absence of the essential nucleophile Cys thiolate, little oxidation of UDP-glucose by UGDH occurs, regardless of whether the source is bovine [18], human [26] or bacterial [22]; moreover, the putative intermediates (UDP-Glc-6-CHO and the first NADH) are barely detectable [19,26,36]. Therefore, the essential thiolate is strongly suggested to be involved in the first oxidation. Furthermore, this hypothesis has been demonstrated to be reasonable by Egger et al. using a stopped-flow kinetic method [26], although their proposal is inconsistent with the failed detection of the aldehyde in the absence of the essential Cys.
(ii) The first oxidation should not involve the classic hydride transfer
An analog of UDP-glucose, UDP-6S-6C-methylglucose (i.e., pro-S H substituted with a -CH3, Figure S4), can be used to verify whether the classic H-transfer (i.e., transfer of pro-R H and C6-OH proton to NAD+ and the proton acceptor, respectively) occurs during the first oxidation because the group at the pro-S site is not involved in the first oxidation. If the classic H-transfer occurs in the first oxidation, a corresponding carbonyl intermediate (a ketone, in this case) will be readily detected (Figure S4). However, the oxidation rate of this analog by SpUGDH was found to be 10,000-fold slower than that of UDP-glucose oxidation [21], indicating that hydride transfer is unlikely to occur in the first oxidation.
(iii) The acceptor of C6-OH of UDP-glucose should be an ordered H2O
Campbell et al. revealed the binding interactions between SpUGDH and UDP-glucose using a crystallographic method and proposed that the acceptor of C6-OH is either an ordered H2O (H-bonded to Thr118, Figure S5A) or an unprotonated Lys-NH2 (Figure S5B) [30]. A primary kinetic isotope effect is observed during the oxidation of UDP-glucose by the Thr118Ala (Thr118 replaced by Ala) mutant of SpUGDH [19], suggesting that Thr118 is involved in this oxidation, thus supporting the hydride transfer pathway in which this ordered H2O molecule serves as the acceptor of C6-OH (Figure S5A). Moreover, this speculation is consistent with a theoretical computation [37] indicating that Lys204 is protonated to stabilize the oxygen rather than to act as the proton acceptor.
(iv) The second NADH should be released after hydrolysis of the thioester intermediate
Incubation of the Cys276Ser mutant of human UGDH with UDP-glucose in the presence of NAD+ results in a covalent bond between Ser276 and acylated UDP-GlcUA [26], demonstrating that the net 4-electron oxidation has been completed, i.e., the second NADH can be generated by Cys276Ser. Cys276Ser has been independently validated to release only one NADH [26,38]; therefore, the second NADH is still in the center of the mutant. The thioester intermediate in the native UGDH is hydrolyzed to produce the UDP-GlcUA product; thus, the second NADH should be released after the hydrolysis of the thioester to enable the second round of catalysis. Note that the Cys260Ser mutant of SpUGDH also forms an ester with UDP-GlcUA [22]; hence, this release order is presumably applicable to SpUGDH.
(v) An NAD+-dependent bimolecular nucleophilic substitution (SN2) reaction is suggested for the first-step oxidation of UGDH
As the first oxidation is expected to involve the nucleophilic addition of Cys-S‒ rather than the classic H-transfer, the nucleophilic addition of Cys-S‒ and the departure of pro-R H (in the form of NADH) should be coupled, i.e., the breakage of the old bond between pro-R H and C-6 is synchronous with the formation of the new bond between Cys-S and C-6. Note that the leaving group (pro-R H) cannot depart all alone, but is abstracted with the 'help' of NAD+. The first oxidation is thus an NAD+-dependent bimolecular nucleophilic substitution (SN2) reaction, generating thiohemiacetal intermediate 2 via a transition state (Figure 2).
The new catalytic mechanism of UGDH. The nucleophilic addition of Cys-S- and the removal of pro-R H (in NADH form) are simultaneous in the first oxidation, giving rise to thiohemiacetal 2. This step is essentially an NAD+-dependent SN2 reaction. Thiohemiacetal 2 is then oxidized to thioester 3 through hydride transfer. The hydrolysis of 3 generates UDP-GlcUA 4, which is followed by the release of NADH and 4 to the solution.
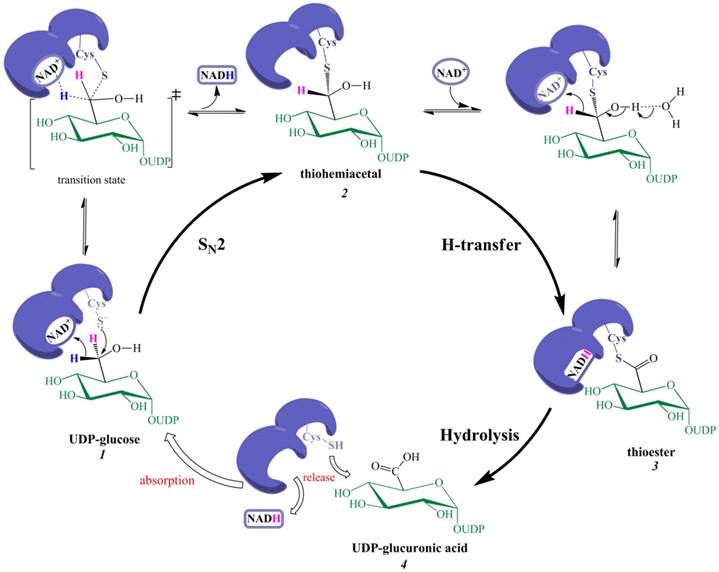
For the second oxidation, the classic H-transfer is reasonable because the hydroxyl proton of a thiohemiacetal (similar to a gem-diol or aldehydrol) is easier to remove than an alcoholic hydroxyl proton; e.g., the hydrated form of acetaldehyde is present at the high ratio of approximately 50%, and the solution is slightly acidic, indicating that removal of an aldehydrol proton by H2O is intrinsically feasible. Moreover, the removal of pro-R H (by NAD+) and abstraction of the C6‒OH proton are mechanistically synergetic, thereby favoring the occurrence of the second oxidation.
A single-step NAD+-assisted SN2 reaction consequently replaces the putative two steps (the first hydride transfer and the nucleophilic addition), whereas the remaining steps are not revised except that the second NADH is released after the hydrolysis of the thioester 3.
Proteinase K digestion and ninhydrin reaction suggest that C260A released a product
The previous mechanisms (Figure 1) suggest that the S-3 strain should produce HA, and the deduced NAD+-dependent SN2 mechanism (Figure 2) shows that the first oxidation cannot occur in the absence of the essential nucleophile Cys thiolate, i.e., S-3 should produce HA. However, there are three main nucleophilic reagents (H2O, -OH, and NH2R) in this case. H2O molecules should be neglected due to their weak nucleophilic ability. The nucleophilic capacity of -OH (>:NH2R) is sufficient, and the resultant product is a hydrated aldehyde (Figure S6). The hydrated aldehyde is readily oxidized [23], but the concentration of -OH is too low (10-6 M when pH is 8), which is one of the reasons that the Cys260Ala mutant of SpUGDH [22] or the Cys276Ala mutant of human UGDH [26] catalyzes the two-fold oxidation exceedingly slowly. Therefore, the primary amine serves as a key nucleophilic reagent in the attack of C-6 of UDP-glucose. According to the NAD+-dependent SN2 mechanism, hydramine intermediate 2′ is generated in the first oxidation (Figure S7). The second oxidation (hydride transfer) can proceed favorably because the tetrahedron geometry at C-6 is similar to that of thiohemiacetal 3, which is generated by native UGDH according to all mechanisms including the new one. Finally, amide product 3′ is generated. The entire catalytic process of C260A does not involve covalent catalysis, as occurs in native UGDH, and 3′ (UDP-Glc-6-amide) could therefore be released from the mutant center upon formation, skipping the hydrolysis step, as occurs in native UGDH.
UDP-Glc-6-amide, UDP-GlcNAc and UDP-GlcUA are incorporated by HA synthase to yield the derivative HAd (Figure S8). The formation of HAd supports the SN2 mechanism and, thus, refutes those previously proposed mechanisms (Figure 1) in which the first oxidation generates UGD-Glc-6-CHO or a Schiff base. It can also be taken into consideration that primary amines are covalently bound to HA. The incorporation of primary amines into HA was verified based on the following findings: (1) S-3 medium became significantly clearer after proteinase K digestion for 12 h (Figure S3); (2) HAd powder isolated from S-3 medium could be digested by proteinase K, due to a striking increase of the solubility; and (3) the digested HAd solution showed a chromogenic reaction [39] with ninhydrin, whereas neither the controls (H2O or undigested HAd) nor the digested solution subjected to purification to remove small molecules (e.g., amino acid and oligopeptide) turned purple (Figure S9).
FT-IR and NMR spectroscopy suggest that C260A released an amide derivative of UDP-GlcUA
Proteinase K does not change the main-chain structure of HAd because it can only cut the peptide bonds of side chains, but it can significantly increase the solubility of HAd, making it possible to better characterize its structure. The 1H NMR spectrum (Figure S10) of digested HAd showed that there were two broad low-intensity peaks at 7.11 and 7.75 ppm (d6-DMSO as solvent). These peaks corresponded to two amide protons. The peak at 7.75 ppm should be assigned to the amide proton of CH3CONH‒ of the GlcNAc subunit [40], and the peak at 7.11 ppm suggested that a primary amine formed an amide bond with C-6 of UDP-glucose. It is difficult to precisely assign each peak in the spectrum because of the various primary amine species present (e.g., approximately 20 single amino acids and innumerable oligopeptides) and the random incorporation of the corresponding amide derivative (UDP-Glc-6-amide) into the backbone of HA by HA synthase. Nevertheless, the FT-IR spectrum of thoroughly digested HAd (denoted by HAd-K, Figure S11) strongly suggested that the linkage is an amide bond, as the benzylamide derivative of HA shows a highly similar broad peak at 1537 cm-1, which is attributed to bending in the NH plane (i.e., the amide band) [41]; moreover, the spectra of HAd and HAd-K are nearly identical, consistent with the above speculation that proteinase K does not change the main-chain structure of HAd.
Conclusion
This NAD+-dependent SN2 reaction indicates that UDP-Glc-6-CHO and the first NADH cannot be detected in the absence of the essential Cys-S- because the first oxidation does not occur, rather than because the already formed aldehyde and NADH cannot be released by Cys260Ala [19]. Moreover, it is compatible with the results of 18O incorporation experiments [19,28], as only one oxygen atom of the carboxylate group in the product UDP-GlcUA should originate from the solvent.
The C260A mutant releases an amide derivative of UDP-GlcUA (UDP-Glc-6 amide), which is incorporated into the backbone of HA, as validated by proteinase K digestion, ninhydrin reaction, FT-IR, and NMR. Therefore, the formation of HAd refutes the previously proposed mechanisms (Figure 1) and instead supports the new mechanism (Figure 2).
To the best of our knowledge, this NAD+-dependent SN2 mechanism has not been reported before, although many other enzymes are known to involve an SN2 reaction, e.g., haloalkane dehalogenase [42-45], uridine diphosphate glucuronosyltransferase [46], sorbitol dehydrogenase [47], and uridine phosphorylase [48-50]. These SN2 reactions are however independent of the cofactor NAD+.
UGDH belongs to the family of oxidoreductases that catalyze a net four-electron oxidation. The NAD+-dependent SN2 mechanism is probably also suitable for other members of this family, including histidinol dehydrogenase [51,52], UDP-ManNAc dehydrogenase [53], and GDP-mannose dehydrogenase [54,55].
Materials and Methods
Streptococcus zooepidemicus ATCC 39920 was purchased from the American Type Culture Collection (ATCC). Todd-Hewitt broth (g/l: peptone 20.0, heart extract powder 3.1, dextrose 2.0. sodium chloride 2.0, disodium hydrogen phosphate 0.4, and sodium bicarbonate 2.5, pH 7.8 ± 0.2) was purchased from Nissui Biotechnologies (Qingdao, China). Hyaluronan was purchased from FREDA (Ji'nan, China). The strains, plasmids and primers used in this study are listed in Table S1.
Site-directed mutagenesis of the hasB gene, which encodes UDP-glucose dehydrogenase
The genomic DNA of S. zooepidemicus ATCC 39920 was extracted according to the instructions from the Ezup Column Bacteria Genomic DNA Purification Kit (Sangon). The hasB gene was amplified with the primers hasB-F and hasB-R, digested with Kpn I and BamH I, and then ligated into the plasmid pUC57 to generate pUC57-hasB. Site-directed mutagenesis was conducted with a PCR-based method [34]. Briefly, pUC57-hasB was amplified as the template using a pair of mismatched primers (hasB*-F and hasB*-R). The mixture was then digested with Dpn I and transformed into competent DH5α cells. The resulting plasmid (pUC57-hasB*) was extracted from a positive clone. The hasB* gene, verified by sequencing (Sangon), was ligated into pKSV7 to generate pKSV7-hasB*.
Electrotransformation of pKSV7-hasB* into Streptococcus zooepidemicus ATCC 39920
Competent S. zooepidemicus was obtained based on a previous method [35]. A 100-µl aliquot of the bacterial suspension was blended with approximately 5 μg of DNA (pKSV7-hasB*) in a cuvette (2 mm). The mixture was subjected to a pulse of 12.5 kV/cm, 25 µF, and 200 Ω (time constant 4.8 ms). After electroporation, the suspension was transferred to 1 ml of fresh Todd-Hewitt medium and incubated on ice for 30 min. Then, it was cultured at 37 °C for 1 h. The suspension was spread on a Todd-Hewitt plate with 25 mg/l of chloramphenicol (Chl) at 37 °C for 48 h. The positive clone was labeled S-2.
Single-crossover homologous recombination
A 100 μl aliquot of a bacterial (S-2) culture was transferred to 20 ml of fresh Todd-Hewitt medium containing 5 mg/l of Chl. The solution was cultured at 42 °C. The medium became turbid after approximately 3 days, indicating that the single-crossover homologous recombination strain was successfully screened. This mixture was spread onto a plate with 5 mg/l of Chl, generating the targeted strain S-3.
Extraction of the HAd product from S-3 cultivation medium
The cultivation medium of S-3 was sterilized at 121 °C for 20 min, followed by centrifugation at 10, 000×g for 2 min. The supernatant was subsequently collected and mixed with three volumes of ethanol (3V). This mixture was placed at 4 °C for approximately 1 h and then centrifuged at 10,000×g for 2 min. The sediment was washed with 75% (V/V) ethanol twice, dried in air (or in an oven at 60 °C), and then resuspended with deionized water (1V). Heating (at 55 °C) and shaking were recommended. The supernatant was collected from the suspension and then mixed with ethanol (3V). After centrifugation at 10, 000×g for 5 min, the supernatant was collected again. Then, a moderate volume of saturated NaCl solution was added until the turbidity of the solution did not increase further. The turbid solution was stored at 4 °C for at least 1 h (or overnight). The sediment was collected after centrifuge at 10,000×g for 5 min and then dried in air.
Proteinase K digestion
Proteinase K (1 mg/ml) was dissolved in phosphate buffer (50 mM, pH 7.0). A 100 µl aliquot of proteinase K solution was added to 3 ml of S-3 medium with 10 mM urea. The medium became significantly clearer after incubation at 55 °C for 12 h. Additionally, 5 mg of HAd powder isolated from S-3 medium was suspended with 2 ml of H2O, and then a 20 µl aliquot of proteinase K solution was added into the suspension. The suspension became clear after incubation at 55 °C for approximately 5 min. The thoroughly digested solution (for 24 h) was then subjected to the ninhydrin reaction.
Ninhydrin chromogenic reaction
Chromogenic reactions were performed according to Lee et al. [39]. Briefly, 0.1 ml of reaction solution (or the control) was added into 1.9 ml of a ninhydrin-citrate-glycerol mixture containing 0.5 ml of 1% ninhydrin solution in 0.5 M citrate buffer (pH 5.5), 1.2 ml of glycerol, and 0.2 ml of 0.5 M citrate buffer (pH 5.5). The mixture was shaken sufficiently and then heated in a boiling water bath for 12 min. The reaction was stopped by cooling under flowing tap-water until it reached room temperature. The HAd suspension (2.5 mg/ml) was thoroughly digested by proteinase K. The digested HAd (labeled as HAd-K) was isolated from the digestion solution via ethanol precipitation (see the above extraction method) and then dissolved with water (1V). The resulting HAd-K solution, as one control, was subjected to the ninhydrin reaction.
FT-IR and NMR spectroscopy
The thoroughly digested HAd solution was subjected to ultrafiltration with a molecular weight cut-off of 3000 Da (Minipore) to remove small molecules. The retention was recovered and lyophilized. The resulting HAd-K, HAd, and HA were subjected to FT-IR (Nicolet iS10 spectrometer). HAd-K was dissolved with DMSO-d6 (approximately 5 mg/ml) and then subjected to 1H NMR on a Bruker AVANCE III 600 MHz spectrometer.
Abbreviations
Ala: alanine; Cys: cysteine; FT-IR: Fourier transform infrared spectrum; GDP: guanine diphosphate; HA: hyaluronan; Lys: lysine; NADH/ NAD+: reduced and oxidized form of nicotinamide-adenine dinucleotide, respectively; NMR: nuclear magnetic resonance; Ser: serine; SN2: biomolecular nucleophilic substitution reaction; Thr: threonine; UDP: uridine diphosphate; UGDH: UDP-glucose dehydrogenase.
Supplementary Material
Supplementary figures and table.
Acknowledgements
We thank Professor Heng'an Wang (Shanghai Jiao Tong University) for the gift of the pKSV7 plasmid.
Funding
Funding was provided by the National High Technology Research and Development Program of China (No: 2014AA022107) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (No: 2011837).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Strominger JL, Kalckar HM, Axelrod J. et al. Enzymatic oxidation of uridine diphosphate glucose to uridine diphosphate glucuronic acid. J Am Chem Soc. 1954;76:6411-6412
2. Walsh EC, Stainier DY. UDP-glucose dehydrogenase required for cardiac valve formation in zebrafish. Science. 2001;293:1670-1673
3. Johansson H, Sterky F, Amini B. et al. Molecular cloning and characterization of a cDNA encoding poplar UDP-glucose dehydrogenase, a key gene of hemicellulose/pectin formation. Biochim Biophys Acta. 2002;1576:53-58
4. Tenhaken R, Thulke O. Cloning of an enzyme that synthesizes a key nucleotide-sugar precursor of hemicellulose biosynthesis from soybean: UDP-glucose dehydrogenase. Plant Physiol. 1996;112:1127-1134
5. Griffith CL, Klutts JS, Zhang L. et al. UDP-glucose dehydrogenase plays multiple roles in the biology of the pathogenic fungus Cryptococcus neoformans. J Biol Chem. 2004;279:51669-51676
6. Hacker UT, Gerner FM, Büning H. et al. Standard heparin, low molecular weight heparin, low molecular weight heparinoid, and recombinant hirudin differ in their ability to inhibit transduction by recombinant adeno-associated virus type 2 vectors. Gene Ther. 2001;8:966-968
7. Jayson GC, Gallagher JT. Heparin oligosaccharides: inhibitors of the biological activity of bFGF on Caco-2 cells. Br J Cancer. 1997;75:9-16
8. Parish CR. The role of heparan sulphate in inflammation. Nature Reviews Immunology. 2006;6:633-643
9. Kitagawa H. Chondroitin sulfate is indispensable for pluripotency and differentiation of mouse embryonic stem cells. Sci Rep. 2014;4:3701-3714
10. Jung SH, Lee HC, Yu DM. et al. Heparan sulfation is essential for the prevention of cellular senescence. Cell Death Differ. 2015;23:417-429
11. Spinelli FM, Vitale DL, Demarchi G. et al. The immunological effect of hyaluronan in tumor angiogenesis. Clinical & Translational Immunology. 2015;4:e52
12. Rogerson SJ, Brown GV. Chondroitin sulphate A as an adherence receptor for Plasmodium falciparum infected erythrocytes. Parasitol Today. 1997;13:70-75
13. Turnbull J, Powell AS. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001;11:75-82
14. Bao X, Moseman EA, Saito H. et al. Endothelial heparan sulfate controls chemokine presentation in recruitment of lymphocytes and dendritic cells to lymph nodes. Immunity. 2010;33:817-829
15. Muzzarelli RA, Greco F, Busilacchi A. et al. Chitosan, hyaluronan and chondroitin sulfate in tissue engineering for cartilage regeneration: a review. Carbohydr Polym. 2012;89:723-739
16. Chen YL, Lee HP, Chan HY. et al. Composite chondroitin-6-sulfate/dermatan sulfate/chitosan scaffolds for cartilage tissue engineering. Biomaterials. 2007;28:2294-2305
17. Nelsestuen GL, Kirkwood S. The mechanism of action of uridine diphosphoglucose dehydrogenase: uridine diphosphohexodialdoses as intermediates. J Biol Chem. 1971;246:3828-3834
18. Ridley W, Houchins J, Kirkwood S. Mechanism of action of uridine diphosphoglucose dehydrogenase. Evidence for a second reversible dehydrogenation step involving an essential thiol group. J Biol Chem. 1975;250:8761-8767
19. Ge X, Penney LC, van de Rijn I. et al. Active site residues and mechanism of UDP-glucose dehydrogenase. Eur J Biochem. 2004;271:14-22
20. Ordman AB, Kirkwood S. Mechanism of action of uridine diphoglucose dehydrogenase. Evidence for an essential lysine residue at the active site. J Biol Chem. 1977;252:1320-1326
21. Campbell RE, Tanner ME. UDP-Glucose analogues as inhibitors and mechanistic probes of UDP-glucose dehydrogenase. J Org Chem. 1999;64:9487-9492
22. Ge X, Campbell RE, van de Rijn I. et al. Covalent adduct formation with a mutated enzyme: Evidence for a thioester intermediate in the reaction catalyzed by UDP-glucose dehydrogenase. J Am Chem Soc. 1998;120:6613-6614
23. Campbell RE, Tanner ME. Uridine diphospho-α-D-gluco-hexodialdose: synthesis and kinetic competence in the reaction catalyzed by UDP-glucose dehydrogenase. Angew Chem Int Ed. 1997;36:1520-1522
24. Campbell RE, Sala RF, van de Rijn I. et al. Properties and kinetic analysis of UDP-glucose dehydrogenase from group A Streptococci: irreversible inhibition by UDP-chloroacetol. J Biol Chem. 1997;272:3416-3422
25. Egger S, Chaikuad A, Klimacek M. et al. Structural and kinetic evidence that catalytic reaction of human UDP-glucose 6-dehydrogenase involves covalent thiohemiacetal and thioester enzyme intermediates. J Biol Chem. 2012;287:2119-2129
26. Egger S, Chaikuad A, Kavanagh KL. et al. Structure and mechanism of human UDP-glucose 6-dehydrogenase. J Biol Chem. 2011;286:23877-23887
27. Nelsestuen G, Kirkwood S. The mechanism of action of uridine diphosphoglucose dehydrogenase uridine diphosphohexodialdoses as intermediates. J Biol Chem. 1971;246:3828-3834
28. Schiller JG, Bowser AM, Feingold DS. Studies on the mechanism of action of UDP-D-glucose dehydrogenase from beef liver. II. Carbohydr Res. 1972;25:403-410
29. Ordman AB, Kirkwood S. Udpglucose dehydrogenase kinetics and their mechanistic implications. Biochim Biophys Acta. 1977;481:25-32
30. Campbell RE, Mosimann SC, van de Rijn I. et al. The first structure of UDP-glucose dehydrogenase reveals the catalytic residues necessary for the two-fold oxidation. Biochemistry. 2000;39:7012-7023
31. Turner W, Botha FC. Purification and kinetic properties of UDP-glucose dehydrogenase from sugarcane. Arch Biochem Biophys. 2002;407:209-216
32. Stewart DC, Copeland L. Uridine 5'-diphosphate-glucose dehydrogenase from soybean nodules. Plant Physiol. 1998;116:349-355
33. Blank LM, Hugenholtz P, Nielsen LK. Evolution of the hyaluronic acid synthesis (has) operon in Streptococcus zooepidemicus and other pathogenic streptococci. J Mol Evol. 2008;67:13-22
34. Liu H, Naismith JH. An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 2008;8:91-100
35. Zhang J, Hao N, Chen GQ. Effect of expressing polyhydroxybutyrate synthesis genes (phbCAB) in Streptococcus zooepidemicus on production of lactic acid and hyaluronic acid. Appl Microbiol Biotechnol. 2006;71:222-227
36. Strominger JL, Maxwell ES, Axelrod J. et al. Enzymatic formation of uridine diphosphoglucuronic acid. J Biol Chem. 1957;224:79-90
37. Huang W, Llano J, Gauld JW. A DFT study on the catalytic mechanism of UDP-glucose dehydrogenase. Can J Chem. 2010;88:804-814
38. Sommer BJ, Barycki JJ, Simpson MA. Characterization of human UDP-glucose dehydrogenase: CYS-276 is required for the second of two successive oxidations. J Biol Chem. 2004;279:23590-23596
39. Lee YP, Takahashi T. An improved colorimetric determination of amino acids with the use of ninhydrin. Anal Biochem. 1966;14:71-77
40. Scott JE, Heatley F, Moorcroft D. et al. Secondary structures of hyaluronate and chondroitin sulphates. A 1H n.m.r. study of NH signals in dimethyl sulphoxide solution. Biochem J. 1981;199:829-832
41. Bellini D, Topai A. Amides of hyaluronic acid and the derivatives thereof and a process for their preparation. United States Patent. 2013 US 8575129 B2
42. Hur S, Kahn K, Bruice TC. Comparison of formation of reactive conformers for the SN2 displacements by CH3CO2- in water and by Asp124-CO2- in a haloalkane dehalogenase. Proc Natl Acad Sci U S A. 2003;100:2215-2219
43. Olsson MH, Warshel A. Solute solvent dynamics and energetics in enzyme catalysis: the SN2 reaction of dehalogenase as a general benchmark. J Am Chem Soc. 2004;126:15167-15179
44. Devi-Kesavan LS, Gao J. Combined QM/MM study of the mechanism and kinetic isotope effect of the nucleophilic substitution reaction in haloalkane dehalogenase. J Am Chem Soc. 2003;125:1532-1540
45. Lightstone FC, Zheng YJ, Maulitz AH. et al. Non-enzymatic and enzymatic hydrolysis of alkyl halides: a haloalkane dehalogenation enzyme evolved to stabilize the gas-phase transition state of an SN2 displacement reaction. Proc Natl Acad Sci U S A. 1997;94:8417-8420
46. Yin H, Bennett G, Jones JP. Mechanistic studies of uridine diphosphate glucuronosyltransferase. Chem Biol Interact. 1994;90:47-58
47. Lindstad RI, Mckinley-Mckee JS. Stereo-selective affinity labelling of sheep liver sorbitol dehydrogenase by chloro-substituted analogues of 2-bromo-3-(5-imidazolyl)propionic acid. Biochim Biophys Acta. 1996;1293:267-271
48. Komissarov AA, Moltchan OK, Romanova DV. et al. Enzyme-catalyzed uridine phosphorolysis: SN2 mechanism with phosphate activation by desolvation. FEBS Lett. 1994;355:192-194
49. Silva RG, Vetticatt MJ, Merino EF. et al. Transition-state analysis of Trypanosoma cruzi uridine phosphorylase-catalyzed arsenolysis of uridine. J Am Chem Soc. 2011;133:9923-9931
50. Silva RG, Schramm VL. Uridine phosphorylase from Trypanosoma cruzi: kinetic and chemical mechanisms. Biochemistry. 2011;50:9158-9166
51. Adams E. The enzymatic synthesis of histidine from histidinol. J Biol Chem. 1954;209:829-846
52. Adams E. L-Histidinal, a biosynthetic precursor of histidine. J Biol Chem. 1955;217:325-344
53. Kawamura T, Ishimoto N, Ito E. Enzymatic synthesis of uridine diphosphate N-acetyl-D-mannosaminuronic acid. J Biol Chem. 1979;254:8457-8465
54. Snook CF, Tipton PA, Beamer LJ. Crystal structure of GDP-mannose dehydrogenase: a key enzyme of alginate biosynthesis in P. aeruginosa. Biochemistry. 2003;42:4658-4668
55. Naught LE, Gilbert S, Imhoff R. et al. Allosterism and cooperativity in Pseudomonas aeruginosa GDP-mannose dehydrogenase. Biochemistry. 2002;41:9637-9645
Author contact
![]() Corresponding author: Tel/Fax: +86-25-84315945; E-mail: yshulinedu.cn.
Corresponding author: Tel/Fax: +86-25-84315945; E-mail: yshulinedu.cn.