Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Natural history of NAFLD
Adipose tissue in the...
NAFLD and cancer: the...
Conclusion
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(3):610-616. doi:10.7150/ijbs.29599 This issue Cite
Review
Obesity, Nonalcoholic Fatty Liver Disease and Adipocytokines Network in Promotion of Cancer
Rosa Divella1, Antonio Mazzocca2, Antonella Daniele1, Carlo Sabbà2, Angelo Paradiso3 ![]()
1. Department of Clinical Pathology Laboratory. IRCCS - Istituto Tumori Giovanni Paolo II, Viale Orazio Flacco 65, 70124 Bari, Italy
2. Interdisciplinary Department of Medicine, University of Bari School of Medicine, Piazza G. Cesare, 11, 70124 Bari, Italy
3. Experimental Medical Oncology, IRCCS - Istituto Tumori Giovanni Paolo II, Viale Orazio Flacco 65, 70124 Bari, Italy
Received 2018-8-30; Accepted 2018-10-27; Published 2019-1-1
Abstract

Western populations are becoming increasingly sedentary and the incidence of nonalcoholic fatty liver disease (NAFLD) is increasing and becoming one of the most common causes of liver disease worldwide. Also, NAFLD is considered one the new emerging risk factors for development of tumors of the gastro-intestinal tract, particularly hepatocellular carcinoma (HCC). Visceral obesity is an important risk factor for the onset of NAFLD. An accumulation of ectopic fat, including visceral obesity and fatty liver leads to a dysfunction of the adipose tissue with impaired production of adipocytokines which, in turn, favor an increase in pro-inflammatory cytokines. In this review, we discuss how the obesity-related chronic state of low-grade inflammation and the presence of NAFLD lead to the emergence of a microenvironment favorable to the development of cancer.
Keywords: NAFLD, adipocytokines, inflammation, obesity, cancer
Introduction
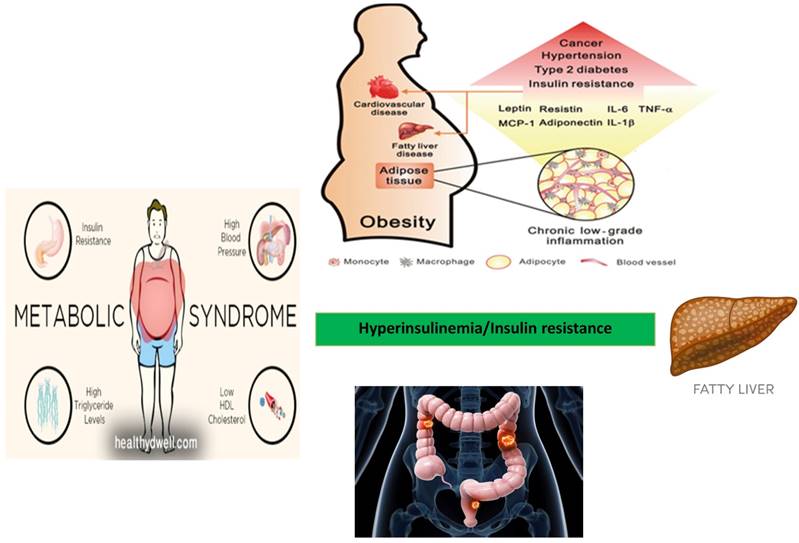
Fatty liver (triglyceride content > 5% of organ weight) is the most common liver disease found in the obese. It is classified as non-alcoholic fatty liver disease (NAFLD) if independent from the alcohol consumption. In addition, NAFLD is strongly associated with features of the metabolic syndrome, including obesity, insulin resistance, type 2 diabetes (DM2) and dyslipidemia [1,2]. NAFLD represents a new emerging risk factors for the onset of extra-hepatic tumors, particularly in the development of tumors of the gastro-intestinal tract [3,4]. Risk factors associated with the development of NAFLD include bad eating habits associated with a sedentary lifestyle. Therefore, patients with NAFLD are strongly recommended to follow a regimen including dietary restrictions associated with physical activity. Bad eating habits and sedentary lifestyle are increasing in Western Countries and NAFLD is accordingly becoming one of the most common causes of liver disease worldwide [5,6]. The prevalence of NAFLD is 15-30% of the general population and it is close to 50-90% in obese subjects. This prevalence correlates with the obesity rate. In fact, hepatic steatosis is found in 65% of subjects with grade I-II obesity (BMI = 30-39.9 kg/m2) and in 85% of patients with grade III obesity (BMI = 40-59 kg/m2). The prevalence of NAFLD and its inflammatory progression into non-alcoholic steatohepatitis (NASH) varies according to age, gender and degree of obesity [7]. Since NAFLD represents a spectrum of disease and histological patterns, a staging encompassing both the histological findings and the prognostic aspects has been proposed (Table 1). In the stages of NAFLD progression, classes I and II are considered substantially benign and reversible if the patients are adequately treated. Despite considered a benign condition in most cases, NAFLD can progress towards cirrhosis and liver failure (class III and IV) in a discrete percentage of subjects over a period of about two decades [8]. It is worth mentioning that a significant proportion of patients develop NAFLD despite having a normal body mass index (BMI) and few metabolic syndrome features. Patients with NAFLD are generally asymptomatic and NAFLD is often diagnosed during an occasional ultrasound evaluation of the abdomen. Indeed, several cases of NAFLD are associated with other metabolic disorders, including hypertriglyceridaemia, hypercholesterolemia, and hypertension, thus configuring the clinical pattern of the so-called "metabolic syndrome" [9]. Visceral obesity is an important risk factor for the onset of NAFLD. An accumulation of ectopic fat, including visceral obesity and fatty liver leads to dysfunction of adipose tissue with impaired production of adipocytokines. This in turn will favor an increase in pro-inflammatory cytokines such as TNF-α (Tumor Necrosis Factor-alfa) and a decrease in anti-inflammatory adipokines such as adiponectin [10]. Thus, the prevalence of NAFLD increases with the increase of the body mass index (BMI). NAFLD is currently considered the liver manifestation of the metabolic syndrome (MS) which represents, as shown by numerous studies, a condition that increases the risk of cancer, particularly for the gastrointestinal tract [11,12]. It is becoming clear that insulin resistance associated with the metabolic syndrome and the consequent activation of the inflammatory cascade jointly with the development of NAFLD may act as promoting factors for cancer development [13,14]. Thus, insulin resistance and metabolic syndrome represent common risk factors for both NAFLD and cancer (Figure 1).
Histological classification of NAFLD
| Class | Histological framework |
|---|---|
| Class I | Simple fatty liver disease |
| Class II | Fatty liver with lobular inflammation |
| Class III | Fatty liver with lobular inflammation and balloniform hepatocytes |
| Class IV | Fatty liver with lobular inflammation, balloniform hepatocytes, Mallory bodies and fibrosis (stage 1-4) |
Relationship between metabolic syndrome, NAFLD, and development of colorectal adenoma and carcinoma.
Natural history of NAFLD
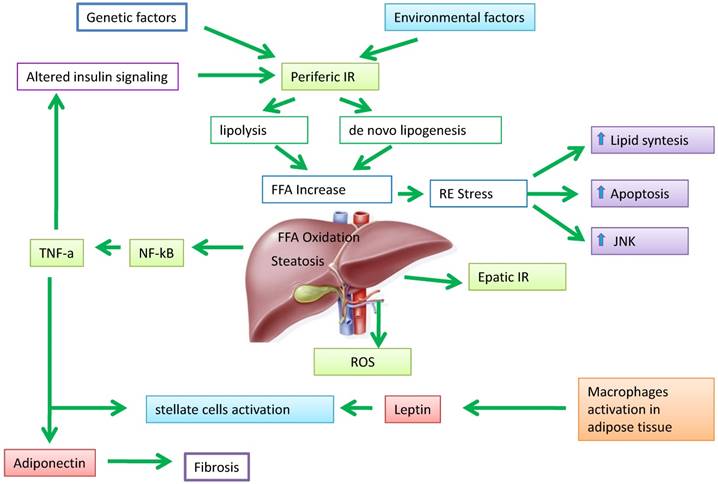
Hepatic steatosis develops when the amount of fatty acids that accumulate in liver cells exceeds the consumption. This imbalance between inputs and outputs, with an intrahepatic deposit of triglycerides, can occur as a result of four mechanisms: i) increased inflow of fatty acids (FA) from the peripheral circulation; ii) increased hepatic synthesis of FA (de novo lipogenesis); iii) reduced intrahepatic and peripheral FA oxidation; IV) reduced triglycerides release in the circulation through the VLDL. All these mechanisms contribute to the pathogenesis of hepatic steatosis supported by metabolic alterations [15,16]. The actors involved in this process are numerous, but the primary role is played by the adipose tissue expansion and particularly by hypertrophic adipocyte and insulin resistance generated at hepatic and muscular level [17]. Elevated serum levels of free fatty acids (FFAs) and cholesterol, insulin resistance, and adipocyte proliferation are generally present in obesity associated with bad dietary habits and bad lifestyle. Insulin resistance worsens adipocyte function and induces hepatic de novo lipogenesis. In addition, insulin resistance promotes the release of proinflammatory adipokines including interleukin (IL)-6, IL-1β and tumor necrosis factor (TNF)-α. These adipokines can in turn worsen the state of insulin resistance [18,19]. This condition results in an increased of hepatic flux of FFAs and leads to accumulation of triglycerides (TGs), mitochondrial dysfunction, reactive oxygen species (ROS) production, and eventually hepatic inflammation and fibrogenesis [20]. Recent findings support the evidence that increased FFA levels leads to an accumulation of certain types of lipid derivatives including ceramides, which inhibit the action of insulin [21]. Ceramides can be considered mediators of insulin resistance and lipotoxicity. Elevated plasma levels of ceramide are detectable in obese subjects and may contribute, for example, to insulin resistance through activation of TNF-α and other inflammatory mediators [22]. This corroborates the role of ceramide in the pathogenesis of NAFLD with a direct pro-inflammatory effects. In addition, the development of liver dysfunction, hepatic insulin resistance and steatosis in rodents are associated with increased levels of ceramide in both liver and peripheral blood [23]. Additionally, adiponectin lowers the hepatic content of ceramide by improving the blood glucose homeostasis through its receptor-associated ceramidase activity [24]. Ectopic fat deposition is “lipotoxic” and has been linked to the severity of insulin resistance. Holland and colleagues demonstrated that ceramide metabolism influences the insulin-sensitizing effects of adiponectin on the liver which is its primary target tissue [25]. Adiponectin displays anti-lipotoxic effects that are thought to protect tissues (i.e. the liver and muscle) from the accumulation of triglycerides. Decreased adiponectin levels may impair ceramide clearance by facilitating ceramide accumulation within the tissue [26]. An increase in hepatic ceramides is associated to elevated levels of plasma ceramides in obese patients and may promote insulin resistance at level of skeletal muscle. Taken together, these data indicate that insulin resistance represents one of the key factors in the pathogenesis of NAFLD (Figure 2).
The pathogenesis of NAFLD. Peripheral insulin resistance (IR) promotes increased production of free fatty acids (FFA) direct in the liver, resulting in an imbalance between oxidation/divestiture and uptake/synthesis of FFA, and then steatosis liver. The fabric fat secretes adipokines such as leptin, able to adjust the adipocyte metabolism and numerous insulin-mediated processes. Adiponectin plays a role in anti-inflammatory and anti-steatotic; its secretion It is in part regulated by TNF-α under the control of NF-kB.
Adipose tissue in the pathogenesis of NAFLD
In patients with NAFLD, the total amount of triglyceride pool in the liver originates from three sources: i) adipose tissue (60%); ii) remnant chylomicrons (15%) mostly from dietary fats; iii) hepatic lipogenesis or de novo lipogenesis (approximately 25%). Regarding the aliquot from adipocytes, 75-80% of triglyceride originates from the subcutaneous adipose tissue whereas the remaining portion is derived from the visceral adipose tissue and conveyed to the liver via the portal vein (the portal hypothesis) [27,28]. According to this hypothesis, the omental fat, which drains directly into the portal vein, contributes significantly to the flow of FFA to the liver as a consequence of the high lipolytic activity of the fat present therein. From the pathogenic point of view, the portal hypothesis is intriguing for several reason. For example, the visceral adipose tissue has a secretory profile different from that of subcutaneous adipose tissue characterized by abundant release of inflammatory cytokines. These cytokines are drained directly from the portal vein and poured out in the liver before reaching any other organ [29]. Therefore, the anatomical contiguity places the liver as the first barrier to the visceral adipose tissue. In physiological conditions, the liver is already subject to a high metabolic activity from which derives a large release of free radicals. In the presence of fatty liver, the oxidative metabolism increases further and even more if hyperglycemia and insulin resistance are present. The abnormal release of free radicals can be compensated until saturation is reached by the antioxidant barrier [30]. When the protective systems are exhausted, ROSs begin to oxidize hepatocytes and other hepatic cells, which in turn activate protective mechanisms by triggering the inflammatory response [31]. This results in an increased expression of TNF-α, IL-1, IL-6 and other inflammatory cytokines which contribute to acceleration of the damage and the more rapid progression of the disease [32,33].
NAFLD and cancer: the adipocytokine network
Based on their effect on NAFLD, adipokines are generally divided into adipokines promoting the onset of NAFLD and adipokines inhibiting the development of NAFLD. A crosstalk between these two groups of adipokines can result either in a beneficial or harmful effect on NAFLD progression [34,35]. However, the ultimate effect is generally temporary because both endogenous factors (e.g. genetic components) and exogenous factors (e.g. eating habits and lifestyles) determine continuous changes in the metabolic environment followed by dynamic changes of the adipokine profile with different effects on NAFLD evolution. For example, an antagonist relationship exists between adiponectin (with inhibitory action) and TNF-α (with promoting action) showing opposite effects on insulin resistance and NAFLD [36,37]. Under normal conditions, there is an equilibrium between the two adipokine groups. This homeostatic equilibrium can be altered by hypertrophy or hyperplasia of adipocytes, and can lead to a chronic state of inflammation, insulin resistance and development of NAFLD [38]. The expansion of adipose tissue, independently of other concomitant factors, deprives patients with NAFLD of the anti-inflammatory and antifibrotic effects of adiponectin, effects which are inversely related to total adiposity [39]. Adiponectin is a potent cytokine exerting insulin sensitizing, anti-inflammatory and anti-steatotic activity in hepatocytes through increasing oxidation of free fatty acids and decreasing gluconeogenesis, FFA flow and de novo lipogenesis [40,41]. In addition, adiponectin protects hepatocytes from apoptosis in the NAFLD-associated hepatic inflammatory microenvironment [42]. TNF-α is another important pro-inflammatory cytokine in NAFLD which promotes insulin resistance [43]. The excessive release of TNF-α from both the hypertrophic adipocytes and Kuppfer cells is one of the early events of hepatic damage. In fact, TNF-α stimulates the production of other chemo-attractant cytokines in the hepatocytes, which in turn recruit inflammatory cells [44,45]. High levels of TNF-α and low levels of adiponectin are favorable conditions for the development of insulin resistance and NAFLD [46]. These two adipokines involved in the pathogenesis of NAFLD mutually inhibit the synthesis and activity of each other by targeting metabolic equilibrium [47]. However, these adipokines are thought to be involved in the link between obesity, NAFLD and development of both intra and extra hepatic tumors [48-50]. In particular, adiponectin exerts antitumor activity by activating the AMP protein kinase (AMPK), an inhibitor of tumor cell growth that stimulates the caspase signaling pathway leading to apoptosis of endothelial cells [51,52]. In addition, adiponectin exerts its anti-tumor action by inhibiting TNF-α, involved in both tumor proliferation and angiogenesis [53,54]. Accordingly, insulin is becoming a relevant factor in the regulation of cell proliferation, apoptosis and tumor angiogenesis [55,56]. Leptin is another adipocitokine involved in the onset of NAFLD [57]. Leptin plays a dual role in NAFLD. On one hand, it seems to have a protective role for hepatic steatosis, especially in the early stages of the disease. On the other hand, it could act as an inflammatory and fibrinogenic factor as the disease progresses. As visceral adipose tissue increases, leptin levels concomitantly increase. This should limit the expansion of fat mass in order to avoid the formation of insulin resistance. In this sense, leptin should exert an anti-steatotic effect on the hepatocytes [58,61]. If adipose tissue expands, a state of leptin resistance occurs. Accordingly, leptin is no longer able to compensate insulin resistance and the progression of steatosis [62]. In this context, leptin may exert non-beneficial effects by acting as a fibrinogenic and pro-inflammatory adipokine. Leptin is considered an adipocytokine acting as molecular link between obesity and cancer [63-65]. Low levels of adiponectin and high levels of leptin are associated with the processes of mitogenesis, tumor growth and cell motility when adipose tissue dysfunction occurs. Furthermore, an increase in leptin stimulates the release of pro-inflammatory cytokines such as TNF-α and IL-6, which in turn give rise to an inflamed microenvironment which favors tumor development and up-regulation of angiogenesis promoting factors (i.e. VEGF and HIF-1α) [66-68].
Conclusion
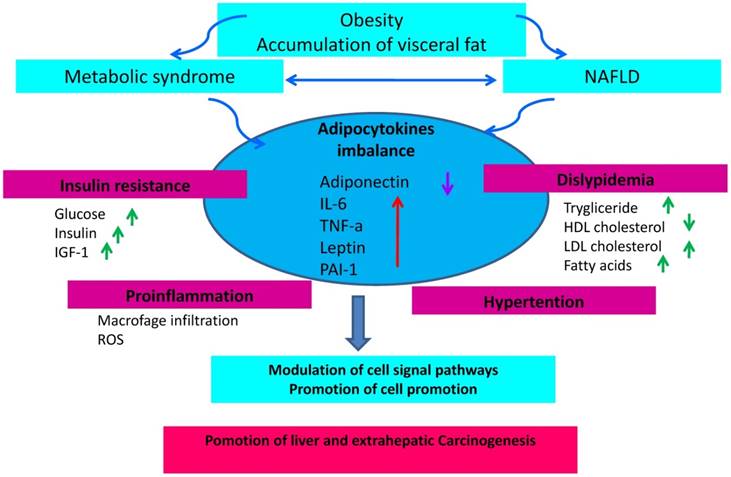
The incidence of NAFLD is increasing in accordance with the increasing prevalence of metabolic syndrome and obesity. In addition, NAFLD and predisposition to cancer is still matter of debate. Consequently, the question arises as to whether the increasing NAFLD incidence accounts for the increase of certain type of cancer (particularly liver and colon cancer). NAFLD is frequently associated with visceral obesity, diabetes and dyslipidemia, and it is related to clinical and biological markers of insulin resistance. Insulin resistance associated with the metabolic syndrome and the consequent activation of the inflammatory cascade underlying the development of NAFLD represents a favorable background for cancer development (Fig. 3). The prevention of an excessive increase of adipose tissue is therefore important to avoid the promotion of a pro-inflammatory activity sustained by an adverse adipokine profile predisposing to an increased risk of cancer. In conclusion, in overweight or obese patients with NAFLD, a program of cancer prevention aimed at quenching the inflammatory cascade and improve insulin sensitivity should be mandatory. It is therefore advisable for these subjects to adopt correct lifestyles including healthy eating habits and avoid sedentariness.
The state of chronic low-grade inflammation due to obesity and the presence of NAFLD leads to the emergence of a micro-environment favorable to the development of cancer and the onset of insulin resistance due to activation of the axis that regulates the insulin growth factor-1 IGF-1 and insulin resulting in hyperinsulinemia. Through its proliferative and anti-apoptotic effects, this process can increase mutations that promote carcinogenesis.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Dietrich P, Hellerbrand C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract Res Clin Gastroenterol. 2014;28(4):637-653
2. Milić S, Lulić D, Štimac D. Non-alcoholic fatty liver disease and obesity: biochemical, metabolic and clinical presentations. World J Gastroenterol. 2014;20(28):9330-9337
3. Kim GA, Lee HC, Choe J. et al. Association between non-alcoholic fatty liver disease and cancer incidence rate. J Hepatol. 2017 S0168-8278(17): 32294-32298
4. Derra A, Bator M, Menżyk T, Kukla M. Underrated enemy - from nonalcoholic fatty liver disease to cancers of the gastrointestinal tract. Clin Exp Hepatol. 2018;4(2):55-71
5. Aller R, Fernández-Rodríguez C, Lo Iacono O. et al. Management of non-alcoholic fatty liver disease (NAFLD). Clinical practice guideline. Gastroenterol Hepatol. 2018;41(5):328-349
6. Kwak MS, Kim D. Non-alcoholic fatty liver disease and lifestyle modifications, focusing on physical activity. Korean J Intern Med. 2018;33(1):64-74
7. Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci. 2018 doi: 10.1007 / s00018-018-2860-6. Jun 23
8. Koo SH. Nonalcoholic fatty liver disease: molecular mechanisms for the hepatic steatosis. Clin Mol Hepatol. 2013;19(3):210-215
9. Yki-Järvinen H. Non-alcoholic fatty liver disease as a cause and a consequence of metabolic syndrome. Lancet Diabetes Endocrinol. 2014;2(11):901-910
10. Hohenester S, Christiansen S, Nagel JM. et al. Lifestyle intervention for morbid obesity: effects on liver steatosis, inflammation and fibrosis. Am J Physiol Gastrointest Liver Physiol. 2018. 2018;315(3):G329-G338
11.
12. Brown JC, Harhay MO, Harhay MN. Nonalcoholic fatty liver disease and mortality among cancer survivors. Cancer Epidemiol. 2017;48:104-109
13. Sanna C, Rosso C, Marietti M. et al. Non-Alcoholic Fatty Liver Disease and Extra-Hepatic Cancers. Int J Mol Sci. 2016:17 (5)
14. Del Campo JA, Gallego P, Grande L. Role of inflammatory response in liver diseases: Therapeutic strategies. World J Hepatol. 2018;10(1):1-7
15. Chen Z, Yu R, Xiong Y. et al. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis. 2017;16(1):203
16. Satapathy SK, Sanyal AJ. Epidemiology and Natural History of Nonalcoholic Fatty Liver Disease. Semin Liver Dis. 2015;35(3):221-235
17. Marengo A, Jouness RI, Bugianesi E. Progression and Natural History of Nonalcoholic Fatty Liver Disease in Adults. Clin Liver Dis. 2016;20(2):313-324
18. Itoh H, Kanayama N. Developmental Origins of Nonalcoholic Fatty Liver Disease (NAFLD). Adv Exp Med Biol. 2018;1012:29-39
19. Altamirano-Barrera A, Barranco-Fragoso B, Méndez-Sánchez N. Management strategies for liver fibrosis. Ann Hepatol. 2017;16(1):48-56
20. Lambertucci F, Arboatti A, Sedlmeier MG. et al. Disruption of tumor necrosis factor alpha receptor 1 signaling accelerates NAFLD progression in mice upon a high-fat diet. J Nutr Biochem. 2018;58:17-27
21. Tsochatzis EA, Papatheodoridis GV, Archimandritis AJ. Adipokines in nonalcoholic steatohepatitis: from pathogenesis to implications in diagnosis and therapy. Mediators Inflamm. 2009;2009:831670
22. Apostolopoulou M, Gordillo R, Koliaki C. et al. Specific Hepatic Sphingolipids Relate to Insulin Resistance, Oxidative Stress, and Inflammation in Nonalcoholic Steatohepatitis. Diabetes Care. 2018;41(6):1235-1243
23. Ordoñez M, Presa N, Trueba M. et al. Implication of Ceramide Kinase in Adipogenesis. Mediators Inflamm. 2017;2017:9374563
24. Watt MJ, Barnett AC, Bruce CR. et al. Regulation of plasma ceramide levels with fatty acid oversupply: evidence that the liver detects and secretes de novo synthesised ceramide. Diabetologia. 2012;55(10):2741-2746
25. Holland WL, Miller RA, Wang ZV. et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011;17(1):55-63
26. Holland WL, Xia JY, Johnson JA. et al. Inducible overexpression of adiponectin receptors highlight the roles of adiponectin-induced ceramidase signaling in lipid and glucose homeostasis. Mol Metab. 2017;6(3):267-275
27. Reibe-Pal S, Febbraio MA. Adiponectin serenades ceramidase to improve metabolism. Mol Metab. 2017;6(3):233-235
28. Karim MF, Al-Mahtab M, Rahman S. et al. Non-alcoholic Fatty Liver Disease (NAFLD) A Review. Mymensingh Med J. 2015;24(4):873-880
29. Fuchs CD, Claudel T, Trauner M. Role of metabolic lipases and lipolytic metabolites in the pathogenesis of NAFLD. Trends Endocrinol Metab. 2014;25(11):576-585
30. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015;62(1S):S47-64
31. Masarone M, Rosato V, Dallio M. et al. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid Med Cell Longev. 2018;2018:9547613
32. Perumpail BJ, Khan MA, Yoo ER. et al. Clinical epidemiology and disease burden of nonalcoholic fatty liver disease. World J Gastroenterol. 2017;23(47):8263-8276
33. Abenavoli L, Peta V. Role of adipokines and cytokines in non-alcoholic fatty liver disease. Rev Recent Clin Trials. 2014;9(3):134-140
34. Stojsavljević S, Gomerčić Palčić M, Virović Jukić L. et al. Adipokines and proinflammatory cytokines, the key mediators in the pathogenesis of nonalcoholic fatty liver disease. World J Gastroenterol. 2014;20(48):18070-18091
35. Polyzos SA, Kountouras J, Mantzoros CS. Adipokines in nonalcoholic fatty liver disease. Metabolism. 2016;65(8):1062-1079
36. Jarrar MH, Baranova A, Collantes R. et al. Adipokines and cytokines in non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2008;27(5):412-421
37. Rabe K, Lehrke M, Parhofer KG, Broedl UC. Adipokines and insulin resistance. Mol Med. 2008;14(11-12):741-751
38. Antuna-Puente B, Feve B, Fellahi S. et al. Adipokines: the missing link between insulin resistance and obesity. Diabetes Metab. 2008;34(1):2-11
39. Divella R, De Luca R, Abbate I. et al. Obesity and cancer: the role of adipose tissue and adipo-cytokines-induced chronic inflammation. J Cancer. 2016;7(15):2346-2359
40. Neuman MG, Cohen LB, Nanau RM. Biomarkers in nonalcoholic fatty liver disease. Can J Gastroenterol Hepatol. 2014;28(11):607-618
41. Ma H, You GP, Cui F. et al. Effects of a low-fat diet on the hepatic expression of adiponectin and its receptors in rats with NAFLD. Ann Hepatol. 2015;14(1):108-117
42. Buechler C, Wanninger J, Neumeier M. Adiponectin, a key adipokine in obesity related liver diseases. World J Gastroenterol. 2011;17(23):2801-2811
43. Finelli C, Tarantino G. What is the role of adiponectin in obesity related non-alcoholic fatty liver disease? World J Gastroenterol. 2013;19(6):802-812
44. Nieto-Vazquez I, Fernández-Veledo S, Krämer DK. et al. Insulin resistance associated to obesity: the link TNF-alpha. Arch Physiol Biochem. 2008;114(3):183-194
45. Sato A, Nakashima H, Nakashima M. et al. Involvement of the TNF and FasL produced by CD11b Kupffer cells/macrophages in CCl4-induced acute hepatic injury. PLoS One. 2014:9 (3)
46. Su L, Li N, Tang H. et al. Kupffer cell-derived TNF-α promotes hepatocytes to produce CXCL1 and mobilize neutrophils in response to necrotic cells. Cell Death Dis. 2018;9(3):323
47. Chen Z, Yu R, Xiong Y. et al. A vicious circle between insulin resistance and inflammation in nonalcoholic fatty liver disease. Lipids Health Dis. 2017;16(1):203
48. Adolph TE, Grander C, Grabherr F. et al. Adipokines and Non-Alcoholic Fatty Liver Disease: Multiple Interactions. Int J Mol Sci. 2017:18 (8). pii: E1649
49. Duan XF, Tang P, Li Q. et al. Obesity, adipokines and hepatocellular carcinoma. Int J Cancer. 2013;133(8):1776-1783
50. Booth A, Magnuson A, Fouts J. et al. Adipose tissue, obesity and adipokines: role in cancer promotion. Horm Mol Biol Clin Investig. 2015;21(1):57-74
51. Derra A, Bator M, Menżyk T. et al. Underrated enemy - from nonalcoholic fatty liver disease to cancers of the gastrointestinal tract. Clin Exp Hepatol. 2018;4(2):55-71
52. Nagaraju GP, Aliya S, Alese OB. Role of adiponectin in obesity related gastrointestinal carcinogenesis. Cytokine Growth Factor Rev. 2015;26(1):83-93
53. Zhang L, Wen K, Han X. et al. Adiponectin mediates antiproliferative and apoptotic responses in endometrial carcinoma by the AdipoRs/AMPK pathway. Gynecol Oncol. 2015;137(2):311-320
54. Hajri T, Tao H, Wattacheril J. et al. Regulation of adiponectin production by insulin: interactions with tumor necrosis factor-α and interleukin-6. American Journal of Physiology - Endocrinology And Metabolism. 2011;300(2):E350-E360
55. Huang M, Chen Z, Xu D. et al. Adiponectin inhibits proliferation and induces apoptosis in colorectal cancer HCT116 cells. Chinese Journal of Cellular and Molecular Immunology. 2018;34(3):253-259
56. Djiogue S, Nwabo Kamdje AH, Vecchio L. et al. Insulin resistance and cancer: the role of insulin and IGFs. Endocr Relat Cancer. 2013;20(1):R1-R17
57. Cao Y. Angiogenesis and vascular functions in modulation of obesity, adipose metabolism, and insulin sensitivity. Cell Metab. 2013;18(4):478-489
58. Polyzos SA, Aronis KN, Kountouras J. et al. Circulating leptin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Diabetologia. 2016;59:30-43
59. Boutari C, Tziomalos K, Athyros VG. The adipokines in the pathogenesis and treatment of nonalcoholic fatty liver disease. Hippokratia. 2016;20(4):259-263
60. Boutari C, Perakakis N, Mantzoros CS. Association of Adipokines with Development and Progression of Nonalcoholic Fatty Liver Disease. Endocrinol Metab (Seoul). 2018;33(1):33-43
61. de Luis DA, Perez Castrillon JL, Duenas A. Leptin and obesity. Minerva Med. 2009;100:229-236
62. Asrih M, Veyrat-Durebex C, Poher AL. et al. Leptin as a Potential Regulator of FGF21. Cell Physiol Biochem. 2016;38(3):1218-1225
63. Zhang L, Song H, Ge Y. et al. Temporal relationship between diet-induced steatosis and onset of insulin/leptin resistance in male Wistar rats. PLoS One. 2015:10 (2)
64. Mullen M, Gonzalez-Perez RR. Leptin-Induced JAK/STAT Signaling and Cancer Growth. Vaccines (Basel). 2016:4 (3) pii: E26
65. Ray A, Cleary MP. The potential role of leptin in tumor invasion and metastasis. Cytokine Growth Factor Rev. 2017;38:80-97
66. Ackerman SE, Blackburn OA, Marchildon F. et al. Insights into the Link Between Obesity and Cancer. Curr Obes Rep. 2017;6(2):195-203
67. Park J, Morley TS, Kim M. et al. Obesity and cancer—mechanisms underlying tumour progression and recurrence. Nat Rev Endocrinol. 2014;10(8):455-465
68. Iyengar NM, Gucalp A, Dannenberg AJ. et al. Obesity and Cancer Mechanisms: Tumor Microenvironment and Inflammation. J Clin Oncol. 2016;34(35):4270-4276
69. Himbert C, Delphan M, Scherer D. et al. Signals from the Adipose Microenvironment and the Obesity-Cancer Link-A Systematic Review. Cancer Prev Res (Phila). 2017;10(9):494-506
Author contact
![]() Corresponding author: Rosa Divella, Ph.D., Department of Clinical Pathology Laboratory. IRCCS - Istituto Tumori Giovanni Paolo II, Viale Orazio Flacco 65, 70124 Bari, Italy. Tel. +39 805555259; E-mail: rosadiveit
Corresponding author: Rosa Divella, Ph.D., Department of Clinical Pathology Laboratory. IRCCS - Istituto Tumori Giovanni Paolo II, Viale Orazio Flacco 65, 70124 Bari, Italy. Tel. +39 805555259; E-mail: rosadiveit