Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
DNA damage response
Protection of chromosome ends
Cell cycle regulation
Transcriptional regulation
Tumorigenesis and cancer...
Conclusion remarks and...
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(5):909-918. doi:10.7150/ijbs.31972 This issue Cite
Review
The Functions of DNA Damage Factor RNF8 in the Pathogenesis and Progression of Cancer
Tingting Zhou 1, Fei Yi 1, Zhuo Wang 1, Qiqiang Guo1, Jingwei Liu 1, Ning Bai 1, Xiaoman Li 1, Xiang Dong 1, Ling Ren2, Liu Cao 1 ![]() , Xiaoyu Song 1
, Xiaoyu Song 1 ![]()
1. Institute of Translational Medicine, China Medical University; Key Laboratory of Medical Cell Biology, Ministry of Education; Liaoning Province Collaborative Innovation Center of Aging Related Disease Diagnosis and Treatment and Prevention, Shenyang, Liaoning Province, China
2. Department of Anus and Intestine Surgery, First Affiliated Hospital of China Medical University, Shenyang, Liaoning Province, China
Received 2018-12-3; Accepted 2019-2-8; Published 2019-3-9
Abstract

The really interesting new gene (RING) finger protein 8 (RNF8) is a central factor in DNA double strand break (DSB) signal transduction. DSB damage is the most toxic type of DNA damage to cells and is related to genomic instability. Multiple roles for RNF8 have been identified in DNA damage response as well as in other functions, such as telomere protection, cell cycle control and transcriptional regulation. These functions are closely correlated to tumorigenesis and cancer progression. Indeed, deficiency of RNF8 caused spontaneous tumorigenesis in a mouse model. Deciphering these mechanisms of RNF8 may shed light on strategies for cancer treatment. In this review, we summarize the current understanding of both classical and nonclassical functions of RNF8, and discuss its roles in the pathogenesis and progression of tumor.
Keywords: RNF8, DSB, telomere, cell cycle, transcriptional regulation, tumor
Introduction
The really interesting new gene (RING) finger protein 8 (RNF8) is a member of the RING finger family that was initially identified by Seki in 1998 [1]. The gene encoding RNF8 is located at chromosome 6p21.3 and is expressed ubiquitously in a variety of human tissues except for spleen. It consists of two functional domains, which are N-terminal forkhead-associated (FHA) domain and C-terminal RING finger motif. The FHA domain is responsible for protein-protein interaction, while the RING finger motif is involved in ubiquitin ligase activity. Multiple RING finger proteins exist as a cluster at chromosome 6p21.3, and several RING finger proteins are involved in transformation and tumorigenesis, such as PML and TRIM24 [1]. E3 ubiquitin ligases determine substrate specificity and ubiquitination type, and the RING finger-containing E3 ligase family represents the largest among E3 ubiquitin ligases. RNF8 functions in histone H2A and H2B mono-ubiquitination in the context of DNA damage and facilitates propagating DNA damage signal, which is essential for DNA damage repair and cell cycle checkpoint activation to maintain genomic stability [2, 3]. RNF8 also catalyzes lysine (K) 48 or K63 poly-ubiquitination through its interactions with class III E2 ubiquitin conjugating enzymes (UBE2E2, UbcH6, and UBE2E3) or UBC13, respectively [4-6]. RNF8 is also an essential factor for protection of telomere end integrity and regulates cell cycle progression. Deficiency of RNF8 in mice led to tumorigenesis, which might be caused by genomic instability [7, 8]. However, RNF8 may have dual effects on tumorigenesis, as a recent report showed that RNF8 promoted tumor growth and metastasis through coactivating the transactivation of certain transcription factors [9, 10]. In this review, we discuss the functions of RNF8 that are closely related to tumorigenesis and progression of cancer.
DNA damage response
The main function of RNF8 is to transduce DSB signal (figure 1). DSB is the most toxic DNA damage induced by exogenous and endogenous stimuli, such as ionizing radiation (IR) and reactive oxygen species (ROS), respectively [11]. The two main mechanisms of DSB repair are nonhomologous end joining (NHEJ) and homologous recombinational (HR) repair. In the initial stages of recognizing DSBs, the Mre11-Rad50- Nbs1 (MRN) complex senses the damage and recruits the serine/threonine kinase ATM to damage sites. After activation by autophosphorylation, ATM phosphorylates the C-terminus of histone H2A variant (H2AX), which is referred to as γH2AX [11-13]. MDC1 interacts with γH2AX through its BRCT domain and functions as a platform to recruit downstream signal proteins [11, 14]. RNF8 recognizes phosphorylated MDC1 via its FHA domain [15-18]. However, it may be inaccessible for RNF8 to get access to the C-terminus of H2A around unfolded chromatin. RNF8 recruits NuRD ATPase CHD4 (a chromatin remodeling factor required for efficient DNA damage response) to DSB sites to promote extensive decondensation of chromatin. This local chromatin environment facilitates following ubiquitin conjugation [19-21]. The JMJD1C demethylase interacts with RNF8 and is stabilized, leading to demethylation of MDC1 at K45, which promotes MDC1 association with RNF8 [22]. RNF8, by coordinating with the UBC13 E2 enzyme, catalyzes K63 ubiquitination of histone H2A and H2AX around DSB sites [5, 14, 15]. The ubiquitination chain is amplified by RNF168, another RING finger E3 ubiquitin ligase [23].
The role of RNF8 involves in DSB signaling pathway. The MRN complex senses the DSB and recruits ATM. Upon being activated by autophosphorylation, ATM phosphorylates histone H2AX (γH2AX). MDC1 recognizes γH2AX and is phosphorylated by ATM and then acts as a platform that recruits RNF8. RNF8 binds CHD4 to decondense chromatin and allow RNF8 to access H2A. RNF8 promotes monoubiquitination of H2A/H2AX with the help of DYRK2. Upon phosphorylation by ATM, L3MBTL2 associates with MDC1 and is polyubiquitinated by RNF8. Ubiquitinated L3MBTL2 recruits RNF168, which amplifies the polyubiquitination chain of H2A/H2AX. After being stabilized by RNF8, JMJD1C facilitates the demethylation and ubiquitination of MDC1. This effect promotes the recognition of RAP80 to ubiquitination signal. Subsequently, BRCA1 complex is recruited to RAP80 via CCDC98 and induces the downstream HR repair by Rad51 through PALB2. The RNF8-UBC13 complex also catalyzes K48 polyubiquitination of JMJD2A. The degradation of JMJD2A promotes the interaction of 53BP1 with methylated H3/H4 and induces NHEJ repair. 53BP1 is also recruited to K15 polyubiquitinated H2A conjugated by RNF8 and RNF168. The RNF8 partner DYRK2 also facilitates the recruitment of 53BP1 to DSB sites.
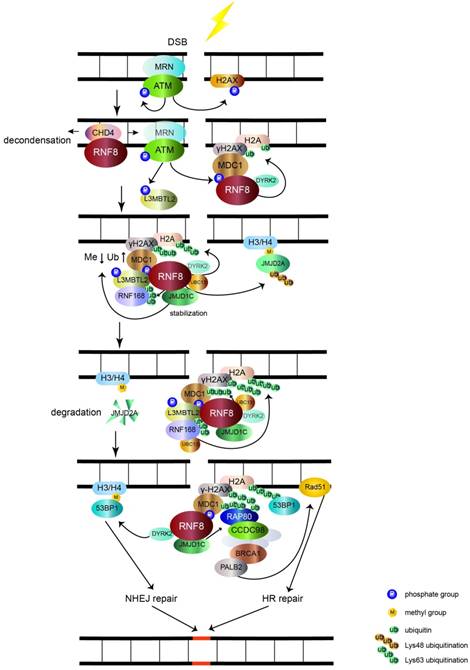
Lethal malignant brain tumor-like protein 2 (L3MBTL2) participates in the DNA damage response pathway and is a link between RNF8 and RNF168. L3MBTL2 is a putative polycomb group protein that plays an essential role in embryonic development and is commonly mutated in various cancers. L3MBTL2 contains an MBT domain that recognizes methylated histones. L3MBTL2 is phosphorylated by ATM and is then recruited by MDC1. Phosphorylated L3MBTL2 is subsequently ubiquitinated by RNF8, which promotes the localization of RNF168 to DSB sites [24]. Therefore, L3MBTL2 dictates the sequential order of RNF8- and RNF168-mediated ubiquitination of histone H2A and H2AX.
By interacting with RNF8, JMJD1C also promotes RNF8-UBC13 mediated K63 polyubiquitination of MDC1 at K1977 [22, 25]. RAP80, which contains ubiquitin-interacting motifs binding to K63 type polyubiquitination chains [26-29], recognizes the ubiquitination signal assisted by JMJD1C, which further recruits BRCA1-BARD1 complex via CCDC98 to DSB sites [25, 30-34]. For further repair, the PALB2 molecular scaffold acts downstream of BRCA1 and recruits the RAD51 recombinase [35], which catalyzes homologous DNA strands to exchange for HR repair. In addition, RNF8 interacts and ubiquitinates a member of the MRN complex, Nbs1, to stabilize its binding to DSB and promote HR repair [36]. RNF8 also participates in ubiquitination and subsequent degradation of the JMJD2A demethylase, which previously binds to methylated histone H3/H4. Ectopic expression of JMJD2A results in abolishing the recruitment of 53BP1 to dimethylated H4K20 [37]. This indicates that RNF8 controls the recruitment of 53BP1 to regulate DNA repair via the degradation of JMJD2A. Dual specificity tyrosine phosphorylation-regulated kinase 2 (DYRK2), which phosphorylates p53 at Ser46 to induce cell death [38], functions as a partner of RNF8 to facilitate γH2AX monoubiquitination and 53BP1 recruitment [39]. The detailed mechanism of DYRK2 in facilitating RNF8-mediated DNA damage response needs further investigation. In addition to recognizing dimethylated H4K20, 53BP1 also acts as a reader of K15 ubiquitination of H2A catalyzed by RNF8 and RNF168 [40]. Subsequently, 53BP1 induces NHEJ repair of DNA damage [41].
In addition to its function in DSB signaling, RNF8 is also involved in UV-induced DNA single-strand break damage. RNF8 coordinates with the E2 ubiquitin-conjugating enzyme UbcH5c to target p12 (a subunit of DNA polymerase δ) for K48-polyubiquitination and subsequent degradation, which switches polymerase δ4 to δ3 for genoprotection [42]. Thus, RNF8 is an essential factor to transduce DNA damage signals and maintain genomic stability.
Protection of chromosome ends
RNF8 also participates in telomere protection to sustain chromosome integrity, which is critical for the maintenance of genomic stability. The telomere is a protective structure on the ends of chromatin that prevents deterioration or improper fusion of chromosomes. The telomere is comprised of stretches of TTAGGG-repeats bound by the shelterin complex (composed of TRF1, TRF2, RAP1, TIN2, TPP1 and POT1) [43]. Loss of telomere protection gives rise to the recognition and recruitment of DSB signaling factors. Shelterin prevents activation of the ATM and ATR response and inhibits inappropriate repair by NHEJ and HR, thus protecting chromosome ends against improper ligation [43, 44]. A previous study showed that silence of TRF2 induces classical NHEJ (C-NHEJ), while inactivation of TPP1-POT1 elicits alternative NHEJ (A-NHEJ) [45]. The resultant dysfunctional chromosomes induce aberrant cell division, which leads to genomic instability.
Peuscher et al. [46] used a system for temperature-sensitive inactivation of TRF2, in which TRF2 detached from telomere at 37~40°C and induced C-NHEJ mediated chromosome end fusion. Under this condition, RNF8 mediated the ubiquitination of H2A on uncapped telomeres, while depletion of RNF8 significantly reduced the level of H2A ubiquitination and the accumulation of 53BP1 and phosphorylated ATM in this region. Furthermore, the accumulation of these two proteins is known to promote C-NHEJ and aggravate genomic instability. These results suggest that RNF8 is required for C-NHEJ-mediated chromosome fusion with uncapped telomeres. Otherwise, the deficiency of RNF8 was not found to affect the accumulation of TRF1, TRF2 and RAP1 on telomeres. Loss of RNF8 reduced the stabilization of TPP1 and the recruitment of TIN2, TPP1 and POT1 on telomeres dependent on the E3 activity of RNF8 [47]. These findings indicate that RNF8 is involved in suppressing A-NHEJ at telomeres, which further protects chromosome end integrity.
Chromosome ends also need protection during telomere replication. Timely resolution of cohesion in G2/M phase is important to protect telomere integrity and inappropriate fusion of chromosome ends [48, 49]. Poly (ADP-ribose) polymerase tankyrase 1 is the essential factor required for timely removal of cohesion at telomeres [50]. RNF8-mediated K63-polyubiquitination of tankyrase 1 promotes its stability and association with telomere, and then facilitates the resolution of sister telomere cohesion [51].
Taken together, these results show that the E3 ligase activity of RNF8 plays an essential role in chromosome end protection by repressing A-NHEJ-mediated telomere fusion and by participating in timely resolution of cohesion at telomeres during replication to maintain chromosome end integrity. However, if the telomere is already uncapped by depletion of TRF2, RNF8 may promote C-NHEJ, which induces telomere fusion and further generates genomic instability.
Cell cycle regulation
Parental cells must faithfully transmit genetic information to daughter cells during cell division. This requires the appropriate coordination of mitosis with cytokinesis, the final event of cell cycle that occurs concurrently with mitosis. Failure of mitotic checkpoint and cytokinesis can result in genomic instability which contributes to cancer development [52, 53]. RNF8 also participates in maintaining genomic stability by regulating mitosis and cytokinesis. RNF8 is the human homolog of Saccharomyces cerevisiae Dma1p/Dma2p and Saccharomyces prombe Dma1p, which share the same domains of N-terminal FHA and C-terminal RING domains [54]. Dma1p/ Dma2p are mitotic checkpoint proteins localized to the midbody that help sustain mitotic arrest when the spindle check point is activated with microtubule damage [55, 56]. The midbody localization of human RNF8 during cytokinesis was confirmed by Plan et al [57], suggesting the regulatory function of RNF8 in cytokinesis. The authors also demonstrated that RNF8 showed cell-cycle dependent turnover, and its expression reached a peak in mitosis and decreased sharply in late mitotic stages. Tuttle and colleagues [54] synchronized RNF8 silenced U2OS cells at the G1-S boundary and treated the cells with microtubule poison nocodazole. The percentage of nocodazole-induced mitotic arrest cells decreased when RNF8 was knockdown, indicating that depletion of RNF8 compromised the ability to maintain mitotic arrest. The mechanism of RNF8 enforcing mitotic arrest in response to nocodazole might involve the inhibition of degradation of the substrates of anaphase-promoting complex (APC), such as cyclin A, cyclin B1, Aurora-A and Aurora-B. APC is required for progression and exit from mitosis, and its substrates decreases when cells exit mitosis [58]. However, the cell cycle inhibitor p27/KIP1 might also plays a role in RNF8 mediating nocodazole-induced mitotic arrest, as p27/KIP1 levels increased when cells exit mitosis and enter G1 [59]. The depletion of RNF8 reduced the protein levels of cyclin A and Aurora-B and maintained the high level of p27/KIP1 in cells treated with nocodazole, suggesting that RNF8 plays an important role in enforcing mitotic arrest in response to nocodazole. Depletion of RNF8 in HeLa cells delayed mitosis exit after release from nocodazole treatment [57]. In addition, overexpression of RNF8 delayed the resolution of cytokinesis, associated with the decreased recruitment of checkpoint protein Mad2 to kinetochores after nocodazole treatment, which was attenuated by diminishing the E3 ligase activity of RNF8. The localization of Mad2 to kinetochores is an early event for spindle assembly checkpoint activation by nocodazole. These results indicates that the E3 ligase activity of RNF8 is required for efficient mitosis exit and appropriate cytokinesis [57]. A function for RNF8 in regulating cytokinesis was also shown by Chahwan et al [60], and RNF8 localized to centrosomes and cell division sites. The authors found that RNF8 ubiquitinated the septin SEPT7, the human homolog of yeast septin Shs1 [61, 62]. Septins are critical factors for cytokinesis. RNF8 mediated-ubiquitination was required for the organization and function of SEPT7, and interference of this process by inactivating RNF8 caused inadequate cell abscission resulting in cytokinesis defects. These observations suggest that RNF8 plays an essential role in the regulation of mitosis and cytokinesis progression. Previous studies showed that cytokinesis failure induced multinucleated cells and further caused genomic instability, which is critical for tumorigenesis [63, 64]. Therefore, precise regulation of the expression of RNF8 at a proper level appears to be required for cellular homeostasis.
Transcriptional regulation
In addition to the roles of RNF8 in DNA damage signal transduction, chromosome end protection and regulation of mitosis and cytokinesis, whose defects account for genomic instability-related tumorigenesis, RNF8 also acts as a transcriptional coregulator in tumor development. RNF8 was initially identified as a binding protein for transcription factor retinoid X receptor α (RXRα) by two-hybrid screening, β-galactosidase assays and GST pull-down assays [65]. RXRα is one subtype of the retinoid X receptor (RXR), whose ligand is retinoid acid (RA). RXRα affects the proliferation and differentiation of various cell types through regulating RA-mediated gene transcription. RNF8 co-localized and interacted with RXRα via its FHA and RING domains. Overexpression of RNF8 dramatically increased RXRα transactivation and the transcription level of its downstream genes RBP2 and RARβ, even without the RA ligand. Furthermore, ligand addition enhanced the ability of RNF8 to upregulate transactivation activity of RXRα [65]. However, RNF8 does not affect the ubiquitination of RXRα, and the mechanism by which RNF8 upregulates RXRα has not been clearly elucidated [65].
Recent studies have demonstrated that RNF8 regulates the transcriptional activity of other transcription factors dependent on its E3 ligase activity. In addition to the role in DSB signal transduction, RNF8-mediated K63-linkage ubiquitination also plays a pivotal function in transcription activation of the epithelial-mesenchymal transition (EMT)-related transcription factor Twist. Twist mainly functions in cell lineage determination and differentiation. Twist overexpression was found to render EMT and cancer stem cell (CSC) features to breast cancer xenografts and a transgenic mouse model [66, 67]. The expression level of Twist was also induced by epidermal growth factor (EGF) [68]. Lee et al. [9] found that RNF8 directly catalyzed K63 ubiquitination of Twist in breast cancer cells with EGF treatment, and the modification further promoted Twist protein stability and nuclear translocation (figure 2). As the repression of E-cadherin gene transcription is a well characterized readout for Twist and EMT activity, a luciferase assay system driven by the E-cadherin promoter was established to elucidate whether the transcriptional activity of Twist was regulated by RNF8 [69]. Overexpression of Twist effectively suppressed E-cadherin transcription, while the deficiency of RNF8 repressed this activity of Twist. These results indicate that RNF8 acts as a transcriptional coactivator of Twist via its E3 ligase activity.
The dual impact of RNF8 on cancer. (1) RNF8 promotes K48 polyubiquitination and degradation of N1ICD, thereby causing the downregulation of Notch signaling. RNF8 also transduces DSB signaling to repair DNA damage. These two pathways help to restrain the tumorigenesis of breast cancer. (2) With EGF- or DSB-inducing drug treatment, RNF8 conjugates K63 polyubiquitination of Twist, which promotes Twist stabilization and activation. This induces CSC selfrenewal and increases cell mobility to render chemoresistance and metastasis capacity to TNBC cells. (3) RNF8 interacts with and promotes monoubiquitination of ERα which partially contributes to the stabilization of ERα. RNF8 enhances the transactivation of ERα and upregulates ERα target genes with estrogen treatment, which increases breast cancer proliferation.
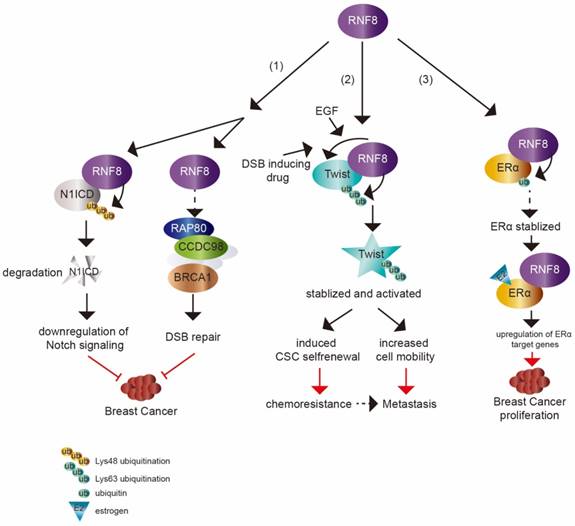
RNF8 is also a coactivator of another transcription factor, estrogen receptor α (ERα) (figure 2), which is a member of the hormone receptor superfamily. Upon ligand binding, ERα is activated and translocates to nucleus to drive the transcription of various target genes involved in cell proliferation, differentiation and breast cancer development [70-72]. Wang et al. [10] showed that RNF8 interacted and co-localized with ERα in an estrogen (E2)-dependent manner in breast cancer cells. Overexpression of RNF8 enhanced ERα transactivation while silencing of RNF8 attenuated ERα transactivation using well characterized ERα luciferase assay system in the presence of E2. The FHA domain and RING finger motif in RNF8 were both required for the effect of RNF8 reinforcing ERα transactivation. The function of RNF8 on upregulation of ERα transcriptional activity was confirmed by testing the transcription level of ERα target genes (MYC, CCND1, E2F1, TERT and TFF1) by knockdown of RNF8. Moreover, the authors found that RNF8 was recruited to the ERα-responsive elements in the MYC and TFF1 promoters by E2 treatment, which further suggests that RNF8 is a coactivator of ERα. Another mechanism of RNF8 upregulating ERα might be due to RNF8 enhancement of ERα stability via promoting its monoubiquitination.
RNF8 mediated K63-linkage polyubiquitination of H2A/H2AX around DSB transduces the DNA damage response signal to downstream repair factors [5, 14, 15, 30-33, 35]. RNF8-mediated K63 polyubiquitination and monoubiquitination have also been found to increase transcriptional activity of Twist and ERα. In addition, RNF8 coordinated with the E2 ubiquitin-conjugating enzyme Ube2S to promote K11 polyubiquitination on H2A/H2AX, which was involved in DNA damage-induced transcriptional silencing [73]. Whether RNF8 upregulates or downregulates transcription in different contexts and whether it participates in the regulation of other transcription factor transactivation or via promoting different types of ubiquitination linkage needs further research.
Tumorigenesis and cancer progression
As an important factor in propagating DSB signaling, RNF8 plays a critical role in maintaining genomic stability as demonstrated by RNF8 knockout mice, which were predisposed for tumorigenesis of lymphoma (50%), thymoma (38%), mammary carcinoma (13%), skin tumor (13%) and sarcoma (13%) [7]. The expression level of tumor suppressor p53, which is the central cellular response regulator to DNA damage, was also increased in RNF8 knockout mice. Additional knockout of p53 increased the level of genomic instability and tumor incidence, suggesting a synergistic role of RNF8 and p53 in maintaining genomic stability and suppressing tumorigenesis [74]. Thus, RNF8 is considered to be a tumor suppressor.
A recent study showed that RNF8 suppressed mammary tumorigenesis [8] (figure 2). The loss of RNF8 in female mouse mammary epithelium cells (MECs) caused spontaneous mammary tumors in mice. RNF8 negatively regulated the Notch signaling pathway through targeting N1ICD degradation by promoting its K48 polyubiquitination, which restrained the expansion of mammary luminal progenitors. N1ICD, the intracellular domain of NOTCH1, is released upon NOTCH1 binding to its ligand and then translocates into nucleus to activate target gene transcription. The release of N1ICD acts as the critical step for Notch signaling activation [75, 76]. It was consistent with the elevated level of N1ICD and activated Notch signaling in MEC specific deficiency of RNF8 in female mice and RNF8 deficient human breast cancer cell lines. Gene expression profile analysis based on The Cancer Genome Atlas (TCGA) data also supported the inverse relationship between RNF8 expression and Notch signaling.
Furthermore, DSB levels and p53 activation were increased in MEC RNF8-defient (RNF8-/-) female mice. Additional homozygous or heterozygous TP53 mutations in MECs increased the mammary tumorigenesis. As a result, RNF8-deficient mice and human breast cancer cell showed enhanced sensitivity to combined treatment of GSI (gamma secretase inhibitor, Notch inhibitor) and PARPi (PARP inhibitor). These results uncover the tumor prevention role of RNF8 and provide insight into treatment to tumors with defective expression or function of RNF8.
In contrast to its tumor suppressive functions, RNF8 may also play positive roles in tumor progression. The mRNA level of RNF8 was found to be higher in colorectal cancer than in normal tissues [77], which suggests RNF8 may promote the survival of certain cancers. The silencing of RNF8 by specific siRNA in nasopharyngeal cancer (NPC) cells enhanced the sensitivity of NPC cells to irradiation. NPC cells silenced for RNF8 also exhibited increased growth inhibition and apoptosis compared with control cells. Furthermore, the expression of checkpoint proteins Chk1 and Chk2 and DNA damage proteins ATM and Nbs1 was reduced in RNF8 knockdown cells with irradiation treatment, which impeded DNA repair function. The repair capacity of irradiation-induced DNA damage is a common factor for NPC recurrence after radiation therapy [78]. These results suggeste that RNF8 is required in NPC cells for resistance to radiation therapy. Consistent with this notion, Zhan et al [79] demonstrated that the absence of RNF8 sensitized bladder cancer cells to radiotherapy, and this result was further confirmed in bladder tumor-implanted nude mice. Taken together, these results suggest that RNF8 may be a potential target for treating radiation-resistant cancers.
A positive relation between RNF8 and breast cancer progression was also recently observed. Breast cancer is the most frequently diagnosed and the leading cause of death among females worldwide [80]. Overexpression of RNF8 in MCF-7 breast cancer cells was found to enhance EMT phenotype, while silencing RNF8 promoted MET in MDA-MB-231 cells. This effect of RNF8 was probably mediated by inactivation of GSK3β and activation of the β-catenin pathway. The inhibition of GSK3β causes the accumulation and nuclear translocation of β-catenin, which activates EMT-related gene transcription [81]. Overexpression of RNF8 in MCF-7 cells promoted the phosphorylation of GSK3β and further inhibited the activity of GSK3β [82]. The phosphorylation of β-catenin decreased and its protein level increased in RNF8 overexpressed cells, while phosphorylated β-catenin increased and its expression decreased in RNF8 silenced cells. These results were also confirmed by the depletion of RNF8 in MDA-MB-231 cells. This indicates that RNF8 may participate in promoting EMT through β-catenin activation.
ERα is expressed at a high level in approximately 70% of breast cancers [10, 83]. The main treatment for ERα-positive luminal breast cancer is through drugs that block its activity, such as hormone therapy (temoxifen) [84]. However, many patients who are treated with temoxifen showed therapy resistance with activated ERα signaling [85-88]. The activity of ERα can be regulated by various cofactors (co-activators or co-repressors) and the aberrant expression of cofactors can render ERα additional agonistic activity, driving breast cancer progression and enhancing drug resistance [89, 90]. Thus, it is necessary to identify ERα cofactors that are abnormally expressed in breast cancer and to understand the mechanisms of regulating ERα activity. RNF8 is a newly identified coactivator of ERα [10]. The expression of RNF8 was found to be higher in malignant breast cancer tissues compared with normal tissues and was strongly correlated to lymph node metastases and poor survival [82]. The increased level of RNF8 in breast cancer samples was positively correlated with ERα expression. RNF8 might promote breast cancer cells proliferation by upregulation of ERα transactivation and its target gene expression [10] (figure 2).
Among breast cancers, approximately 15%~20% cases are triple negative breast cancer (TNBC), which lacks of estrogen receptor, progesterone receptor and shows HER2 overexpression [91]. TNBC is the most aggressive subtype of breast cancer [92]. Although TNBC is initially sensitive to chemotherapy, there is a high risk of relapse in patients with activated EMT characteristic and CSC enrichment [91, 93-95]. EMT program is central to cancer metastasis, which is mediated by EMT-inducing transcription factors (EMT-TFs), such as Slug, Snail, Twist and Zeb1. These EMT-TFs are also linked to CSC property acquisition [96]. Tumor cells that have undergone EMT show enhanced resistance to various therapies [97-99]. Defining the mechanism of EMT regulation is critically required and may provide sight into developing therapies for cancer metastasis and treatment resistance. Recently, RNF8 was found to be involved in the progression of TNBC. Upregulated RNF8 expression in advanced breast cancer was positively correlated to disease progression and poor survival rate. RNF8 enhanced the breast cancer cell EMT and CSC self-renewal by stabilizing Twist protein levels and promoting its nuclear localization and upregulating its transcriptional activity, which endowed chemoresistant capacity to breast cancer cells [9] (figure 2).
Together these results suggest that RNF8 may be a protector in normal cells from DNA damage induced by exogenous or endogenous stimuli, and the absence of RNF8 would confer high cancer risk. In contrast, RNF8 may have malignant activities in cancer by facilitating DNA damage repair after radiotherapy or by activating β-catenin signal and transcription factors Twist as well as ERα.
Conclusion remarks and perspectives
The RNF8 E3 ligase, initially identified as a DSB signal transductor, conjugates mono and polyubiquitination chains to histone H2A/H2AX around DSB to transduce damage signals to repair proteins involved in DSB repair [5, 14, 15, 30-33, 35]. Failure of DSB repair results in genomic instability, and indeed, RNF8 knockout mice developed several kinds of tumors [7]. RNF8 also functions in the protection of chromosome ends integrity. RNF8 plays an important role in suppressing A-NHEJ, which inhibits the improper fusion of telomeres [47], and facilitates timely resolution of cohesions during telomere replication [48]. These activities protect the integrity of chromosome ends and genomic stability. RNF8 also participates in mitosis regulation. Defect in this function results in cytokinesis failure, which causes multinucleated cells and genomic instability [57, 60, 100]. Thus, RNF8 plays essential roles in protecting genomic stability. Genomic instability is known as a pivotal cause of tumorigenesis [64]. Furthermore, female mice with deficiency of RNF8 in MECs exhibit spontaneous mammary tumors, since the loss of RNF8 results in impaired DSB repair and sustained activation of Notch signaling [8]. These findings indicate that RNF8 probably protects against tumorigenesis in some contexts.
In contrast, the DNA damage response function of RNF8 in certain cancers is involved in radiation therapy resistance [78, 79]. When the telomere was already uncapped by deletion of TRF2, the E3 ligase activity of RNF8 in promoting H2A ubiquitination helped to induce C-NHEJ and further caused genomic instability [46]. Furthermore, expression of RNF8 was found to be higher in breast cancer than in normal tissues and was correlated to poor prognosis [82]. RNF8 activated the β-catenin pathway via its E3 ligase activity and promoted EMT phenotype of breast cancer cell [82]. RNF8 also acted as a coactivator of transcription factor Twist and ERα and enhanced breast cancer cell growth and metastasis via upregulation of these two transcription factors [9, 10]. Together these observations suggest that RNF8 also shows roles in promoting tumor development.
These results suggest a “double-edged sword” role of RNF8 in tumorigenesis and leads to important questions. In addition to its effect on genomic stability, does RNF8 play other negative roles in tumor development? Does RNF8 impact tumor development dependent on different stage of progression or different type of tumors? Does RNF8 acts as a coregulator of other transcription factors or participate in the other cell signal pathway? Future studies are required to address these questions and decipher the paradoxical roles of RNF8 in different types of tumors and under different conditions.
Abbreviations
RNF8: ring finger protein 8; DSB: DNA double strand break; FHA: forkhead-associated; RING: really interesting new gene; K: lysine; NHEJ: nonhomologous end joining; HR: homologous recombination; MRN: Mre11-Rad50-Nbs1 complex; γH2AX: phosphorylated histone H2A variant; L3MBTL2: lethal malignant brain tumor-like protein 2; DYRK2: dual specificity tyrosine phosphorylation-regulated kinase 2; C-NHEJ: classical NHEJ; A-NHEJ: alternative NHEJ; RXRα: retinoid X receptor α; RA: 9-cis-retinoid acid; EMT: epithelial-mesenchymal transition; CSC: cancer stem cells; EGF: epidermal growth factor; ERα: estrogen receptor α; E2: estradiol; MEC: mammary epithelium cell; NPC: nasopharyngeal cancer; TNBC: triple negative breast cancer.
Acknowledgements
We received funding from National Natural Science Foundation of China to Tingting Zhou (81502438); National Key R&D Program of China (2016YFC1302400); The Ministry of education innovation team development plan to Liu Cao (IRT_17R107&IRT13101); Key project of the National Natural Science Foundation to Liu Cao (81130042, 31171323, 81770001, 2015225003); National Natural Science Foundation of China to Xiaoyu Song (31300963, LFWK201725), Fei Yi (81502414), Qiqiang Guo (81502400).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Seki N, Hattori A, Sugano S, Suzuki Y, Nakagawara A, Ohhira M. et al. Isolation, tissue expression, and chromosomal assignment of a novel human gene which encodes a protein with RING finger motif. J Hum Genet. 1998;43:272-4
2. Bohgaki T, Bohgaki M, Hakem R. DNA double-strand break signaling and human disorders. Genome Integr. 2010;1:15
3. Wu J, Huen MS, Lu LY, Ye L, Dou Y, Ljungman M. et al. Histone ubiquitination associates with BRCA1-dependent DNA damage response. Mol Cell Biol. 2009;29:849-60
4. Ito K, Adachi S, Iwakami R, Yasuda H, Muto Y, Seki N. et al. N-Terminally extended human ubiquitin-conjugating enzymes (E2s) mediate the ubiquitination of RING-finger proteins, ARA54 and RNF8. Eur J Biochem. 2001;268:2725-32
5. Plans V, Scheper J, Soler M, Loukili N, Okano Y, Thomson TM. The RING finger protein RNF8 recruits UBC13 for lysine 63-based self polyubiquitylation. J Cell Biochem. 2006;97:572-82
6. Ma T, Keller JA, Yu X. RNF8-dependent histone ubiquitination during DNA damage response and spermatogenesis. Acta Biochim Biophys Sin (Shanghai). 2011;43:339-45
7. Li L, Halaby MJ, Hakem A, Cardoso R, El Ghamrasni S, Harding S. et al. Rnf8 deficiency impairs class switch recombination, spermatogenesis, and genomic integrity and predisposes for cancer. J Exp Med. 2010;207:983-97
8. Li L, Guturi KKN, Gautreau B, Patel PS, Saad A, Morii M. et al. Ubiquitin ligase RNF8 suppresses Notch signaling to regulate mammary development and tumorigenesis. J Clin Invest. 2018;128:4525-42
9. Lee HJ, Li CF, Ruan D, Powers S, Thompson PA, Frohman MA. et al. The DNA Damage Transducer RNF8 Facilitates Cancer Chemoresistance and Progression through Twist Activation. Mol Cell. 2016;63:1021-33
10. Wang S, Luo H, Wang C, Sun H, Sun G, Sun N. et al. RNF8 identified as a co-activator of estrogen receptor alpha promotes cell growth in breast cancer. Biochim Biophys Acta. 2017;1863:1615-28
11. Yan J, Jetten AM. RAP80 and RNF8, key players in the recruitment of repair proteins to DNA damage sites. Cancer Lett. 2008;271:179-90
12. Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739-45
13. Kim JE, Minter-Dykhouse K, Chen J. Signaling networks controlled by the MRN complex and MDC1 during early DNA damage responses. Mol Carcinog. 2006;45:403-8
14. Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C. et al. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887-900
15. Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB. et al. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901-14
16. Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci U S A. 2007;104:20759-63
17. Kolas NK, Chapman JR, Nakada S, Ylanko J, Chahwan R, Sweeney FD. et al. Orchestration of the DNA-damage response by the RNF8 ubiquitin ligase. Science. 2007;318:1637-40
18. Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213-26
19. Luijsterburg MS, Acs K, Ackermann L, Wiegant WW, Bekker-Jensen S, Larsen DH. et al. A new non-catalytic role for ubiquitin ligase RNF8 in unfolding higher-order chromatin structure. EMBO J. 2012;31:2511-27
20. Larsen DH, Poinsignon C, Gudjonsson T, Dinant C, Payne MR, Hari FJ. et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J Cell Biol. 2010;190:731-40
21. Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J. 2010;29:3130-9
22. Watanabe S, Watanabe K, Akimov V, Bartkova J, Blagoev B, Lukas J. et al. JMJD1C demethylates MDC1 to regulate the RNF8 and BRCA1-mediated chromatin response to DNA breaks. Nat Struct Mol Biol. 2013;20:1425-33
23. Campbell SJ, Edwards RA, Leung CC, Neculai D, Hodge CD, Dhe-Paganon S. et al. Molecular insights into the function of RING finger (RNF)-containing proteins hRNF8 and hRNF168 in Ubc13/Mms2-dependent ubiquitylation. J Biol Chem. 2012;287:23900-10
24. Nowsheen S, Aziz K, Aziz A, Deng M, Qin B, Luo K. et al. L3MBTL2 orchestrates ubiquitin signalling by dictating the sequential recruitment of RNF8 and RNF168 after DNA damage. Nat Cell Biol. 2018;20:455-64
25. Strauss C, Halevy T, Macarov M, Argaman L, Goldberg M. MDC1 is ubiquitylated on its tandem BRCT domain and directly binds RAP80 in a UBC13-dependent manner. DNA Repair (Amst). 2011;10:806-14
26. Yan J, Kim YS, Yang XP, Albers M, Koegl M, Jetten AM. Ubiquitin-interaction motifs of RAP80 are critical in its regulation of estrogen receptor alpha. Nucleic Acids Res. 2007;35:1673-86
27. Cho HJ, Lee S, Kim H. The linker connecting the tandem ubiquitin binding domains of RAP80 is critical for lysine 63-linked polyubiquitin-dependent binding activity. BMB Rep. 2009;42:764-8
28. Sato Y, Yoshikawa A, Mimura H, Yamashita M, Yamagata A, Fukai S. Structural basis for specific recognition of Lys 63-linked polyubiquitin chains by tandem UIMs of RAP80. EMBO J. 2009;28:2461-8
29. Sims JJ, Cohen RE. Linkage-specific avidity defines the lysine 63-linked polyubiquitin-binding preference of rap80. Mol Cell. 2009;33:775-83
30. Yan J, Kim YS, Yang XP, Li LP, Liao G, Xia F. et al. The ubiquitin-interacting motif containing protein RAP80 interacts with BRCA1 and functions in DNA damage repair response. Cancer Res. 2007;67:6647-56
31. Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP. et al. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194-8
32. Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B. et al. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198-202
33. Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202-5
34. Strauss C, Goldberg M. Recruitment of proteins to DNA double-strand breaks: MDC1 directly recruits RAP80. Cell Cycle. 2011;10:2850-7
35. Zhang F, Bick G, Park JY, Andreassen PR. MDC1 and RNF8 function in a pathway that directs BRCA1-dependent localization of PALB2 required for homologous recombination. J Cell Sci. 2012;125:6049-57
36. Lu CS, Truong LN, Aslanian A, Shi LZ, Li Y, Hwang PY. et al. The RING finger protein RNF8 ubiquitinates Nbs1 to promote DNA double-strand break repair by homologous recombination. J Biol Chem. 2012;287:43984-94
37. Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G. et al. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012;31:1865-78
38. Taira N, Nihira K, Yamaguchi T, Miki Y, Yoshida K. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol Cell. 2007;25:725-38
39. Yamamoto T, Taira Nihira N, Yogosawa S, Aoki K, Takeda H, Sawasaki T. et al. Interaction between RNF8 and DYRK2 is required for the recruitment of DNA repair molecules to DNA double-strand breaks. FEBS Lett. 2017;591:842-53
40. Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CC, Huang H. et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50-4
41. Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15:7-18
42. Zhang S, Zhou Y, Sarkeshik A, Yates JR 3rd, Thomson TM, Zhang Z. et al. Identification of RNF8 as a ubiquitin ligase involved in targeting the p12 subunit of DNA polymerase delta for degradation in response to DNA damage. J Biol Chem. 2013;288:2941-50
43. Jacobs JJ. Fusing telomeres with RNF8. Nucleus. 2012;3:143-9
44. de Lange T. How telomeres solve the end-protection problem. Science. 2009;326:948-52
45. Rai R, Zheng H, He H, Luo Y, Multani A, Carpenter PB. et al. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J. 2010;29:2598-610
46. Peuscher MH, Jacobs JJ. DNA-damage response and repair activities at uncapped telomeres depend on RNF8. Nat Cell Biol. 2011;13:1139-45
47. Rai R, Li JM, Zheng H, Lok GT, Deng Y, Huen MS. et al. The E3 ubiquitin ligase Rnf8 stabilizes Tpp1 to promote telomere end protection. Nat Struct Mol Biol. 2011;18:1400-7
48. Hsiao SJ, Smith S. Sister telomeres rendered dysfunctional by persistent cohesion are fused by NHEJ. J Cell Biol. 2009;184:515-26
49. Canudas S, Smith S. Differential regulation of telomere and centromere cohesion by the Scc3 homologues SA1 and SA2, respectively, in human cells. J Cell Biol. 2009;187:165-73
50. Dynek JN, Smith S. Resolution of sister telomere association is required for progression through mitosis. Science. 2004;304:97-100
51. Tripathi E, Smith S. Cell cycle-regulated ubiquitination of tankyrase 1 by RNF8 and ABRO1/BRCC36 controls the timing of sister telomere resolution. EMBO J. 2017;36:503-19
52. Potapova TA, Zhu J, Li R. Aneuploidy and chromosomal instability: a vicious cycle driving cellular evolution and cancer genome chaos. Cancer Metastasis Rev. 2013;32:377-89
53. Cullati SN, Gould KL. Spatiotemporal regulation of the Dma1-mediated mitotic checkpoint coordinates mitosis with cytokinesis. Curr Genet. 2019
54. Tuttle RL, Bothos J, Summers MK, Luca FC, Halazonetis TD. Defective in mitotic arrest 1/ring finger 8 is a checkpoint protein that antagonizes the human mitotic exit network. Mol Cancer Res. 2007;5:1304-11
55. Guertin DA, Venkatram S, Gould KL, McCollum D. Dma1 prevents mitotic exit and cytokinesis by inhibiting the septation initiation network (SIN). Dev Cell. 2002;3:779-90
56. Fraschini R, Bilotta D, Lucchini G, Piatti S. Functional characterization of Dma1 and Dma2, the budding yeast homologues of Schizosaccharomyces pombe Dma1 and human Chfr. Mol Biol Cell. 2004;15:3796-810
57. Plans V, Guerra-Rebollo M, Thomson TM. Regulation of mitotic exit by the RNF8 ubiquitin ligase. Oncogene. 2008;27:1355-65
58. Castro A, Bernis C, Vigneron S, Labbe JC, Lorca T. The anaphase-promoting complex: a key factor in the regulation of cell cycle. Oncogene. 2005;24:314-25
59. Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A. et al. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999;13:1181-9
60. Chahwan R, Gravel S, Matsusaka T, Jackson SP. Dma/RNF8 proteins are evolutionarily conserved E3 ubiquitin ligases that target septins. Cell Cycle. 2013;12:1000-8
61. Mostowy S, Cossart P. Septins: the fourth component of the cytoskeleton. Nat Rev Mol Cell Biol. 2012;13:183-94
62. Kim MS, Froese CD, Estey MP, Trimble WS. SEPT9 occupies the terminal positions in septin octamers and mediates polymerization-dependent functions in abscission. J Cell Biol. 2011;195:815-26
63. Fujiwara T, Bandi M, Nitta M, Ivanova EV, Bronson RT, Pellman D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature. 2005;437:1043-7
64. Shi Q, King RW. Chromosome nondisjunction yields tetraploid rather than aneuploid cells in human cell lines. Nature. 2005;437:1038-42
65. Takano Y, Adachi S, Okuno M, Muto Y, Yoshioka T, Matsushima-Nishiwaki R. et al. The RING finger protein, RNF8, interacts with retinoid X receptor alpha and enhances its transcription-stimulating activity. J Biol Chem. 2004;279:18926-34
66. Morel AP, Hinkal GW, Thomas C, Fauvet F, Courtois-Cox S, Wierinckx A. et al. EMT inducers catalyze malignant transformation of mammary epithelial cells and drive tumorigenesis towards claudin-low tumors in transgenic mice. PLoS Genet. 2012;8:e1002723
67. Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q. et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell. 2014;25:210-25
68. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871-90
69. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C. et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927-39
70. Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G. et al. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535-65
71. Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ. et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389-93
72. Yang J, AlTahan A, Jones DT, Buffa FM, Bridges E, Interiano RB. et al. Estrogen receptor-alpha directly regulates the hypoxia-inducible factor 1 pathway associated with antiestrogen response in breast cancer. Proc Natl Acad Sci U S A. 2015;112:15172-7
73. Paul A, Wang B. RNF8- and Ube2S-Dependent Ubiquitin Lysine 11-Linkage Modification in Response to DNA Damage. Mol Cell. 2017;66:458-72 e5
74. Halaby MJ, Hakem A, Li L, El Ghamrasni S, Venkatesan S, Hande PM. et al. Synergistic interaction of Rnf8 and p53 in the protection against genomic instability and tumorigenesis. PLoS Genet. 2013;9:e1003259
75. Fryer CJ, Lamar E, Turbachova I, Kintner C, Jones KA. Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev. 2002;16:1397-411
76. Fryer CJ, White JB, Jones KA. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol Cell. 2004;16:509-20
77. Sasaki Y, Osada S, Mori R, Imai H, Tanaka Y, Matsuhashi N. et al. Determining timing of hepatectomy for colorectal cancer with distant metastasis according to imaging-based tumor shrinkage ratio. Int J Med Sci. 2013;10:1231-41
78. Wang M, Chen X, Chen H, Zhang X, Li J, Gong H. et al. RNF8 plays an important role in the radioresistance of human nasopharyngeal cancer cells in vitro. Oncol Rep. 2015;34:341-9
79. Zhao MJ, Song YF, Niu HT, Tian YX, Yang XG, Xie K. et al. Adenovirus-mediated downregulation of the ubiquitin ligase RNF8 sensitizes bladder cancer to radiotherapy. Oncotarget. 2016;7:8956-67
80. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394-424
81. Valenta T, Hausmann G, Basler K. The many faces and functions of beta-catenin. EMBO J. 2012;31:2714-36
82. Kuang J, Li L, Guo L, Su Y, Wang Y, Xu Y. et al. RNF8 promotes epithelial-mesenchymal transition of breast cancer cells. J Exp Clin Cancer Res. 2016;35:88
83. Sflomos G, Dormoy V, Metsalu T, Jeitziner R, Battista L, Scabia V. et al. A Preclinical Model for ERalpha-Positive Breast Cancer Points to the Epithelial Microenvironment as Determinant of Luminal Phenotype and Hormone Response. Cancer Cell. 2016;29:407-22
84. Manavathi B, Dey O, Gajulapalli VN, Bhatia RS, Bugide S, Kumar R. Derailed estrogen signaling and breast cancer: an authentic couple. Endocr Rev. 2013;34:1-32
85. Michalides R, Griekspoor A, Balkenende A, Verwoerd D, Janssen L, Jalink K. et al. Tamoxifen resistance by a conformational arrest of the estrogen receptor alpha after PKA activation in breast cancer. Cancer Cell. 2004;5:597-605
86. Fan W, Chang J, Fu P. Endocrine therapy resistance in breast cancer: current status, possible mechanisms and overcoming strategies. Future Med Chem. 2015;7:1511-9
87. Gao A, Sun T, Ma G, Cao J, Hu Q, Chen L. et al. LEM4 confers tamoxifen resistance to breast cancer cells by activating cyclin D-CDK4/6-Rb and ERalpha pathway. Nat Commun. 2018;9:4180
88. Zheng L, Meng X, Li X, Zhang Y, Li C, Xiang C. et al. miR-125a-3p inhibits ERalpha transactivation and overrides tamoxifen resistance by targeting CDK3 in estrogen receptor-positive breast cancer. FASEB J. 2018;32:588-600
89. Wade MA, Jones D, Wilson L, Stockley J, Coffey K, Robson CN. et al. The histone demethylase enzyme KDM3A is a key estrogen receptor regulator in breast cancer. Nucleic Acids Res. 2015;43:196-207
90. Giamas G, Filipovic A, Jacob J, Messier W, Zhang H, Yang D. et al. Kinome screening for regulators of the estrogen receptor identifies LMTK3 as a new therapeutic target in breast cancer. Nat Med. 2011;17:715-9
91. Lee A, Djamgoz MBA. Triple negative breast cancer: Emerging therapeutic modalities and novel combination therapies. Cancer Treat Rev. 2018;62:110-22
92. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA. et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429-34
93. Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F. et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13:2329-34
94. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y. et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750-67
95. Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist. 2010;15(Suppl 5):39-48
96. Chaffer CL, San Juan BP, Lim E, Weinberg RA. EMT, cell plasticity and metastasis. Cancer Metastasis Rev. 2016;35:645-54
97. Kurrey NK, Jalgaonkar SP, Joglekar AV, Ghanate AD, Chaskar PD, Doiphode RY. et al. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells. 2009;27:2059-68
98. Zhang P, Wei Y, Wang L, Debeb BG, Yuan Y, Zhang J. et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat Cell Biol. 2014;16:864-75
99. Smith BN, Bhowmick NA. Role of EMT in Metastasis and Therapy Resistance. J Clin Med. 2016:5
100. Yoshioka T, Kimura M, Saio M, Era S, Okano Y. Plk1 is negatively regulated by RNF8. Biochem Biophys Res Commun. 2011;410:57-61
Author contact
![]() Corresponding authors: Xiaoyu Song, e-mail: xysongedu.cn and Liu Cao, e-mail: lcaoedu.cn. Key Laboratory of Medical Cell Biology, Ministry of Education; Institute of Translational Medicine, China Medical University; Collaborative Innovation Center of Aging Related Disease Diagnosis and Treatment and Prevention, Shenyang, Liaoning Province, 110122, China. Tel: +86 24 31939636, Fax: +86 24 31939636.
Corresponding authors: Xiaoyu Song, e-mail: xysongedu.cn and Liu Cao, e-mail: lcaoedu.cn. Key Laboratory of Medical Cell Biology, Ministry of Education; Institute of Translational Medicine, China Medical University; Collaborative Innovation Center of Aging Related Disease Diagnosis and Treatment and Prevention, Shenyang, Liaoning Province, 110122, China. Tel: +86 24 31939636, Fax: +86 24 31939636.