ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2019; 15(3):617-627. doi:10.7150/ijbs.29689 This issue Cite
Research Paper
Activation of AMPK by metformin promotes renal cancer cell proliferation under glucose deprivation through its interaction with PKM2
Meihan Liu1, Zhuo Zhang2, Hui Wang1, Xiaoliang Chen2 ![]() , Chunxiang Jin1
, Chunxiang Jin1 ![]()
1. Department of Ultrasonography, China-Japan Union Hospital, Jilin University, Changchun, China
2. Department of Urology, China-Japan Union Hospital, Jilin University, Changchun, China
Abstract

Metformin, a common therapeutics for type 2 diabetics, was recently demonstrated to possess antitumor activity in various cancer types. However, its therapy effect in renal cell carcinoma (RCC) still remains controversial. In this study, we found that metformin treatment in RCC cells lead to activation of AMPK, which suppressed the cell proliferation under normal condition, but enhanced cell proliferation under glucose deprivation (GD) condition. Depletion of AMPK by siRNA abolished the proliferation effect of MF under GD condition. Mechanistic investigations revealed that the effect of AMPK on cell proliferation under GD condition is dependent on its nuclear translocation. Moreover, the nuclear AMPK recruits PKM2 and β-Catenin to form a complex, which promotes the transcription of cell proliferation related genes, including CCND1 and c-Myc. Furthermore, depletion of PKM2 or β-Catenin abrogated the proliferative effects of metformin under GD condition. And inhibition of PKM2 also re-sensitized the A498 xenograft in response to metformin treatment. Together, our results suggested that combined of AMPK activation and PKM2 depletion or inhibition can achieve better therapeutic effect for RCC patients.
Keywords: AMPK, renal cancer, PKM2, β-Catenin
Introduction
Renal cell carcinoma (RCC) is the most common and lethal cancer of kidney cancer, which accounts for 85% of all kidney cancers [1]. The increasing incidence of kidney cancer is mostly attributed to the wide use of diagnostic imaging and the increasing rates of hypertension and obesity [2]. In addition, it has been demonstrated that renal cancer patients with diabetes mellitus have poorer survival compared with those without diabetes mellitus [3]. Aerobic glycolysis plays an important role in glucose metabolism in various cancer cells. Recent insights into the biology of renal cancer suggest that renal cancer is fundamentally a metabolic disease. The mutations of the renal cancer genes related to disorders in oxygen, iron, nutrient and/or energy sensing frequently happen in renal cancer syndromes [4]. Therefore, a better understanding of the metabolic changes in renal cancer might lead to innovative therapeutic strategies to treat this disease.
The AMP-activated protein kinase (AMPK) is a cellular energy sensor which reflects cellular energy status by undergoing phosphorylation and increasing activity when AMP levels increase and ATP levels decrease, which is an indicative marker of energy deficiency [5]. Although RCC tumor cells might be expected to show AMPK activation because of their dependence on less efficient glycolysis and their rapid consumption of ATP during growth, unexpectedly, the reverse was observed, with markedly reduced phospho-AMPK levels, and reduced levels of AMPK subunits AMPKα and AMPKβ1 at both transcriptional and protein levels[6]. Therefore, activation of AMPK might be an attractive strategy for RCC treatment. Exactly, Metformin (MF), an AMPK agonist, has shown antitumor activity in various cancers in multiple preclinical studies using cultured cancer cells and mouse models [7,8]. For example, in differentiated and undifferentiated thyroid cancer cells, Chen et al demonstrated that in vitro activation of AMPK with MF inhibited cell growth, induced cell cycle arrest and apoptosis, inhibited colony sphere formation, and chemosensitized cells to doxorubicin [9]. The administration of MF was associated with favorable overall survival (OS), cancer-specific survival (CSS), and disease-free survival (DFS) of RCC patients [10], and lower risk of mortality in the processes of therapy [11]. Although MF use on RCC patients was in favor of better CSS and DFS, it did not benefit the patients with metastatic RCC [12]. Therefore, it remains controversial about whether use of MF will achieve better survival outcomes in patients with renal cancer.
The role of AMPK as a modulator of energy use is well established [13,14]. The activated AMPK can either maintain intracellular energy homeostasis by down-regulating energy consuming (anabolic) pathways under metabolic stress, or up-regulated energy producing (catabolic) pathways to replenish cellular ATP stores when there is nutrient deprivation [15]. We therefore hypothesized that the poor outcome of MF in RCC patient might related to activation of AMPK under conditions of nutrient deprivation, since cancer cells usually encounters nutrient limitation [16]. In this study, we used a model of glucose deprivation to study the effect of nutrient limitation in RCC cells under MF treatment. Our results indicated that activation of AMPK by MF suppressed the RCC cell proliferation under normal condition, but enhanced the cell proliferation under GD condition. Mechanically, the activation of AMPK recruits PKM2 and β-catenin into nuclear and triggers the transcription of several proliferstion related genes, including cyclin D1 (CCND1), c-Myc. Our results suggested inhibition of PKM2 or β-catenin could be an alternative strategy to overcome the RCC proliferation caused by AMPK activation under GD condition.
Materials and methods
Cell culture
RCC cell lines (A498 and GRC-1) were obtained from the Shanghai Cell Bank, Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Dulbecco minimum essential medium (DMEM) (Invitrogen Life Technologies, Carlsbad, CA, USA) with the supplements of 10% heat-inactivated fetal bovine serum (Invitrogen Life Technologies), 100× penicillin-streptomycin solution (Invitrogen Life Technologies), and incubated in an incubator (Thermo Fisher Scientific Inc., Waltham, MA, USA) set to 37°C, 100% humidity, and 5% CO2.
Glucose restriction and drug treatment
For the glucose deprivation (GD) treatment, cells were seeded overnight and transferred to DMEM with 2.5 mM glucose (treatment group) or with 25 mM glucose (control group). The high glucose medium was purchased from Thermo Fisher (11965118, Waltham, MA, USA) and diluted with glucose-free DMEM (11966025) to 2.5 mM GD medium. AMPK activator metformin (MF, PHR1084) and AMPK inhibitor (Compound C, 171261) were purchased from Sigma-Aldrich (St. Louis, MO, USA).
siRNA transfection
siRNA of AMPK, PKM2 and β-Catenin was synthesized by Shanghai GenePharma, Co., Ltd. The target sequences of these genes were as follows, AMPK: GTGGCTTATCATCTTATCATT and GAAGGTTGTAAACCCATATTA, PKM2: CCATAATCGTCCTCACCAA, β-Catenin: CATGTGTTGGTAAGCTCTA, and control: ATGCTGATCAGTGTCGATT. The transfection of siRNA was conducted by lipofectamin 2000 reagent (Thermo Fisher Scientific, Shanghai, China) according to the manufacturer's instruction.
Western blotting and immunoprecipitation
Cells were lysed for 30 min on ice in IP buffer with protease inhibitors cocktail (SRE0055-1BO, Sigma-Aldrich). Samples were quantified with BCA assays (23225, Thermofisher) and analyzed by western blotting. For IP, cell lysates were incubated with protein-specific antibodies or normal rabbit IgG (sc-2027, Santa Cruz Biotechnology, Dallas, TX, USA) overnight at 4 °C, followed by incubation with protein A/G agarose beads (Millipore; IP10) for 3 h and washed in lysis buffer supplemented with protease inhibitors. Antibodies for western blotting were: AMPK (SAB4502329), p-AMPK (Thr172, SAB4503754), Ki-67 (SAB5500134), Actin (A5441) (Sigma-Aldrich), tubulin (SC-8035, Santa Cruz, Dallas, TX, USA), PKM2 (4053, Cell Signaling, Danvers, MA, USA), H3 (ab1791), β-Catenin (ab32572) (Abcam, Cambridge, UK).
Cell fraction assay
Cells were lysed using membrane lysis buffer (10 mM HEPES, pH 8.0, 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT and 1% Igepal CA-630), then vortexed and centrifuged at 13200 rpm for 5 min. The supernatant was collected as the cytoplasmic fraction. The pellets were re-suspended with RIPA buffer (Millipore) and then sonicated twice on ice and centrifuged at 13200 rpm for 15 min. The supernatant was the nuclear fraction. The cytoplasmic fraction and the nuclear fraction were analyzed by Western blotting.
RNA extraction and quantitative real-time PCR
Total RNA was isolated from thyroid cancer cells using RNeasy Mini Kit (74104, Qiagen, Hilden, Gemany) according to the manufacturer's protocol. SYBR Green-based qPCR master mixes (330600) were obtained from Qiagen. Expression of Cyclin D1 (CCND1), c-Myc were examined using specific primers as following: CCND1, 5′-CCCTCGGTGTCCTACTTCAAA-3′ and 5′ CCAGGTTCCACTTGAGCTTGT-3′, c-Myc, 5′-CCTCAACGTTAGCTTCACCAA-3′ and 5′-TTTGATGAA GGTCTCGTCGTC-3′.
Cell proliferation assays
RCC cells were treated with drugs for 48 h, and then pulsed with 5-Bromo-2-deoxyuridine (BrdU) for an additional 8 h. Cell proliferation was determined by BrdU incorporation assay according to the manufacturer's instructions (11647229001, Roche Diagnostics GmbH, Roche Applied Science, Germany). The absorbance at 450nm was detected. Cell viability was assayed by using 3-(4,5)-dimethylthiahiazo(-z-y1)-3,5-di-phenytetrazoliumromide (MTT, G4000, Promega, Madison, MI, USA). After treatment, 10 µL MTT (5 mg/mL) was added into cultured medium in each well for 2-4 h until purple precipitate is visible. After the removal of culture medium, 75 µL dimethyl sulphoxide was added to each well, leaving the cells at room temperature in the dark for 2 h. The absorbance at 570 nm was detected.
ATP production and glucose uptake assays
The level of intracellular ATP was determined by ATP colorimetric assay kit (K354, Bio vision, Milpitas, CA, USA). Glucose uptake was measured by fluorimetric cell-based glucose uptake assay kit (#EFGU-100, Bioassay systems, Hayward, CA, USA). All the measurements were normalized to cell numbers and protein concentration.
Xenograft model
BALB/C nude mice provided by SLAC Laboratory Animal (Shanghai, China), were studied after approval from the Medical ethics committee of College of Basic Medical Sciences, Jilin University. 6- to 8-week-old mice were maintained in high-efficiency particulate air-filtered cages in a pathogen-free facility. A498 cells were washed once and resuspended in serum-free medium. 1×106 cells in matrigel (BD Biosciences, San Jose, CA) were injected into the neck region; mice were examined the day after injection. 7 days after tumor cell injection, mice were treated continuously with MF in drinking water (200 μg/ml), or injected with SKN (4 mg/kg) by (intraperitoneal) I.P. injection for 2 weeks. Control mice were injected with the same volume of PBS. Tumor size was measured with a caliper each week for 5 weeks; the tumor volume was determined by measuring the maximal (a) and minimal (b) diameters using a caliber and calculated by using the formula a×b2. For the food starvation, short-term fasting was implemented 32 h prior and 12 h post-injection. During the treatment, animals were monitored routinely for body weight loss and general behavior.
Statistical Methods
For comparison of the data presented statistics were done using a two group, unpaired Student's t-test, while for the comparison of three groups, one-way ANOVA were performed via GraphPad Prism software.
Results
Activation of AMPK promotes RCC cell proliferation under glucose deprivation
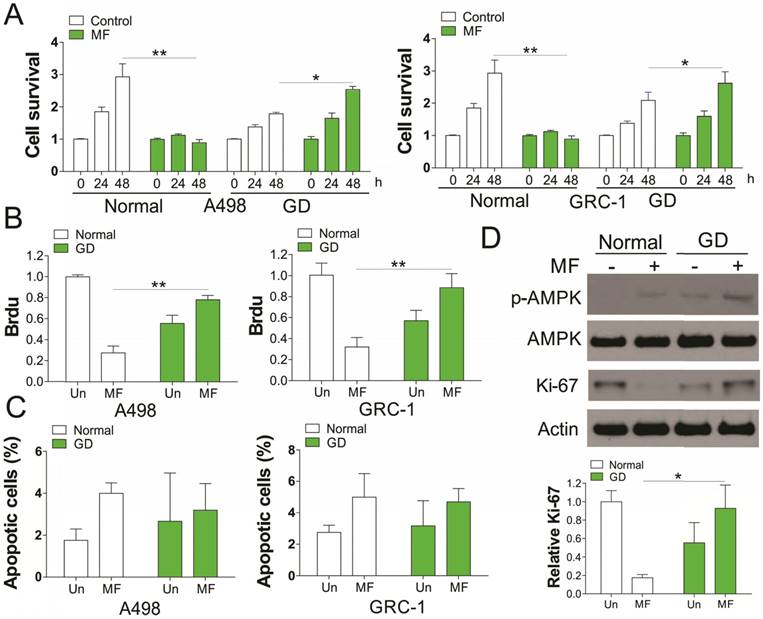
We first investigated the in vitro effect of AMPK agonists, MF (3 mM) on renal cancer cell (RCC) proliferation with or without glucose deprivation (GD). Our results indicated that supplement of MF in A498 and GRC-1 cells substantially suppressed the cell grow under normal condition (Fig. 1A). However, MF treatment did not have the similar effect on RCC cell growth under GD condition, but promoted the cell growth, although RCC cells grew slower under GD condition (Fig. 1A). The Brdu assay results suggested that MF treatment suppressed the RCC cell proliferation under normal condition, but enhanced the cell proliferation under GD condition (Fig. 1B). In contrast, no obvious apoptosis was found in RCC cells in response to MF treatment in normal or GD conditions (Fig. 1C). Since MF is an AMPK agonist, we further investigate the activation of AMPK, and found that MF treatment induced AMPK phosphorylation in both normal and GD condition (Fig. 1D). Interestingly, the GD condition also increases the phosphorylation of AMPK, which was reinforced by MF treatment (Fig. 1D). We also tested the expression of proliferation marker, Ki-67, and found that MF treatment suppressed the expression of Ki-67 in normal condition, but induced its expression under GD condition (Fig. 1D). Therefore, our results suggested that MF treatment increased the RCC cell proliferation under glucose deprivation.
Activation of AMPK by metformin (MF) promotes RCC cell proliferation under glucose deprivation (GD) condition. (A) A498 and GRC-1 cells were treated Metformin (MF, 3 mM) for indicated time point under normal cell culture or GD condition. The cell viability was analyzed by MTT assay. (B) The cell proliferation of A498 and GRC-1 cells treated with MF under GD condition was analyzed by Brdu assay. (C) The cell apoptosis of A498 and GRC-1 cells treated with MF under GD condition was analyzed by hoechst 33342 staining. (D) Western blot of indicated proteins in A498 cells treated with MF under normal or GD conditions. Every experiment was repeated for 3 times. *, p<0.05; **, p<0.01.
AMPK modulates the RCC proliferation independent of its kinase activity
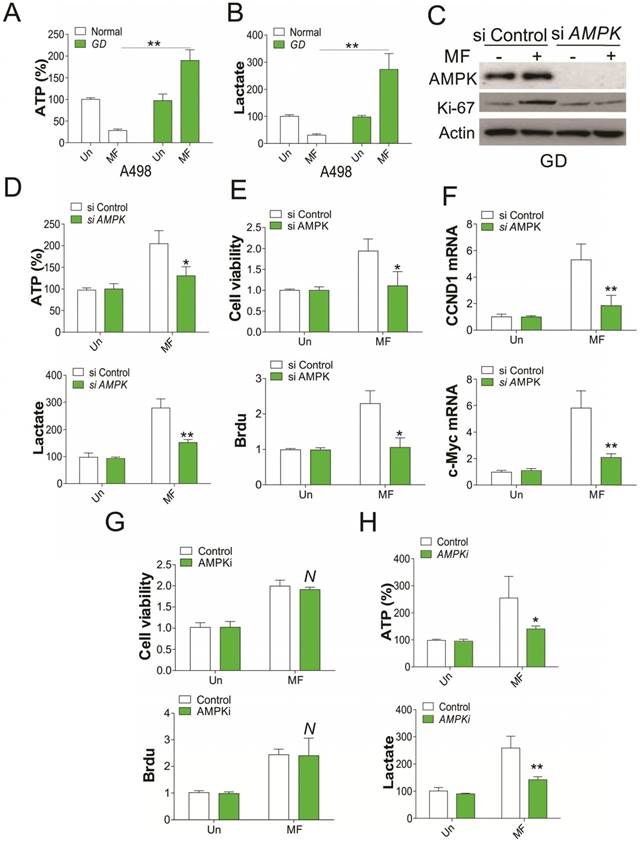
Since AMPK activation is correlated to glucose consumption, we therefore tested the ATP and lactate level in RCC cells after MF treatment. We found that MF treatment reduced the ATP and lactate level in RCC cells under normal condition, but enhanced ATP and lactate level under GD condition (Fig. 2A, B). Depletion of AMPK by shRNA in RCC cells under GD condition compromised the induction of Ki67 by MF treatment (Fig. 2C), and the ATP and lactate secretion (Fig. 2D). Furthermore, silence of AMPK also reduced the RCC cell proliferation under GD condition (Fig. 2E), and suppressed the mRNA induction of CCND1 and c-Myc by MF (Fig. 2F), which were reported to modulated the cell proliferation. However, combined treatment with AMPK inhibitor did not compromised the cell proliferation induced by MF treatment (Fig. 2G), although it reduced the ATP and lactate secretion in RCC cells under GD condition (Fig. 2H). These results collectively suggested that AMPK modulated the RCC cell proliferation under GD condition, which is independent of its kinase activity.
Nuclear AMPK is important for RCC cell proliferation by recruiting β-catenin
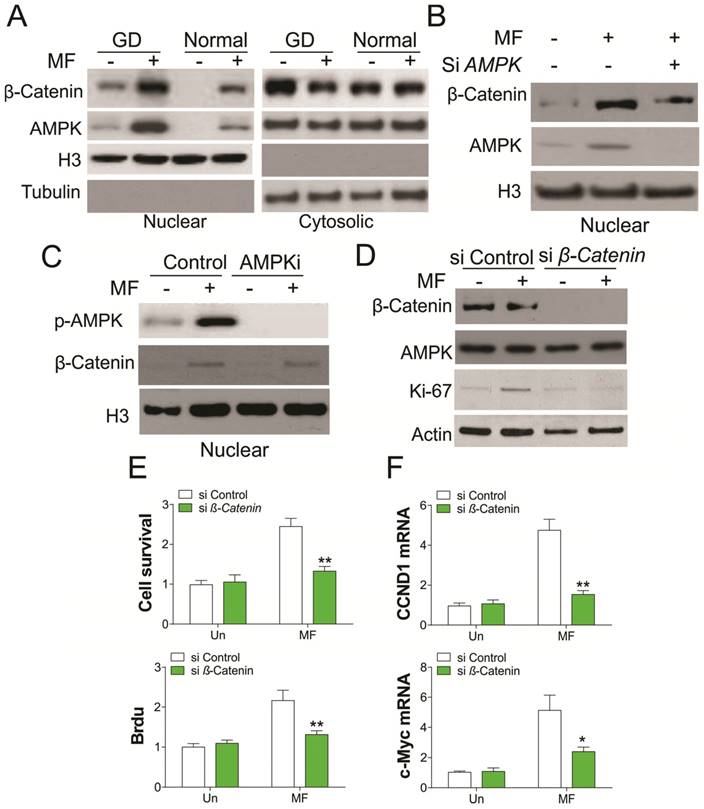
AMPK is a crucial nutrient sensor, and previous studies have demonstrated that it is translocated into the nucleus under energy restriction stress [17]. AMPK was reported to promote β-catenin nuclear translocation, which mediates cancer cell migration [18,19], we therefore tested whether GD condition promotes nuclear translocation of AMPK and β-catenin. As predicted, MF treatment in A498 cells promotes AMPK and β-catenin nuclear translocation under both normal and GD conditions (Fig. 3A). However, the nuclear translocation of AMPK and β-catenin under GD condition was much higher than that in normal condition, which is positively proportional with the AMPK activation (Fig. 1D). Depletion of AMPK abolished the nuclear translocation of β-catenin (Fig. 3B). However, inhibition of AMPK did not change the nuclear translocation of β-catenin promoted by MF treatment (Fig. 3C), suggesting the nuclear translocation of β-catenin is AMPK kinase independent. To further confirm the role of β-catenin in AMPK activation mediated cell proliferation, we utilized the siRNA to knockdown β-catenin upon MF treatment in A498 cells. Silence of β-catenin in A498 cells treated with MF suppressed the induction of Ki-67, which is a proliferation marker (Fig. 3D). Accordingly, depletion of β-catenin also inhibited cell proliferation caused by MF treatment in A498 cells (Fig. 3E), as well as the mRNA level of CCND1 and c-myc (Fig. 3F). Therefore, our results suggested activation of AMPK promotes RCC cell proliferation by recruiting β-catenin into nuclear.
AMPK mediates RCC cell proliferation under GD condition. (A) The ATP level of A498 cells treated with MF for 48 h under normal or GD conditions. (B) The lactate level of A498 cells treated with MF for 48 h under normal or GD conditions. (C) Western blot of indicated proteins in A498 cells transfected with control or AMPK siRNA and treated with MF for 48 h under GD condition. (D) The ATP and lactate level in A498 cells treated as in (C). (E) The cell viability and Brdu analysis of cell proliferation in A498 cells treated as in (C). (F) The mRNA level of CCND1 and c-Myc in A498 cells treated as in (C). (G) The cell viability and Brdu analysis of cell proliferation in A498 cells treated with MF with or without AMPK inhibitor (AMPKi, 5 µM) under GD condition. (H) The ATP and lactate level in A498 cells treated as in (G). Every experiment was repeated for 3 times. *, p<0.05; **, p<0.01.
Nuclear AMPK promotes RCC cell proliferation by transcription of β-Catenin under GD condition. (A) The nuclear location of AMPK and β-Catenin upon MF treatment under normal or GD conditions was analyzed by cellular fractionation followed by western blot. (B) The nuclear location of β-Catenin in A498 cells transfected with control or AMPK siRNA upon MF treatment for 48 h under GD condition. (C) The nuclear location of β-Catenin in A498 cells treated with MF in combined with or without AMPKi (5 µM) for 48 h. (D) A498 cells transfected with control or β-Catenin siRNA were treated with MF for 48 h under GD condition. The expression of indicated proteins was analyzed by western blot. (E) The cell viability and Brdu analysis of A498 cells treated as in (D). (F) The mRNA level of CCND1 and c-Myc in A498 cells treated as in (D). Every experiment was repeated for 3 times. *, p<0.05; **, p<0.01.
AMPK promotes β-catenin nuclear translocation by binding with PKM2
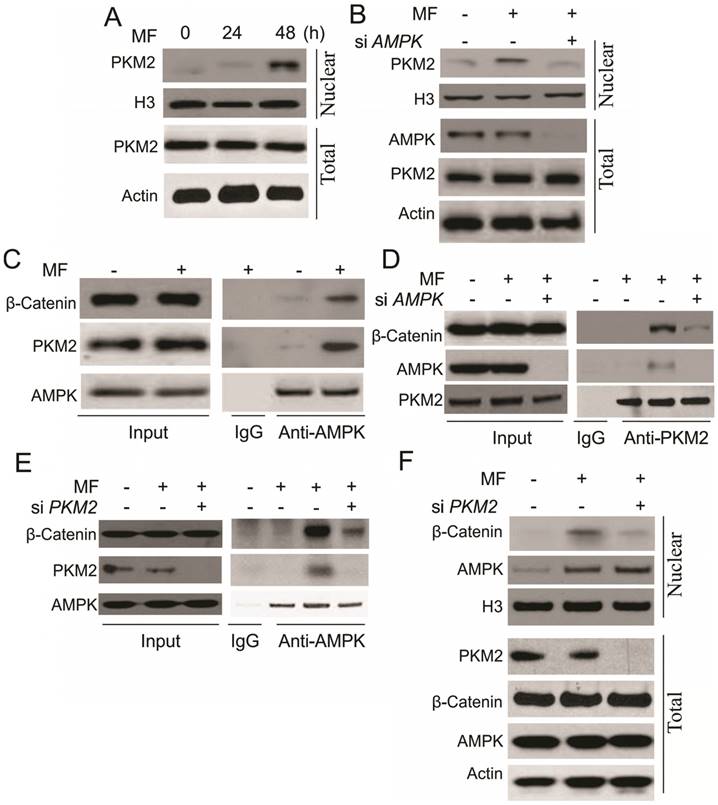
In the next step, we are interested to understand the underlying mechanism for β-catenin nuclear translocation by AMPK. It was reported that activation of AMPK interacts with PKM2 [17], which can promote the transcription activity of β-catenin, and trigger cell proliferation [20]. In this study, we found that MF treatment increased the nuclear translocation of PKM2 in A498 cells (Fig. 4A). Depletion of AMPK suppressed the nuclear translocation of PKM2 (Fig. 4B), suggesting the activity of PKM2 might be involved in the AMPK-induced RCC cell proliferation. We further study the interaction of AMPK, PKM2 and β-catenin, and found that MF treatment assembles these three proteins together (Fig. 4C). Depletion of AMPK or PKM2 by siRNA abolished the complex formation with β-catenin (Fig. 4D, E). However, depletion of PKM2 attenuated the nuclear translocation of β-catenin, but did not affect the AMPK translocating into nulcear (Fig. 4F). Therefore, our results suggested that AMPK promotes β-catenin nuclear translocation by recruiting PKM2 into nuclear and forming a complex with β-catenin.
AMPK form a complex with PKM2 and β-Catenin in nuclear in response to MF treatment under GD condition. (A) The nuclear location of PKM2 in A498 cells upon MF treatment for 48 h under GD condition. (B) The nuclear location of PKM2 in A498 cells transfected with control or AMPK siRNA upon MF treatment for 48 h under GD condition. (C) The interaction of AMPK and PKM2 and β-Catenin was analyzed by immunoprecipitation. (D) The interaction of PKM2 and β-Catenin in A498 cell transfected with control or AMPK siRNA was analyzed by immunoprecipitation. (E) The interaction of AMPK and β-Catenin in A498 cell transfected with control or PKM2 siRNA was analyzed by immunoprecipitation. (F) The nuclear location of AMPK and β-Catenin in A498 cells transfected with control or PKM2 siRNA upon MF treatment for 48 h under GD condition. Every experiment was repeated for 3 times.
PKM2 is necessary for AMPK mediated RCC cell proliferation
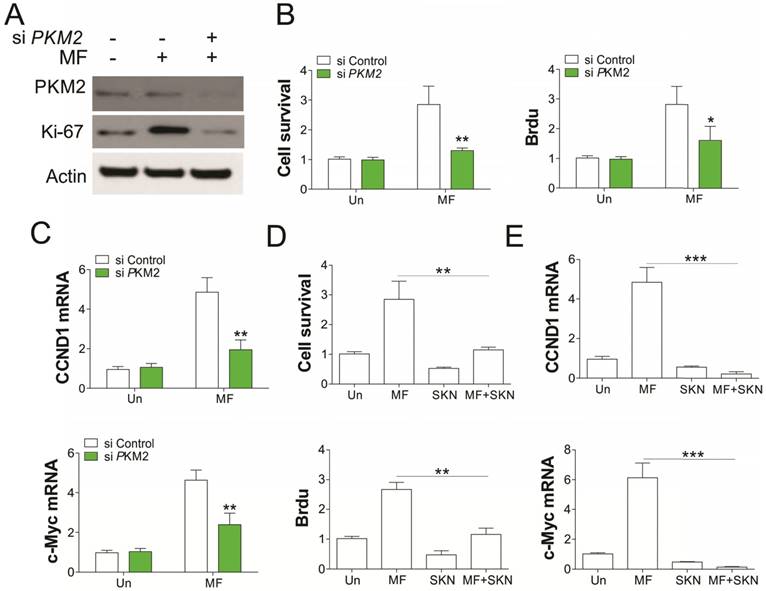
To further confirm the role of PKM2 in AMPK modulated RCC cell proliferation under GD condition, we depleted PKM2 by siRNA transfection. Absence of PKM2 in A549 cells abrogated the RCC cell proliferation induced by MF treatment in A549 cells (Fig. 5A, B). Accordingly, PKM2 knockdown also suppressed the induction of CCND1 and c-Myc in mRNA level (Fig. 5C), suggesting the inhibition of β-catenin transcription activity. Inhibition of PKM2 by shikonin (SKN) also suppressed the cell proliferation, and induced loss of cell survival, suggesting the cell death upon SKN treatment (Fig. 5D). Furthermore, SKN treatment also suppressed the induction of CCND1 and c-Myc by MF (Fig. 5E). Therefore, our results suggested that PKM2 is necessary for AMPK recruits β-catenin into nuclear, and promotes transcription of several genes related to cell proliferation.
Inhibition of PKM2 overcome the cell proliferation induced by MF under glucose deprivation in vivo
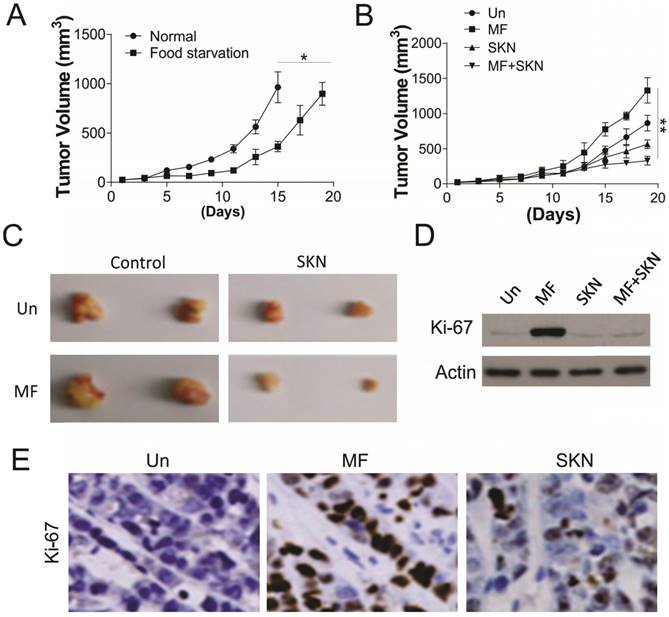
To further confirm the effect of PKM2 inhibition on AMPK activation induced proliferation, the in vivo xenograft experiment was conducted. Firstly, we tested the effect of food starvation on tumor growth in the nude mice injected with 1X106 A498 cells, which was supposed to mimic the effect of glucose deprivation. Our results suggested that food starvation actually slow the tumor growth, but did not have suppression effects (Fig. 6A). We next tested the MF and/or SKN treatment in nude mice under glucose starvation. We found that MF treatment did not suppressed the tumor growth, but increased the tumor volume (Fig. 6B,C). However, SKN treatment actually limited the tumor growth, and exerted maximal inhibitory effect when combined with MF treatment (Fig. 6B, C). Furthermore, the western blot for Ki-67 also revealed that MF treatment promoted the cell proliferation in vivo, and SKN combination can suppressed the induction of Ki-67 (Fig. 6D, E). Collectively, our in vivo results suggested that inhibition of PKM2 can reverse the proliferation effect of MF under food starvation.
PKM2 mediated the proliferation induced by MF under GD condtion. (A) A498 cells transfected with control or PKM2 siRNA were treated with MF for 48 h under GD condition. The expression of indicated proteins was analyzed by western blot. (B-D) The cell viability and proliferation (B) and mRNA level of CCND1 and c-Myc (C) of A498 cells treated as in (A) was analyzed. (D) The cell viability and Brdu analysis of A498 cells treated with MF in combined with or without PKM2 inhibitor (Shikonin, SKN, 0.2 µM) under GD condition. (E) The mRNA level of CCND1 and c-Myc in A498 cells treated as in (D). Every experiment was repeated for 3 times. *, p<0.05; **, p<0.01; ***, p<0.001.
SKN sensitized the RCC xenograft to MF treatment under food starvation. (A) The tumor growth in the control mice and food starvation (FS) group. (B) The tumor growth in the mice received MF (200 μg/ml) and/or injected with SKN (4 mg/kg) under food starvation. (C) The representative tumors. (D,E) The expression of Ki-67 in different groups of tumors was analyzed by western blot (D) and immunochemistry staining (E). N=6 for (A) and (B). *, p<0.05; **, p<0.01.
Discussion
AMPK was originally reported as a tumor suppressor [21], therefore, previous studies primarily focused on the role of AMPK in suppression of cell growth. The AMPK activators, MF, have been used for cancer prevention and therapy. It was recently found that the type 2 diabetes patients treated with MF had lower cancer risk and lower cancer-related mortality rates compared with patients treated with other therapeutics [7]. However, a large controversy exists in the literature related to the AMPK pathway involvement in tumorigenesis and cancer progression, since recent studies suggest that AMPK activation does not decrease risk of colorectal, lung, breast, and prostate cancer [17]. In the present study, we demonstrate the ability of a novel function of AMPK activator in promoting cell growth in RCC cell lines in vitro. Although activation of AMPK suppresses the proliferation of RCC cells in normal condition, it also promotes RCC cell proliferation under GD condition, which might lead to unfavorable clinical outcomes for RCC patient. We found that treatment with an AMPK activator no only enhances its kinase activity, but also lead to its nuclear translocation. The nuclear AMPK therefore recruits PKM2 and β-catenin into a complex in the nuclear, which activate the β-catenin downstream genes' transcription.
AMPK has been classically defined as a master regulator of cellular metabolism in which the activation of AMPK by energetic stress leads to an overall increase in catabolic processes [14]. Several strategies are currently being employed to modulate AMPK activity in cancer. Choi et al. observed that AICAR, an AMPK activator, suppressed cell proliferation rate and apoptosis in thyroid cancer cell line expressing either wild-type BRAF or V600E-mutant BRAF [22]. The anti-proliferative effects of MF in cancer cells are highly dependent on the glucose concentration in the extracellular milieu [23]. Consistent with these previous studies about the anti-proliferative role of AMPK, we also confirmed that activation of AMPK by MF actually inhibited the RCC cell proliferation under normal culture condition. However, different studies demonstrated that AMPK activation mediated by lower ATP/AMP ratio promotes cellular survival under stressful metabolic condition, a characteristic tumor microenvironment [24,25]. Moreover, antagonists of AMPK such as compound C inhibit prostate cancer cell proliferation [26,27]. Therefore, it is unclear whether agonists or antagonists should be used given AMPK's context-dependent role (oncogenic versus tumor suppressive). In our study, we firstly clarified the function of AMPK in RCC cell proliferation. Although AMPK activation processes the tumor suppression function under normal condition, it also enhances RCC cell proliferation under GD condition. Consistent with the previous discoveries that AMPK acts as a nutrient sensor in the cytoplasm [26], our study revealed that the activation of AMPK promotes RCC cell proliferation under stressful metabolic condition. However, the function of AMPK is independent of its kinase activity, instead, the nuclear translocation of AMPK mediated the RCC cell proliferation, which provides a novel function of AMPK. Our results reveal a novel role for AMPK in significantly enriching cancer proliferation properties upon activation that involves a novel nuclear translocation mechanism for PKM2. We demonstrated the physiological interaction between AMPK, PKM2, and β-Catenin, and showed how the protein complexes played an instrumental role in regulating nuclear accumulation of PKM2 and β-Catenin. We also offered evidence suggesting depletion or inhibition of PKM2 can suppress the RCC cell proliferation induced by AMPK activation. Thus, orchestrating the signal pathway of AMPK and PKM2 is important for the interplay between tumor progression and suppression, and will benefit for RCC patient therapy.
PKM2 is the predominant pyruvate kinase isoform in the initiation of leukemia. PKM2 has been demonstrated an important function in promoting cancer cell metabolism and growth, since inhibition of PKM2 by peptide aptamer inhibited cell growth [28], and PKM2 knockdown by siRNA or replacement of PKM2 with PKM1 [29] significantly reduced the tumor volume in nude mice. PKM2 is necessary for aerobic glycolysis of cancer cells, which is a hallmark of cancer metabolism and the major energy source for cancer cell growth and survival [29]. Therefore, PKM2 is a potential therapy target for disrupting glucose metabolism in cancer cells. Though the kinase activity of PKM2 is associated with modulation of aerobic glycolysis in cancer cells, the translocation of PKM2 into the nucleus also play an important role in the expression of the β-catenin target gene, c-Myc [20,30]. Nuclear PKM2 also mediates the epithelial-mesenchymal transition (EMT) of cancer cells upon TGF-β and EGF activation, resulting in the suppression of E-Cadherin transcription [30]. These findings suggested the pivotal role of nuclear PKM2 in the cancer Warburg effect and tumorigenesis. In our study, we observed that PKM2 translocated into the nucleus during AMPK activation in RCC. These results suggest that nuclear translocation of PKM2 was the reason for cancer proliferation upon AMPK activation under GD condition. It has been demonstrated that nuclear PKM2 expression correlates with cancer grading and poor prognosis of patients with glioma and esophageal squamous cell carcinoma [20]. Thus, targeting nuclear translocation of PKM2 in combination with AMPK activation drugs may lead to a novel method of RCC cancer treatment.
Recent evidence indicates that long-term exposure to the metabolic stress caused by nutrient restriction helps cancer cells to adapt and survive, but some of the mechanisms remain elusive [17]. Here, our results revealed a dual role of AMPK in mediating RCC cell proliferation under normal and GD conditions. These important roles of AMPK in different physiological processes may ultimately preclude AMPK itself from ever being a viable therapeutic target. Future RCC cancer treatments may, therefore, be able to target the inhibition of nuclear PKM2 or related complex formation to reduce the side effects caused by AMPK activation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Linehan WM, Rouault TA. Molecular pathways: Fumarate hydratase-deficient kidney cancer-targeting the Warburg effect in cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013;19:3345-52
2. Chow WH, Dong LM, Devesa SS. Epidemiology and risk factors for kidney cancer. Nature reviews Urology. 2010;7:245-57
3. Habib SL, Prihoda TJ, Luna M, Werner SA. Diabetes and risk of renal cell carcinoma. Journal of Cancer. 2012;3:42-8
4. Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nature reviews Urology. 2010;7:277-85
5. Hardie DG. AMP-activated protein kinase: a cellular energy sensor with a key role in metabolic disorders and in cancer. Biochemical Society transactions. 2011;39:1-13
6. Yuan P, Ito K, Perez-Lorenzo R, Del Guzzo C, Lee JH, Shen CH, Bosenberg MW, McMahon M, Cantley LC, Zheng B. Phenformin enhances the therapeutic benefit of BRAF(V600E) inhibition in melanoma. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:18226-31
7. Pollak MN. Investigating metformin for cancer prevention and treatment: the end of the beginning. Cancer discovery. 2012;2:778-90
8. Zhou K, Bellenguez C, Sutherland C, Hardie G, Palmer C, Donnelly P, Pearson E. The role of ATM in response to metformin treatment and activation of AMPK. Nature genetics. 2012;44:361-2
9. Chen G, Xu S, Renko K, Derwahl M. Metformin inhibits growth of thyroid carcinoma cells, suppresses self-renewal of derived cancer stem cells, and potentiates the effect of chemotherapeutic agents. The Journal of clinical endocrinology and metabolism. 2012;97:E510-20
10. Nayan M, Macdonald EM, Juurlink DN, Austin PC, Finelli A, Kulkarni GS, Hamilton RJ, Canadian Drug S, Effectiveness Research N. Medication use and survival in diabetic patients with kidney cancer: A population-based cohort study. Pharmacological research. 2016;113:468-74
11. Keizman D, Ish-Shalom M, Sella A, Gottfried M, Maimon N, Peer A, Hammers H, Eisenberger MA, Sinibaldi V, Neiman V, Rosenbaum E, Sarid D, Mermershtain W, Rouvinov K, Berger R, Carducci MA. Metformin Use and Outcome of Sunitinib Treatment in Patients With Diabetes and Metastatic Renal Cell Carcinoma. Clinical genitourinary cancer. 2016;14:420-5
12. Cheng JJ, Li H, Tan HS, Tan PH, Ng LG, Ng QS, Toh CK, Kanesvaran R, Tan MH. Metformin Use in Relation With Survival Outcomes of Patients With Renal Cell Carcinoma. Clinical genitourinary cancer. 2016;14:168-75
13. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nature reviews Molecular cell biology. 2007;8:774-85
14. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell metabolism. 2005;1:15-25
15. Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS letters. 2003;546:113-20
16. Palorini R, Votta G, Pirola Y, De Vitto H, De Palma S, Airoldi C, Vasso M, Ricciardiello F, Lombardi PP, Cirulli C, Rizzi R, Nicotra F, Hiller K, Gelfi C, Alberghina L, Chiaradonna F. Protein Kinase A Activation Promotes Cancer Cell Resistance to Glucose Starvation and Anoikis. PLoS genetics. 2016;12:e1005931
17. Yang YC, Chien MH, Liu HY, Chang YC, Chen CK, Lee WJ, Kuo TC, Hsiao M, Hua KT, Cheng TY. Nuclear translocation of PKM2/AMPK complex sustains cancer stem cell populations under glucose restriction stress. Cancer letters. 2018;421:28-40
18. Zhao JX, Yue WF, Zhu MJ, Du M. AMP-activated protein kinase regulates beta-catenin transcription via histone deacetylase 5. The Journal of biological chemistry. 2011;286:16426-34
19. Chen X, Song X, Yue W, Chen D, Yu J, Yao Z, Zhang L. Fibulin-5 inhibits Wnt/beta-catenin signaling in lung cancer. Oncotarget. 2015;6:15022-34
20. Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011;480:118-22
21. Liang J, Mills GB. AMPK: a contextual oncogene or tumor suppressor?. Cancer research. 2013;73:2929-35
22. Choi HJ, Kim TY, Chung N, Yim JH, Kim WG, Kim JA, Kim WB, Shong YK. The influence of the BRAF V600E mutation in thyroid cancer cell lines on the anticancer effects of 5-aminoimidazole-4-carboxamide-ribonucleoside. The Journal of endocrinology. 2011;211:79-85
23. Bikas A, Jensen K, Patel A, Costello J Jr, McDaniel D, Klubo-Gwiezdzinska J, Larin O, Hoperia V, Burman KD, Boyle L, Wartofsky L, Vasko V. Glucose-deprivation increases thyroid cancer cells sensitivity to metformin. Endocrine-related cancer. 2015;22:919-32
24. Luo Z, Zang M, Guo W. AMPK as a metabolic tumor suppressor: control of metabolism and cell growth. Future oncology. 2010;6:457-70
25. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nature reviews Cancer. 2011;11:85-95
26. Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661-5
27. Frigo DE, Howe MK, Wittmann BM, Brunner AM, Cushman I, Wang Q, Brown M, Means AR, McDonnell DP. CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer research. 2011;71:528-37
28. Spoden GA, Mazurek S, Morandell D, Bacher N, Ausserlechner MJ, Jansen-Durr P, Eigenbrodt E, Zwerschke W. Isotype-specific inhibitors of the glycolytic key regulator pyruvate kinase subtype M2 moderately decelerate tumor cell proliferation. International journal of cancer. 2008;123:312-21
29. Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230-3
30. Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nature cell biology. 2012;14:1295-304
Author contact
![]() Corresponding authors: Chunxiang Jin, Department of ultrasonography, China-Japan Union Hospital, Jilin University, No.126 Xiantai Str., Changchun, 130000, China. E-mail: jinchunxiang0115com, Tel: +86 431-84995627. Xiaoliang Chen, Department of Urology, China-Japan Union Hospital, Jilin University, No.126 Xiantai Str., Changchun, 130000, China. E-mail: xlchen15jlu.edu.cn, Tel: +86 431-84997589
Corresponding authors: Chunxiang Jin, Department of ultrasonography, China-Japan Union Hospital, Jilin University, No.126 Xiantai Str., Changchun, 130000, China. E-mail: jinchunxiang0115com, Tel: +86 431-84995627. Xiaoliang Chen, Department of Urology, China-Japan Union Hospital, Jilin University, No.126 Xiantai Str., Changchun, 130000, China. E-mail: xlchen15jlu.edu.cn, Tel: +86 431-84997589
Received 2018-9-3
Accepted 2018-10-31
Published 2019-1-1