Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(4):800-811. doi:10.7150/ijbs.30356 This issue Cite
Research Paper
Dendropanax morbifera Ameliorates Thioacetamide-Induced Hepatic Fibrosis via TGF-β1/Smads Pathways
Hun Yong Yang1, Kyeong Seok Kim1, Yong Hee Lee1, Jae Hyeon Park1, Jung-Hwan Kim2, Seok-Yong Lee1, Young-Mi Kim3, In Su Kim1, Sam Kacew4, Byung Mu Lee1, Jong Hwan Kwak1, Kyungsil Yoon5 ![]() , Hyung Sik Kim1
, Hyung Sik Kim1 ![]()
1. School of Pharmacy, Sungkyunkwan University, 2066, Seobu-ro, Suwon, 16419, Republic of Korea
2. Department of Pharmacology, College of Medicine, Institute of Health Sciences, Gyeongsang National University, Jinju, Republic of Korea
3. College of Pharmacy and Institute of Pharmaceutical Science and Technology, Hanyang University, Ansan, Gyeonggi-do 15588, Republic of Korea
4. McLaughlin Centre for Population Health Risk Assessment, University of Ottawa, Ottawa, Canada
5. Comparative Biomedicine Research Branch, Division of Translational Science, National Cancer Center, 323 Ilsandong-gu, Goyang-si, Gyeonggi-do, 10408, Republic of Korea
Received 2018-10-2; Accepted 2019-1-9; Published 2019-2-13
Abstract

Hepatic fibrosis, characterized by persistent deposition of extracellular matrix (ECM) proteins, occurs in most types of chronic liver disease. The prevention of liver damage using extract of Dendropanax morbifera has been widely studied, but its molecular mechanism on the therapeutic efficacy of hepatic fibrosis is unclear. The aim of this study was to assess whether aquatic extract (DM) of D. morbifera ameliorates thioacetamide (TAA)-induced hepatic fibrosis. Hepatic fibrosis was induced by an intraperitoneal (i.p.) injection (150 mg/kg, twice per week) of TAA for 6 weeks. DM (50 mg/kg/day) or silymarin (50 mg/kg/day) was administered daily for 6 weeks. DM markedly reduced serum AST, ALT, ALP, and r-GTP in TAA-treated rats. DM significantly ameliorated the total glutathione (GSH), superoxide dismutase (SOD), and catalase (CAT) activity in TAA-treated rats. In particular, DM significantly reduced expression of α-SMA, type I collagen, vimentin, TGF-β1 and p-Smad2/3 in hepatic fibrosis rats. The protective effects of DM on progression of hepatic fibrosis were clearly shown by detecting 4-hydroxyproline concentration and histopathological examination in the liver. Therefore, our data suggest that DM dramatically prevented hepatic fibrosis by inhibiting oxidative stress and the TGF-β1/Smads signaling pathways.
Keywords: hepatic fibrosis, thioacetamide, TGF-β1, α-smooth muscle actin, Dendropanax morbifera
Introduction
Hepatic fibrosis is characterized by excessive accumulation of extracellular matrix (ECM) proteins resulting from chronic liver damage due to viral hepatitis, or alcoholic or non-alcoholic steatohepatitis [1-3]. Hepatic fibrosis is considered a reversible process, whereas the irreversible results of hepatic dysfunction are usually end-stage liver fibrosis and subsequent mortality [4]. According to the Global Burden of Disease Study in 2013, cirrhosis was among the top 10 most common causes of death in various regions of the world and was the sixth most common cause of death in developed countries [5, 6]. However, the mortality rate associated with liver cirrhosis in developing countries is gradually increasing [7]. As effective anti-fibrotic drugs have not been developed by pharmaceutical companies, hepatic fibrosis is expected to remain an important clinical challenge in the 21st century.
The ultimate manifestations of hepatic fibrosis are characterized by scar accumulation and nodule formation encapsulated by ECM [8, 9]. Overproduction of collagen I play a direct causative role in liver fibrogenesis [1, 3, 10]. In general, activation of hepatic stellate cells (HSCs) leads to excessive ECM production, as well as the secretion of cytokines or chemokines by Kupffer cells and inflammatory cells that may be activated during hepatic fibrosis [11, 12]. Transforming growth factor beta (TGF-β) is the most important pro-fibrotic cytokine because it stimulates ECM synthesis and inhibits ECM degradation [13, 14]. TGF-β1 appears to stimulate collagen synthesis and the differentiation of fibroblasts into myofibroblasts [13, 15]. TGF-β1 also stimulates fibroblasts by activating HSCs through the TGF-β1/Smad signaling pathway [16, 17]. Therefore, compounds capable of suppressing TGF-β1-induced collagen production represent good candidate drugs that can interfere with development of hepatic fibrosis. In addition, the c-Jun N-terminal kinase/stress-activated protein kinase (JNK)/p38 signaling cascade, a non-Smad pathway, is activated by TGF-β1. However, stimulated JNK/p38 acts cooperatively with Smad to regulate apoptosis and the epithelial to mesenchymal transition (EMT) through the activation of transcription factors [18, 19].
A rat model of thioacetamide (TAA)-induced fibrosis was used to study fibrosis regression because of its reproducibility, ease of use, and appropriate simulation of the mechanisms involved in human liver fibrosis [20, 21]. In general, TAA-treated animals induced acute liver failure upon bioactivation into sulfene or sulfone metabolites of TAA via the CYP450 system [20]. The reactive radicals of intermediate metabolites of TAA induce oxidative stress in hepatocytes, resulting in necrosis of the lobular center and liver damage [21, 22]. Therefore, it is generally accepted that oxidative stress is an important phenomenon for regulation of liver fibrosis [23]. Furthermore, oxidative agents and lipid peroxidation products contribute to release of profibrogenic growth factors, cytokines, and prostaglandins [24, 25]. Thus, reactive oxygen species (ROS) play a critical role in initiation of fibrosis through the secretion of various profibrotic factors.
Recently, plant-derived natural products have emerged as compounds with potential to effectively prevent liver fibrosis since there is no specific antifibrotic therapy. Therefore, further research has been extensively studied on the complementary or alternative medicine for prevention of liver fibrosis. Dendropanax morbifera is used in folk remedies to treat migraines, menstrual pain, and wind-dampness [26, 27]. The roots, leaves, and stems of this plant contain a variety of biologically active compounds, including polyphenol and polyacetylene compounds, which exhibit potent antioxidant, anti-inflammatory, anti- cancer, anti-complement, and anti-diabetic properties [28-30]. A number of studies have reported that methanolic extracts of D. morbifera displayed potent antioxidant activity [30, 31]. However, the molecular mechanism of water extract from D. morbifera against hepatic fibrosis is not well understood. Thus, the aim of the present study was to investigate the potential role of water extract from D. morbifera in hepatic fibrosis induced by TAA-treated animal model. To the best of our knowledge, this investigation is the first report to examine the molecular mechanisms underlying the effects of D. morbifera on the prevention and treatment of hepatic fibrosis. In the present study, we included silymarin as a positive control for the protective effects on TAA-induced liver fibrosis, because it is used clinically as a hepatoprotective drug in Europe and Asia [32].
Materials and methods
Materials
TAA and silymarin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Assay kits used to measure aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP), and r-glutamyl transferase (GPT) were purchased from Abcam (Cambridge, UK). Malondialdehyde (MDA) assay kit was purchased from the R&D Systems (Minneapolis, MN, USA) and total glutathione (GSH) assay kit was purchased from Enzo Life Sciences (NY, USA). All other chemicals used in this study were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Plant Material and Preparation of Aquatic Extract from D. morbifera
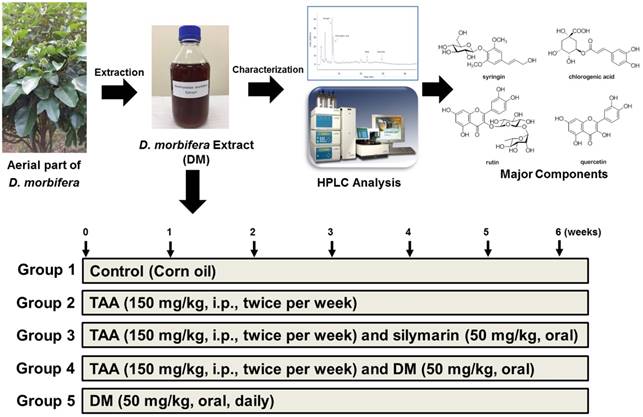
The aerial parts of D. morbifera were collected at Gwangyang, Jeollanam-do, Korea in September 2016. A voucher specimen was deposited at the School of Pharmacy, Sungkyunkwan University (SKKU-Ph-16- 031). The dried aerial parts of D. morbifera (100 g) were extracted twice with water (1 L) at 90oC for 5 h. The extracts were combined, and were concentrated under reduced pressure to prepare a water extract (1 L volume) of D. morbifera. The water extract (DM, 1 L) was stored at 4oC until use.
HPLC analysis for Water Extract of D. morbifera
To characterize DM, quantitative analysis of main components was performed on a reverse phase C18 column using a gradient elution. HPLC analysis was carried out by using a Knauer Smartline system (a Manager 5000, two Pump 1000 and an UV Detector 2500) equipped with a Phenomenex Kinetex C18 column (5 µm, 100 A, 150 × 4.6 mm). The mobile phases were 0.1% formic acid in water (A) and 0.1% formic acid in methanol (B). The gradient profile was as follows: 0-5, isocratic elution with 80% A in B; 5-30 min, linear change from 80% to 50% A in B; 30-40 min, isocratic elution with 50% A in B; 40-45 min, linear change from 50% to 0% A in B. The flow rate and column oven temperature were set at 1 mL/min and 30°C, respectively. UV absorption was measured at a wavelength of 254 nm. The four main components detected as major peaks at 254 nm were quantified using standard curves for each compound.
Experimental design
Eight-week-old male Sprague-Dawley rats (body weight approximately 250 ± 5 g) were purchased from Charles River (Japan) and housed in an air- conditioned room at 21°C with a relative humidity of 50 ± 5% and subjected to a 12 h light/dark cycle. The rats were provided with food (PMI, Brentwood, MO) and tap water ad libitum. All animals were acclimated for 2 weeks prior to experimental use. TAA was injected to induce hepatic fibrosis in accordance with the method of Starkel and Leclercq [33], with slight modifications. The rats were randomly allocated to control and drug treatment groups of six rats per group: 1) Control; 2) TAA only; 3) TAA followed by silymarin; 4) TAA followed by DM; 5) DM alone. All experiments were conducted in accordance with the Ministry of Food and Drug Safety Health guideline for the care and use of Laboratory animals (MIDF Publications No. 152785, revised 2018) and were approved by the Institutional Animal Care and Use Committee of the Sungkyunkwan University (Approval Number SKKUIACUC-17-8-15).
To induce hepatic fibrosis, TAA (150 mg/kg, dissolved in normal saline) was injected intraperitoneally (i.p.) to rats twice per week for 6 weeks. DM (50 mg/kg/day) were daily administered by oral gavage for 6 weeks. The dosage of DM used in this study was calculated based upon the daily consumption for human intake as determined from folk medicine in Korea. The TAA + DM group received i.p. injections of TAA followed by oral administration of DM for 6 weeks. The silymarin group received i.p. injections of TAA followed by administration of silymarin (50 mg/kg/day) by oral gavage for 6 weeks (Fig 1). After the final treatment, all animals were anesthetized with CO2. Blood was collected from the abdominal aorta into heparinized tubes; plasma samples were obtained after centrifugation at 3,000 g for 15 min. Liver samples were collected for histological and molecular analysis. All samples were immediately stored at -80°C until analysis.
The procedure of water extraction of D. morbifera and characterization of major components. Animal experimental design.
Serum biochemical analysis
Serum samples were aliquoted into sterile tubes and frozen at -80°C within 2 h of collection for subsequent analysis. The activities of AST, ALT, ALP, and r-glutamyl transferase (r-GTP) were analyzed by using a VetScan analyzer (Abaxis, Inc., Union City, CA).
Histopathological examination and Masson's trichrome staining
After the rats were sacrificed, the livers were excised, fixed overnight in 10% neutral buffered formalin, and dehydrated with 70% ethanol. After liver tissue was embedded in paraffin, 5 μm-thick sections were cut and mounted on slides. The slide sections were stained with hematoxylin & eosin (H&E) or Masson's trichrome (MT). Photomicrographs of liver sections were taken at 200× magnification using a light microscope (Zeiss Axiophot, Oberkochen, Germany) fitted with a Sony 3CCD camera (AVT Horn, Aalen, Germany).
Measurement of total GSH content
A portion of the liver tissue was washed with cold PBS containing 0.16 mg/mL heparin to prevent coagulation. Total glutathione (GSH) content was measured by using an assay kit in accordance with manufacturer instructions (Enzo Life Sciences (NY, USA). Tissue samples (100 mg) were homogenized in ice-cold 5% (w/v) meta-phosphoric acid and centrifuged at 12,000 g for 10 min at 4°C. The reaction mixture was added to the diluted samples (10 µL) and measured using a VetScan analyzer (Abaxis, Union City, CA). Total GSH was expressed as nmol/mg protein and quantitated using a standard curve.
Assay of superoxide dismutase activity
Superoxide dismutase (SOD), which catalyzes transformation of superoxide anion into H2O2 and O2, was measured using a colorimetric SOD assay kit (Cayman Chemical Co., Ann Arbor, MI) in accordance with manufacturer instructions. Liver tissue (100 mg) was homogenized in cold HEPES buffer (pH 7.2) containing 1 mM EGTA, 210 mM mannitol, and 70 mM sucrose. The tissue homogenate was collected by centrifugation at 1,500 × g for 5 min at 4°C; subsequently, the pellet was discarded. The supernatant (10 µL) was mixed with a diluted radical detector (200 µL) and the reaction was initiated by addition of 20 µL diluted xanthine oxidase. SOD activity was measured at 450 nm. SOD activity was expressed as U/mg protein and quantitated using a standard curve.
Assay of catalase activity
Catalase (CAT) is a ubiquitous antioxidant enzyme involved in detoxification of hydrogen peroxide (H2O2), a toxic product of both normal aerobic metabolism and pathogenic ROS production [34]. The peroxidase function of CAT was measured utilizing a colorimetric CAT assay kit (Cayman Chemical Co., Ann Arbor, MI) in accordance with manufacturer instruction. Liver tissue (100 mg) was homogenized in cold buffer (pH 7, 50 mM potassium phosphate with 1 mM EDTA) and centrifuged at 10,000 × g for 15 min at 4°C. The samples were added to a 96-well plate with 30 µl methanol and 100 µl assay buffer in each well. The catalytic reaction was initiated by addition of 5 µl diluted (10x H2O2). The plate was placed on a shaker for 20 min at room temperature, after which the reaction was terminated by addition of 30 µL potassium hydroxide to each well. After addition of 30 µL CAT solution, the plate was incubated for 10 min at room temperature. Finally, 10 µL potassium periodate was added to each well, and the plate was placed on a shaker for 5 min at room temperature. The samples were analyzed using a microplate reader at 540 nm. CAT activity was expressed as nmol/mg protein, and the values were calculated using a standard curve.
Measurement of malondialdehyde (MDA)
The concentration of MDA, an index of lipid peroxidation, was determined based on production of thiobarbituric acid reactive species (TBARS). MDA concentrations were calculated using a TBARS assay kit (cat. no. KGE013) in accordance with manufacturer instructions (R&D Systems, Inc) and normalized to protein concentration. In brief, equal volumes (100 μL) of sample and SDS were added to a 5 mL conical vial. After vortex mixing, the samples were mixed with 0.4 mL of 1% thiobarbituric acid in 50 mm NaOH and 0.2 mL (20%) H3PO4. The mixture was heated to 100˚C for 15 min. After 10 min incubation on ice, the vials were centrifuged at 1,600 × g for 10 min at 4°C. The samples (100 µL) were loaded onto 96-well assay plates, and the absorbance of each well was measured at 540 nm using a microplate reader.
Western blotting analysis
For western blot analysis, liver samples were homogenized in lysis buffer (Cell Signal Technology Inc., Danvers, MA) and centrifuged at 20,000 × g for 60 min at 4°C. Supernatants were considered to represent total liver proteins and were analyzed using western blotting. Protein concentrations were determined using Bio-Rad protein assay, and equal amounts of the extracted proteins were denatured by boiling for 5 min in sample buffer. Equal amounts of protein (10 mg) were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) at 100 V for 90 min on a 6-12% gradient. Proteins were electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (Merck) at 100 V for 90 min in transfer buffer. Membranes were blocked in TBS-T (50 mM Tris-HCl, pH 7.6, 200 mM NaCl and 0.1% Tween-20) with 5% skim milk for 1 h. Specific proteins were then detected via incubation with the corresponding primary antibodies (α-SMA, vimentin, type I collagen, p-Smad2/3, Smad2/3, p-p38, p38, p-JNK, JNK, Bcl-2, Bax, caspase 3, cleaved-caspase 3, p53, TGF-β1, and β-actin) in TBS-T containing 2% BSA for 2 h at room temperature. After washing for 60 min with Tris- NaCl-Tween (TNT) buffer, membranes were incubated for 30 min with horseradish peroxidase- conjugated goat anti-rabbit IgG or anti-mouse IgG (1:5000), and then washed for 60 min in TNT buffer. Immunoreactivity was visualized using chemiluminescence (ECL) in conjunction with western blotting detection reagents (Amersham Biosciences, Amersham, Buckinghamshire, UK).
Measurement of TGF-β1
The concentration of TGF-β1 in serum was measured using an ELISA kit (R&D Systems, Minneapolis, MN). Before being assayed, samples were activated with equal volumes of 2.5 N acetic acid/10 M urea, followed by 2.7 N NaOH/1 M HEPES, in accordance with the kit protocol. Samples were diluted at a 1:24 ratio to eliminate matrix interference, added to wells coated with a monoclonal antibody specific for TGF-β1, and incubated for 1 h. The plate was washed, leaving only bound TGF-β1 on the plate. Add 100 µL of TGF-β1 conjugate was added to the plate and incubated for 2 h at room temperature. Plate was washed two times with washing buffer. A substrate solution was added to each well and incubated for 30 min at room temperature. Stop solution was added and determine the optical density of each well within 30 min, using a microplate reader set to 450 nm. All samples were assayed in duplicate in a blinded fashion; the mean was used for data analysis.
Measurement of hydroxyproline
To hydrolysis, liver tissues (100 mg) were mixed with 10 ml of 5 N HCl and incubated for 12 h at 120°C. The hydrolyzed sample was passed through a 0.45 µm PVDF syringe filter (Sigma-Aldrich) and evaporated to dryness under vacuum at 70°C for 45 min. Hydroxyproline was measured using a hydroxyproline assay kit (ab222941, Abcam, Cambridge, MA) in accordance with manufacturer's instructions and results expressed as micromoles per mg of protein. All samples were assayed in duplicate in a blinded fashion; the mean was used for data analysis.
Immunohistochemical examination
Three-micrometer-thick liver tissues were embedded in paraffin, deparaffinized, and serially dehydrated in ethanol. The tissue sections were then treated with 5% H2O2 for 10 min to inactivate endogenous peroxidase, blocked for 20 min in PBS containing 10% normal goat serum, and incubated with the appropriate primary antibodies. To verify α-SMA expression, the liver sections were blocked in 3% goat serum containing 0.3% Triton X-100 and incubated overnight with an anti-α-SMA antibody (1:200). After reaction with biotinylated goat anti-mouse IgG (1:500), the sections were treated with HRP-streptavidin and visualized by an Ultravision detection system using a DAB plus substrate system. Photomicrographs of the liver sections were collected at 200× magnification using a Zeiss Axiphot light microscope (Zeiss, Oberkochen, Germany) fitted with a PAX-it!TM (PAXcam, Villa Park, IL, USA).
Statistical analysis
All data are expressed as the means ± SD. The results shown were obtained from at least three independent experiments. Statistical significance was evaluated by one-way analysis of variance (ANOVA) followed by Dunnett's post-hoc multiple comparison test using SigmaStat software package (Jandel Scientific). A value of *p<0.05 was considered to indicate a statistically significant difference.
Results
Phytochemical characterization of aquatic extract from D. morbifera
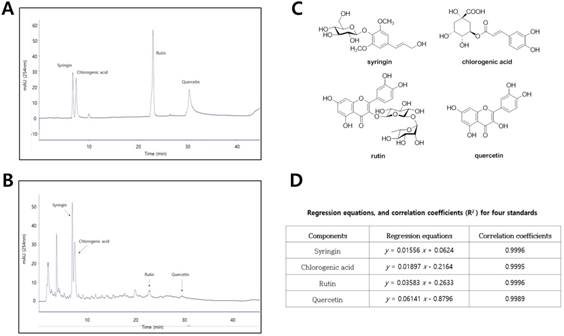
To characterize aquatic extracts of D. morbifera (DM), four compounds were measured by HPLC analysis (Fig. 2A and 2B). The main components in DM, corresponding to the four major peaks in the HPLC-UV chromatogram, were identified as syringin (RT, 6.88 min), chlorogenic acid (RT, 7.36 min), rutin (RT, 19.92 min), and quercetin (RT, 22.92 min) (Fig 2C). Using a standard curve for quantitation, the major components in aquatic extracts of DM were syringin (450.5 μg/mL), chlorogenic acid (391.4 μg/ mL), rutin (21.6 μg/mL), and quercetin (10.7 μg/mL), respectively (Fig. 2D).
Phytochemical characterization of water extracts of D. morbifera (DM) by HPLC analysis. (A) HPLC-UV chromatogram for four standard compounds (syringin, chlorogenic acid, rutin, and quercetin), (B) HPLC-UV chromatogram for water extracts of D. morbifera (DM), (C) Chemical structures of four major compounds in DM, (D) Regression equations and correlation coefficients for four standard compounds. HPLC analysis was carried out using a gradient elution on a Phenomenex Kinetex C18 column (5 µ, 100 A, 150 × 4.6 mm). The flow rate, column oven temperature, and UV wavelength for detection were set at 1 mL/min, 30 °C, and 254 nm, respectively.
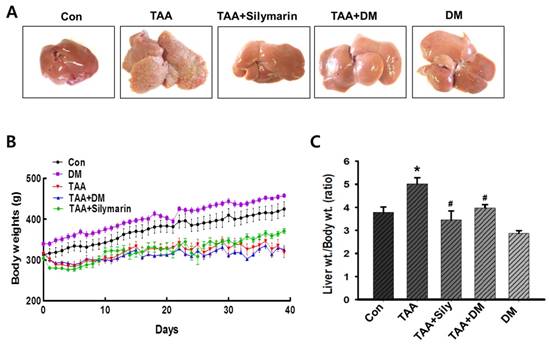
DM attenuated TAA-mediated body and liver weight changes
Injection of TAA for 6 weeks resulted in severe morphological changes in livers of rats, as evidenced by formation of small nodules on the external surface (Fig. 3A). However, treatment with DM or silymarin ameliorated morphological changes in the liver (Fig. 3A). A significant decrease in body weight gain was exhibited in TAA-treated group. TAA and followed by DM or silymarin administration group did not affect the body weight gains (Fig. 3B). In particular, a significant increase in relative liver weight was observed in the TAA group, whereas DM (50 mg/kg) or silymarin (50 mg/kg) dramatically protected the TAA-mediated changes in liver weights. No significant differences in liver weights were observed in rats treated with DM alone (Fig. 3C). Thus, DM or silymarin administration may protect against TAA-induced liver injury.
Inhibitory effects of DM on thioacetamide (TAA)-induced hepatotoxicity. Male Sprague-Dawley rats were injected twice a week with TAA (150 mg/kg) for 6 weeks in the presence or absence of DM or silymarin. DM extracts (50 mg/kg/day) and silymarin (50 mg/kg/day) were administered to rats daily by oral gavage for 6 weeks. (A) Effect of DM on liver morphological changes. Effect of DM or silymarin on body weight changes (B) and relative liver weights (C). The data are expressed as mean ± SD (n = 6 per group). Significant differences from the Con group at p* < 0.05; Significant difference from the TAA group at p# < 0.05. TAA; thioacetamide.
DM protected TAA-induced liver damages
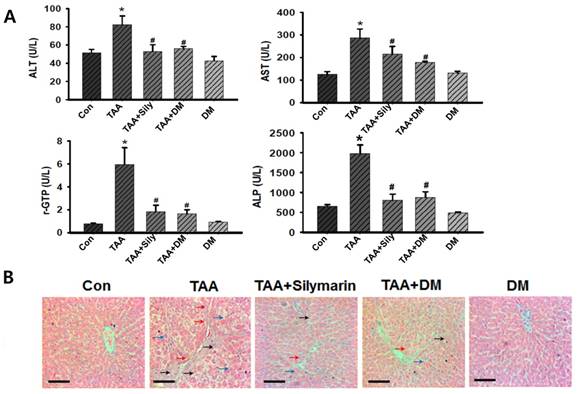
TAA-treated rats exhibited severe liver injury, as evidenced by changes in morphological architecture as shown in Fig. 3A. These findings were also supported by elevated serum biomarkers for liver dysfunction such as AST, ALT, r-GPT, and ALP levels. TAA-treated rats led to significant elevations in ALT, AST, rGPT, and ALP activities compared to those in the control rats (Fig. 4A). However, these liver injury markers were markedly reduced by administration of DM or silymarin in TAA-induced rats. These data indicated that DM or silymarin exerted protective effects against TAA-induced liver injury. In histopathologically finding, the control rats showed normal architecture of the liver lobule with a central vein and portal tracts at the corners of the lobule (Fig. 4B). However, a marked increase in inflammatory infiltrates, degeneration, and necrosis around the central vein or connective tissue was observed in the TAA-treated group. Similar to serum biochemical findings, TAA-induced hepatic injuries were dramatically protected by DM or silymarin administration (Fig. 4B).
Effects of DM on thioacetamide (TAA)-induced hepatotoxicity. Sprague-Dawley male rats were injected twice per week with TAA (150 mg/kg) for 6 weeks in the presence or absence of DM or silymarin. DM extracts (50 mg/kg/day) and silymarin (50 mg/kg/day) were administered to rats daily by oral gavage for 6 weeks. (A) Effects of DM or silymarin on serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), gamma-glutamyltransferase (rGTP), and alkaline phosphatase (ALP) activities. The data are expressed as mean ± SD (n = 6 per group). Significant difference from the control group at p* < 0.05, Significant difference from the TAA group at p# < 0.05. (B) H&E staining analysis of hepatic histological abnormalities showing hepatocellular degeneration (blue arrowheads), infiltration of inflammatory cells (red arrowheads), and vacuolated cells (black arrowheads) (Original magnification: 200×). Scale bars correspond to 100 μm.
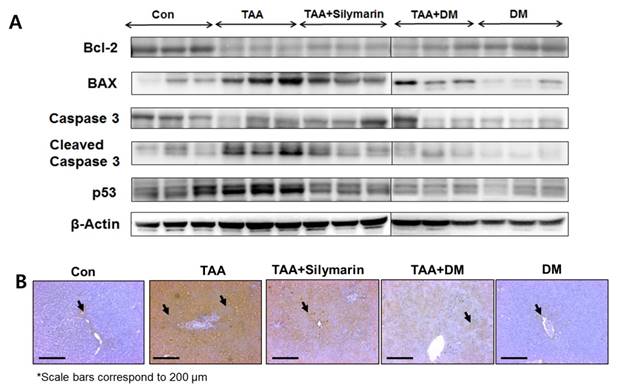
DM prevents TAA-induced liver damage via inhibition of apoptotic cell death
Intermediate metabolites of TAA appear to influence regulation of Bax and Bcl-2 sequences in hepatocytes [35]. In TAA-treated rats, downregulation of Bcl-2 protein and upregulation of Bax, p53, and cleaved caspase-3 were observed in the liver, whereas administration of DM or silymarin markedly protected TAA-induced liver damage via inhibition of apoptosis (Fig. 5A). These results indicated that TAA-induced liver damage was completely protected by DM or silymarin administration. Based on the above observations, we further examined the possible mechanism by which DM attenuates liver injury induced by TAA. The extent of hepatocyte damage such as chromatin condensation were determined by TUNEL assay. As shown in Fig. 5B, massive hepatocyte apoptosis was detected in the liver of rats treated with TAA. DM or silymarin treatment markedly prevented the apoptosis induced by TAA.
Effects of DM on expression of apoptosis-related proteins in thioacetamide (TAA)-induced fibrosis liver. Male Sprague-Dawley rats were injected twice per week with TAA (150 mg/kg) for 6 weeks in the presence or absence of DM or silymarin. DM extracts (50 mg/kg/day) and silymarin (50 mg/kg/day) were administered to rats daily by oral gavage for 6 weeks. (A) Western blotting analysis of Bcl-2, BAX, Caspase 3, Cleaved Caspase 3, and p53 in representative liver tissues was performed. (B) Apoptosis determined by TUNEL staining of liver sections from rats after TAA administration. TUNEL-positive cells were pointed by arrowheads. Original magnification ×100.
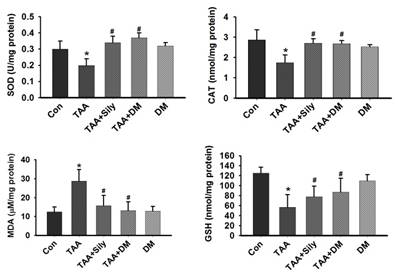
DM induced antioxidant enzyme activities and inhibited MDA concentrations in liver
The antioxidant effects of DM have been reported [27]. Therefore, we hypothesized that DM protected against TAA-induced liver injury through suppression of oxidative stress. In this study, GSH content was significantly reduced in the liver of rats treated with TAA, but rescued by DM or silymarin administration (Fig. 6). Further, TAA injection significantly reduced the activities of SOD and CAT in the liver, whereas the administration of DM or silymarin recovered SOD and CAT activities. Collectively, these data suggested that DM protects TAA-induced liver damage via antioxidant activity. To confirm whether TAA-induced hepatotoxicity was associated with oxidative stress, we measured MDA, an indicator of lipid oxidation, in the liver. TAA injection increased hepatic MDA content by 263.1% in comparison to the control group. However, MDA content was significantly reduced in the groups treated with DM or silymarin by 54.9% and 48.7%, respectively, compared to the TAA- treated group (Fig. 6).
Effects of DM on antioxidant enzyme activities in the liver. Male Sprague-Dawley rats were injected twice per week with TAA (150 mg/kg) for 6 weeks in the presence or absence of DM or silymarin. DM extracts (50 mg/kg/day) and silymarin (50 mg/kg/day) were administered to rats daily by oral gavage for 6 weeks. Activities of GSH, SOD, and catalase, and the content of MDA, were measured in liver tissue. The data are expressed as mean ± SD (n = 6 per group). MDA, Malondialdehyde; GSH, Glutathione; SOD, Superoxide dismutase; CAT, catalase. Significant difference from the control group at p* < 0.05, Significant difference from the TAA-treated group at p# < 0.05.
DM inhibited the production of ECM in hepatic fibrosis models
To assess the effect of DM on hepatic fibrogenesis caused by TAA, liver sections were analyzed using Masson's stain to detect collagen deposition. The results showed that collagen was aggressively deposited in the liver of TAA-injected rats accompanied by nodular formation. This effect was significantly protected by DM or silymarin administration in the TAA-treated rats (Fig. 7A). Similarly, collagen around the extracellular space, especially in the portal triad, significantly increased in livers of TAA-induced rats (Fig. 7A). The lobules in the TAA-treated group were surrounded by a bundle of blue-stained collagen fibers, but this damage was markedly lower in the DM or silymarin treatment groups. In addition, immunohistochemical staining of liver tissue indicated that the number of α-SMA- positive cells was lower in the fibrous septa and the sinusoid in the livers of DM or silymarin administration in TAA-treated rats (Fig. 7A). These results suggested that TAA-mediated ECM production was markedly inhibited by DM or silymarin, which blocked the progression of liver fibrosis.
Effects of DM on the expression of extracellular matrix (ECM) proteins, hydroxyproline content and histopathological changes in thioacetamide (TAA)-induced liver. Sprague-Dawley male rats were injected twice per week with TAA (150 mg/kg) during 6 weeks in the presence or absence of DM or silymarin. DM extracts (50 mg/kg/day) and silymarin (50 mg/kg/day) were administered to rats daily by oral gavage for 6 weeks. (A) DM or silymarin ameliorated pathological changes in liver as shown by immunohistochemistry (IHC) and Masson's trichrome (MT) staining (original magnification: 200×). (B) Western blotting analysis of α-SMA, collagen I, and vimentin. (B) 4-Hydroxyproline content in the livers from treated rats. The data are expressed as the mean ± SD (n = 6 per group). Significant difference from the control group at p* < 0.05, Significant difference from the TAA-treated group at p# < 0.05.
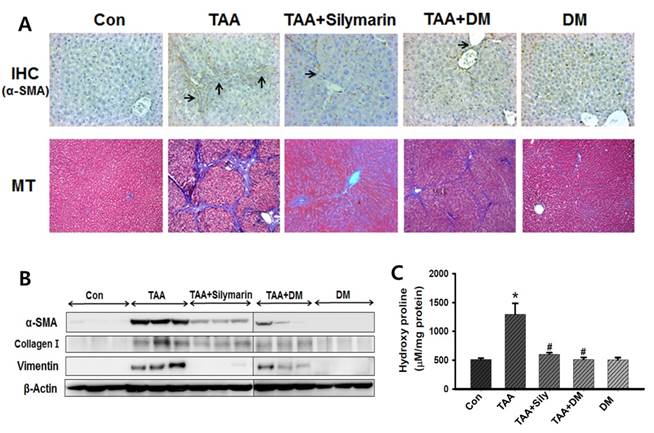
Protein expression of α-SMA, collagen-1, and vimentin was significantly increased in TAA-treated group (Fig. 7B). DM or silymarin caused a significant reduction in the levels of α-SMA, collagen-1, and vimentin expression by western blotting. In general, hydroxyproline, which can reflect the severity of liver fibrosis, is a characteristic amino acid in collagen synthesis [36]. Hydroxyproline levels were significantly higher (by 237%) in livers of TAA-induced rats compared to control rats. Administration of DM or silymarin resulted in a significant reduction in hepatic hydroxyproline levels in the TAA-injected group (Fig. 7C).
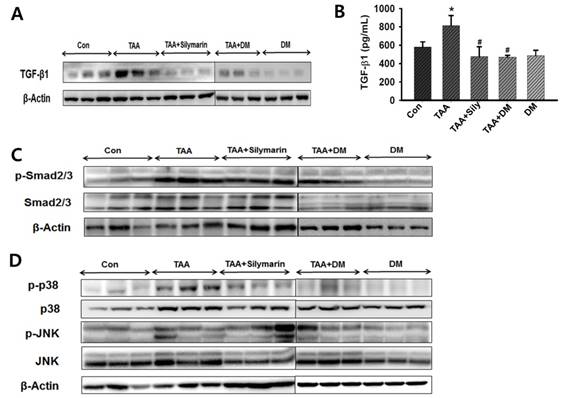
DM attenuated hepatic damage via the TGF-β1/Smads signaling pathway
As TGF-β1 is responsible for increased ECM in the fibrotic liver [17, 37], we measured TGF-β1 expression in serum to investigate the protective effect of DM against hepatic fibrosis. Liver expression of TGF-β1 was measured by western blotting (Fig. 8A). TGF-β1 content was quantified in serum using ELISA (Fig. 8B). Serum content of TGF-β1 was significantly higher in the TAA-treated group than in the control group, but DM or silymarin administration significantly ameliorated serum TGF-β1 levels in the TAA-induced rats. Indeed, Smad2/3 are major factors in the TGF-β1 signaling pathway. Inhibition of Smad2/3 phosphorylation blocks the downstream signaling pathway in development of fibrosis [38], resulting in a subsequent reduction in hepatic fibrosis. To further determine the role of DM on TGF-β and Smad2/3 pathways, we investigated whether DM or silymarin administration modulated the Smad2/3 and MAPKs pathways. As shown in Fig. 8C, p-Smad2/3 expression was increased in the TAA- treated group. In contrast, DM or silymarin administration dramatically reduced phosphorylation of Smad2/3 in liver of TAA-treated rats. MAPKs exert different regulatory actions on cell proliferation. The expression of p-p38 and p-JNK proteins associated with the TGF-β1/non-Smad3 signaling pathway was reduced in the DM-treated group compared to that in the TAA-treated group. Our data also showed that DM or silymarin administration selectively reduced JNK phosphorylation by blocking the TGFβ/Smad pathways during development of TAA-induced liver fibrosis (Fig. 8D).
Effects of DM on expression of TGF-β1, Smad2/3, and MAPKs in livers of Sprague-Dawely rats treated with thioacetamide (TAA). Male rats were injected twice per week with TAA (150 mg/kg) for 6 weeks in the presence or absence of DM or silymarin. DM extracts (50 mg/kg/day) and silymarin (50 mg/kg/day) were administered to rats daily by oral gavage for 6 weeks. (A) Western blotting of TGF-β1 in liver tissue. (B) ELISA assay of TGF-β1 in serum. (C-D) Western blotting of p-Smad2/3, Smad2/3, p38/JNK phosphorylation, non-Smad pathway, in representative liver tissues. The data are expressed as the mean ± SD (n = 6 per group). Significant difference from the control group at p* < 0.05, significant difference from the TAA group at p# < 0.05.
Discussion
It is critical to identify new effective drugs to prevent development of liver fibrosis without adverse effects. The whole plant of DM has been used as an anti-inflammatory and antipyretic drug in many local traditional medicines, including Korean and Chinese herbal medicine. To the best of our knowledge, there has been no study on the protective effect of DM against hepatic fibrosis. Thus, the current study was designed to assess the protective effect of DM against TAA-induced liver fibrosis using animal model.
TAA injection induced a significant increase in oxidative stress and inflammation in livers of rats. Thus, use of this model was suitable for investigating the protective effects of DM against hepatic fibrosis. Previous studies indicated that a decrease in body weight and an increase in liver weight were typical symptoms of TAA-induced hepatic toxicity [21, 22]. These results were similar to our data showing that TAA-injected rats also exhibited liver enlargement as evidenced by liver hyperplasia. Increased liver weight resulting from TAA injection seems to be due to the accumulation of ECM proteins such as collagen [12]. Further, rats exposed to TAA showed a significant elevation of serum enzyme activities (ALT, AST, ALP, or rGTP). However, our data showed that administration of DM produced a significant improvement in hepatic damage markers. Further, silymarin significantly reduced TAA-induced liver enzymes, in agreement with results of other studies [39, 40]. It has been hypothesized that DM and silymarin may stabilize the membrane structure of hepatocytes and prevent cellular damage induced by TAA and its metabolites.
It is widely known that oxidative damage is a cause of liver fibrogenesis. Oxidative stress has been considered as a major molecular mechanism of TAA-induced hepatotoxicity [22]. Oxidative stress plays an important role in TAA-induced liver fibrosis [21, 23]. Several sources of ROS contribute to liver damage, including the epithelium, activated inflammatory cells, and/or microvascular endothelium. Antioxidant enzymes, such as CAT, SOD, and non-enzymatic electron acceptors, such as GSH, are indicators of oxidative stress in liver damage [34]. Recently, various antioxidants were shown to prevent or treat liver damage [41, 42]. We observed that TAA-treated rats exhibited decreased SOD and CAT activities, and decreased content of GSH in the liver. In contrast, treatment with DM significantly increased activities of SOD and CAT, and content of GSH. Therefore, DM can play an antioxidant role in liver fibrosis through helping recover the natural antioxidant defense system of the organism. MDA is a reactive carbon compound that is used as an indicator of lipid peroxidation [42]. MDA levels were significantly increased in liver tissue of TAA-treated rats, demonstrating involvement of ROS-mediated liver injury in the pathogenesis of fibrosis. The protective effect of DM is thought to be connected to its free radical scavenger effect and antioxidant activity, which decreases oxidative stress caused by TAA-mediated ROS generation.
The first stage of liver damage is characterized by increased hepatocyte cell death [43]. We investigated whether DM suppressed apoptosis by examining protein expression of Bcl-2, Bax, and caspase-3, as apoptotic cell death signaling is a key regulator of mitochondrial damage [44]. Moreover, if damage is too extensive to repair and cells must be removed, p53 can induce apoptosis by transcriptional up-regulation of BH3 (Bcl-2 homologous 3)-specific proteins such as Noxa, Puma and/or Bid, and/or the multi-domain protein Bax [45, 46]. The results of this experiment showed modulated Bcl-2 expression, downregulated Bax and p53, and inhibited caspase-3- mediated cell death via Bcl-2/Bax signaling. Western blotting showed that Bcl-2 expression was upregulated and Bax, p53, and cleaved caspase-3 were downregulated in the group treated with TAA in combination with DM or silymarin. These results demonstrated that DM exerted anti-apoptotic effects to protect against liver damage.
To explore potential mechanisms of protective effects of DM in liver fibrosis, we measured the content of 4-hydroxyproline, which represents the majority of collagen content, in liver tissue as a marker for ECM accumulation [47]. Furthermore, hydroxyproline is regarded as a good marker for determination of the extent of fibrosis and evaluation of the efficacy of new antifibrotic agents [45, 46]. Our results implied that DM administration significantly decreased the content of hydroxyproline. Based on this data, we suggested that DM significantly reduced ECM accumulation during chronic liver fibrosis, and that HSCs underwent a process of minimizing intracellular fat droplets and differentiation into myofibroblast-like cells, which induced increased expression of ECM proteins such as α-SMA, collagen I, and vimentin, as indicated in previous studies [48, 49]. Expression of the ECM-related protein, α-SMA, was significantly decreased by DM treatment in liver of TAA-treated rats. It has been demonstrated that DM can act as an anti-fibrotic agent through inhibition of HSC activation and proliferation in results obtained via MT staining and immunohistochemistry (IHC).
Previous studies have demonstrated that inhibition of the TGF-β1 signaling pathway attenuates liver fibrosis [50]. We observed that TGF-β1 expression was significantly elevated in livers of TAA-treated rats and DM markedly reduced TAA-induced TGF-β1 expression. Indeed, TGF-β1 modulates various cellular processes, and its signaling is activated by phosphorylation of Smad2/3 [51, 52]. Our data clearly showed that DM administration significantly reduced the expression of p-Smad 2/3 in TAA-treated rats. However, MAPK pathway was activated by TGF-β1 independently of Smad [53]. Therefore, our data suggested that DM suppressed formation of EMT via TGF-β1 or a non-Smad pathway. However, prior to initiation of clinical investigations, more studies on the molecular mechanism are needed to clarify exactly how DM reduces TGF-β1 levels in the serum of hepatic patients. The MAPK family, including the three major subgroups ERK, p38, and JNK, is involved in proliferation and activation of HSCs, as well as aggravation of hepatic fibrosis. Thus, blocking ERK1/2 can reverse Bcl-2 upregulation and Bax downregulation in activated HSCs [18, 53, 54]. In the present work, DM downregulated levels of p-p38, and p-JNK, which was likely associated with the induction of liver damages, activated hepatic stellate cells. Thus, the present study provided the first in vivo evidence that DM reduced hepatic fibrosis through inhibition of TGF-β1/Smad3 and MAPK signaling in livers of TAA-induced rats.
Taken together, our results indicated that DM markedly protected hepatic dysfunctions in rats damaged by TAA. In particular, hepatic fibrosis markers such as hydroxyproline, type I collagen, and vimentin expression was significantly reduced by DM treatment. DM significantly reduced lipid peroxidetion and increased antioxidant enzyme activities in TAA-treated rats. Therefore, we suggested that the protective effects of DM on hepatic fibrosis were closely associated with inhibition of free radical formation, apoptotic cell death, and TGF-β1 formation. More detailed studies are necessary to completely elucidate the molecular mechanisms behind the protective effect of DM against human hepatic fibrosis.
Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) Grants funded by the Korean Government (NRF-2016R1A4A1011189; NRF-2016R1A4A1011189; NRF-20162017R1D1A1B03032639).
Author Contributions
Conception and design of the experiments: J.H. Kwak, Y.M Kim, B.M. Lee, & H.S. Kim. Collection, analysis, and interpretation of all data: H.Y. Yang, K.S. Kim, Y.H. Lee, J.H. Park, J.H. Kim, & I.S. Kim. Drafting and revising the paper critically for intellectual content: K. Yoon, I.S. Kim, B.M. Lee, S. Kacew, J.H. Kwak and H.S. Kim.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209-18
2. Wynn TA, Ramalingam TR. (2012) Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028-40
3. Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem. 2000;275:2247-50
4. Bae M, Park YK, Lee JY. (2018) Food components with antifibrotic activity and implications in prevention of liver disease. J Nutr Biochem. 2018;55:1-11
5. WHO. Geneva, Switzerland. The top 10 causes of death. Revised 4 February. 2016 http://www.who.int/ mediacentre/ factsheets /fs310/en/index4.html
6. Nilsson E, Anderson H, Sargenti K. et al. Incidence, clinical presentation and mortality of liver cirrhosis in southern Sweden: A 10-year population based study. Aliment Pharmacol Ther. 2016;43:1330-9
7. Mokdad AA, Lopez AD, Shahraz S. et al. Liver cirrhosis mortality in 187 countries between 1980 and 2010: A systematic analysis. BMC Med. 2014;12:145
8. Pinzani M, Rombouts K, Colagrande S. Fibrosis in chronic liver diseases: diagnosis and management. J Hepatol. 2005:42 (Suppl 1); S22-S36
9. Kim B, Farruggia C, Ku CS. et al. Astaxanthin inhibits inflammation and fibrosis in the liver and adipose tissue of mouse models of diet-induced obesity and nonalcoholic steatohepatitis. J Nutr Biochem. 2017;43:27-35
10. Parola M, Robino G. Oxidative stress-related molecules and liver fibrosis. J Hepatol. 2001;35:297-306
11. Seki E, De Minicis S, Osterreicher CH. et al. LR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007;13:1324-32
12. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125-72
13. Yu Y, Sun X, Gu J. et al. Deficiency of DJ-1 ameliorates liver fibrosis through inhibition of hepatic ROS production and inflammation. Int J Biol Sci. 2016;12:1225-35
14. Gressner AM, Weiskirchen R, Breitkopf K. et al. Roles of TGF-beta in hepatic fibrosis. Front Biosci. 2002;7:793-807
15. Garrett Q, Khaw PT, Blalock TD. et al. Involvement of CTGF in TGF-beta1-stimulation of myofibroblast differentiation and collagen matrix contraction in the presence of mechanical stress. Invest Ophthalmol Vis Sci. 2004;45:1109-16
16. Inagaki Y, Okazaki I. Emerging insights into transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut. 2007;56:284-92
17. Feng M, Ding J, Wang M. et al. Kupffer-derived matrix metalloproteinase-9 contributes to liver fibrosis resolution. Int J Biol Sci. 2018;14:1033-40
18. Yu L, Hebert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002;21:3749-59
19. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128-39
20. Koblihová E, Mrázová I, Vernerová Z. et al. Acute liver failure induced by thioacetamide: selection of optimal dosage in Wistar and Lewis rats. Physiol Res. 2014;63:491-503
21. Low TY, Leow CK, Salto-Tellez M, A proteomic analysis of thioacetamide-induced hepatotoxicity, cirrhosis in rat livers. 2004; 4: 3960-74.
22. Wang LC, Kuo IU, Tsai TY. et al. Antrodia camphorata-fermented product cultured in deep ocean water has more liver protection against thioacetamide-induced fibrosis. Appl Microbiol Biotechnol. 2013;97:9955-67
23. Poli G. Pathogenesis of liver fibrosis: role of oxidative stress. Mol Aspects Med. 2000;21:49-98
24. De Minicis S, Marzioni M, Saccomanno S. et al. Cellular and molecular mechanisms of hepatic fibrogenesis leading to liver cancer. Transl Gastrointest Cancer. 2012;1:88-94
25. Crosas-Molist E, Fabregat I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biol. 2015;6:106-11
26. Park BY, Min BS, Oh SR. et al. Isolation and anticomplement activity of compounds from Dendropanax morbifera. J Ethnopharmacol. 2004;90:403-08
27. Hyun TK, Kim MO, Lee H. et al. Evaluation of anti-oxidant and anti-cancer properties of Dendropanax morbifera leveille. Food Chem. 2013;141:1947-55
28. An NY, Kim JE, Hwang D. et al. Anti-diabetic effects of aqueous and ethanol extract of Dendropanax morbifera leveille in streptozotocin-induced diabetes model. J Nutr Health. 2014;47:394-402
29. Choi EH, Lee DY, Kim S. et al. Influence of flavonol-rich excipient food (onion peel and Dendropanax morbifera) on the bioavailability of green tea epicatechins in vitro and in vivo. Food Funct. 2017;8:3664-74
30. Kim M, Park YJ, Lim HS. et al. The clinical effects of Dendropanax morbifera on postmenopausal symptoms. J Menopausal Med. 2017;23:146-55
31. Hyun TK, Ko YJ, Kim EH. et al. Anti-inflammatory activity and phenolic composition of Dendropanax morbifera leaf extracts. Ind Crops Products. 2015;74:263-70
32. Polyak SJ, Ferenci P, Pawlotsky JM. Hepatoprotective and antiviral functions of silymarin components in hepatitis C virus infection. Hepatology. 2013;57:1262-71
33. Starkel P, Leclercq IA. Animal models for the study of hepatic fibrosis. Best Pract Res Clin Gastroenterol. 2001;25:319-33
34. Jurczuk M, Brzoska MM, Moniuszko-Jakoniuk J. et al. Antioxidant enzymes activity and lipid peroxidation in liver and kidney of rats exposed to cadmium and ethanol. Food Chem Toxicol. 2004;42:429-38
35. Chen LH, Hsu CY, Weng CF. Involvement of P53 and Bax/Bad triggering apoptosis in thioacetamide-induced hepatic epithelial cells. World J Gastroenterol. 2006;12:5175-81
36. Lee HS, Shun CT, Chiou LL. et al. Hydroxyproline content of needle biopsies as an objective measure of liver fibrosis: Emphasis on sampling variability. J Gastroenterol Hepatol. 2005;20:1109-14
37. Gressner OA, Lahme B, Demirci I. et al. Differential effects of TGF-beta on connective tissue growth factor [CTGF/CCN2] expression in hepatic stellate cells and hepatocytes. J Hepatol. 2007;47:699-710
38. Liu C, Gaca MD, Swenson ES. et al. Smads 2 and 3 are differentially activated by transforming growth factor-beta (TGF-beta) in quiescent and activated hepatic stellate cells. Constitutive nuclear localization of Smads in activated cells is TGF-beta-independent. J Biol Chem. 2003;278:11721-8
39. Tsai JH, Liu JY, Wu TT. et al. Effects of silymarin on the resolution of liver fibrosis induced by carbon tetrachloride in rats. J Viral Hepat. 2008;15:508-14
40. Clichici S, Olteanu D, Nagy AL. et al. Silymarin inhibits the progression of fibrosis in the early stages of liver injury in CCl4-treated rats. J Med Food. 2015;18:290-8
41. Soylu AR, Aydogdu N, Basaran UN. et al. Antioxidants vitamin E and C attenuate hepatic fibrosis in biliary-obstructed rats. World J Gastroenterol. 2006;12:6835-41
42. Dorğru-Abbasoğlu S, Kanbağli Ö, Balkan J. et al. The protective effect of taurine against thioacetamide hepatotoxicity of rats. Hum Exp Toxicol. 2001;20:23-7
43. Guicciardi ME, Gores GJ. Apoptosis: a mechanism of acute and chronic liver injury. Gut. 2005;54:1024-33
44. Reed JC. Bcl-2 and the regulation of programmed cell death. J Cell Biol. 1994;124:1-6
45. Zhang Y, Miao H, Yan H. et al. Hepatoprotective effect of Forsythiae Fructus water extract against carbon tetrachloride-induced liver fibrosis in mice. J Ethnopharmacol. 2018;218:27-34
46. Saber S, Mahmoud AAA, Helal NS. et al. Renin-angiotensin system inhibition ameliorates CCl4-induced liver fibrosis in mice through the inactivation of nuclear transcription factor kappa B. Can J Physiol Pharmacol. 2018;96:569-76
47. Yum MJ, Koppula S, Kim JS. et al. Protective effects of Ampelopsis brevipedunculata against in vitro hepatic stellate cells system and thioacetamide-induced liver fibrosis rat model. Pharmaceutical Biol. 2017;55:1577-85
48. He J, Bai K, Hong B. et al. Docosahexaenoic acid attenuates carbon tetrachloride-induced hepatic fibrosis in rats. Int Immunopharmacol. 2017;53:56-62
49. Hessin A, Hegazy R, Hassan A. et al. Lactoferrin enhanced apoptosis and protected against thioacetamide-induced liver fibrosis in rats. Open Access Maced J Med Sci. 2015;3:195-201
50. Verrecchia F, Mauviel A. Transforming growth factor-β and fibrosis. World J Gastroenterol. 2007;13:3056-62
51. Meng XM, Huang XR, Chung AC. et al. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol. 2010;21:1477-87
52. Li S, Tan HY, Wang N. et al. The role of oxidative stress and antioxidants in liver diseases. Int J Mol Sci. 2015;16:26087-124
53. Engel ME, McDonnell MA, Law BK. et al. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem. 1999;274:37413-20
54. Yamashita M, Fatyol K, Jin C. et al. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell. 2008;31:918-24
Author contact
![]() Corresponding authors: Hyung Sik Kim, Ph.D., Tel: +82 31 290 7789; Fax: +82 31 290 7748; E-mail: hkimsedu; Kyungsil Yoon, Ph.D., Tel: +82 31 920 2325; Fax: +82 31 920 2337; E-mail: kyoonre.kr
Corresponding authors: Hyung Sik Kim, Ph.D., Tel: +82 31 290 7789; Fax: +82 31 290 7748; E-mail: hkimsedu; Kyungsil Yoon, Ph.D., Tel: +82 31 920 2325; Fax: +82 31 920 2337; E-mail: kyoonre.kr