ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

Int J Biol Sci 2019; 15(5):929-941. doi:10.7150/ijbs.32489 This issue Cite
Research Paper
Feedback Activation of SGK3 and AKT Contributes to Rapamycin Resistance by Reactivating mTORC1/4EBP1 Axis via TSC2 in Breast Cancer
Hongtao Wang1,3*, Fang Huang1*, Zhe Zhang1, Peng Wang1, Yuan Luo2, Hailiang Li4, Na Li1, Jian Wang1, Jianguang Zhou1, Yongan Wang2 ![]() , Shanhu Li1
, Shanhu Li1 ![]()
1. Department of Cell Engineering, Beijing Institute of Biotechnology, Beijing 100850, China.
2. State Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology and Toxicology, Beijing 100850, China.
3. State Key Laboratory of Experimental Hematology, Institute of Hematology and Blood Diseases Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Tianjin 300200, China.
4. Zhuhai People's Hospital, Zhuhai Hospital Affiliated Jinan University, Zhuhai 519000, China.
*These authors contributed equally to this work.
Abstract

The mTORC1 inhibitors, such as rapamycin and its analogs, show limited antitumor activity in clinic, reasons for which have not been clearly elucidated. Here, we undertook an effort to uncover the mechanisms underlying the limited efficacy of rapamycin, and found that the transit suppression of 4EBP1 phosphorylation led to cap-dependent translation and cell proliferation in breast cancer cells. AKT only partially contributed to 4EBP1 re-phosphorylation. By taking advantage of mass spectrometry-based phosphoproteomic analysis, we identified SGK3 as a potent kinase involved in 4EBP1 re-phosphorylation. SGK3 deletion inhibited 4EBP1 phosphorylation and cap-dependent translation. Importantly, 4EBP1 phosphorylation was positively correlated with SGK3 activity in 67 clinical breast cancer specimens. Moreover, SGK3 deletion in combination with AKT inhibition almost blocked the 4EBP1 re-phosphorylation that was induced by rapamycin and profoundly enhanced rapamycin-induced growth inhibition in vitro and in an MCF7 breast cancer mouse xenograft model in vivo. Mechanistically, the feedback activation of SGK3 by rapamycin was dependent on hVps34 and mTORC2, and reactivated mTORC1/4EBP1 axis by phosphorylating TSC2. Collectively, our study reveals a critical role of SGK3 in mediating rapamycin resistance, and provides a rationale for targeting SGK3 to improve mTOR-targeted therapies.
Keywords: SGK3, AKT, rapamycin resistance, 4EBP1, mTOR, breast cancer
Introduction
Mammalian target of rapamycin (mTOR), one of serine/threonine kinases, controls cell proliferation, survival, autophagy, and metabolism by integrating multiple signaling transduction pathways from growth factors and nutrients(1). mTOR exerts its biological functions by forming two distinct complexes; mTORC1 and mTORC2. mTORC2 is composed of mTOR, Rictor, Sin1, and mLST8 and controls cell survival by phosphorylating AKT on Ser-473, which is required for the full activation of AKT. mTORC1 is made up of mTOR, raptor, mLST8, PRAS40, and deptor(2). Following activation by PI3K/AKT pathway, mTORC1 phosphorylates two major downstream messengers, 70 kDa ribosomal protein S6 kinase 1 (p70S6K1) and eukaryotic initiation factor 4E (eIF4E)-binding protein 1 (4EBP1). Hypo-phosphorylated 4EBP1 prevents the formation of the eIF4F translation initiation complex by competing with eIF4G to bind eIF4E, preventing 5' cap-dependent mRNA translation. Phosphorylation of 4EBP1 by mTORC1 promotes its dissociation from eIF4E, leading to the initiation of 5' cap-dependent mRNA translation(3). Accumulating evidence has demonstrated that deregulation of cap-dependent translation promotes cancer initiation and progression by enhancing the expression of a subset of oncogenic proteins such as cyclin D1, survivin, and MMP9 that control cell proliferation, survival, and metastasis, respectively(4).
Breast cancer is the most common malignant cancer type in women and the second lethal form of cancer(5). Despite the benefits of endocrine therapy, many patients develop resistance to endocrine manipulation during treatment(6). The phosphatidyl-inositol-3-kinase (PI3K) activation mutations and phosphatase and tensin homolog (PTEN) loss lead to dysregulation of the mTORC1 pathway in breast cancer. Abnormal activation of mTOR pathway has been implicated in the initiation and progression of breast cancer and the development of endocrine therapy resistance(7). Therefore, targeting mTORC1 is a promising therapeutic strategy for breast cancer patients, in particular those who develop endocrine therapy resistance.
Rapamycin and its analogs, such as Everolimus (RAD001) and temsirolimus, allosterically inhibit mTORC1 activity through binding to intracellular FK506 binding protein 12 (FKBP12) receptors(8). Everolimus exerted promising antitumor activities in several preclinical studies, leading to its approval by the US Food and Drug Administration (FDA) for the treatment of many cancers, such as progressive neuroendocrine tumor of pancreatic origin (pNET), advanced renal cell carcinoma (RCC), and subependymal giant-cell astrocytoma(9-11). In a clinical trial of breast cancer, everolimus was reported to increase the efficacy of hormonal therapy and markedly improved progression free survival (PFS) in patients with HR-positive metastatic breast cancer, suggesting its potential capability in overcoming endocrine resistance(12-14). However, the development of rapamycin resistance limits it use in therapies. It is widely believed that this inadequate therapeutic response mainly results from a concomitant activation of AKT through loss of the mTORC1/S6K-dependent negative feedback loop and weak inhibition of 4EBP1 phosphorylation(15). Although the mechanisms involved in feedback activation of AKT signaling have been well elucidated, the molecular basis underlying the incomplete inhibition of 4EBP1 phosphorylation by rapamycin remains largely undefined. Mi et al. recently reported that AKT feedback activation contributes to weak inhibition of 4EBP1 phosphorylation by rapamycin and combined inhibition of mTORC1 and AKT significantly suppressed tumor progression by inhibiting 4EBP1 phosphorylation and cap-dependent translation, thus establishing the correlation of AKT feedback activation and weak inhibition of 4EBP1 phosphorylation during rapamycin treatment(16). However, whether there are other kinases that contribute to the weak inhibition of 4EBP1 phosphorylation by rapamycin remains unknown.
Serum- and glucocorticoid-inducible kinase 3 (SGK3) belongs to AGC protein kinase family along with two other isoforms, SGK1 and SGK2. Due to containing the same consensus substrate motif, RXRXXS/T, SGK and AKT share many common substrates(17, 18). As with AKT, the full activation of SGK3 requires the phosphorylation of the activation loop residue and the hydrophobic motif, which are phosphorylated by PDK1 and mTORC2, respectively. In contrast to AKT, SGK3 has a unique mechanism of upstream activation. Phosphatidylinositol 3-phosphate (PI(3)P) produced by VPS34 binds to the amino-terminal phox homology (PX) domain of SGK3 and localizes this kinase to endosomal membranes, where PDK1 and mTORC2 activate SGK3 through sequentially phosphorylate the corresponding Thr320 and Ser486 sites(19). SGK3 is amplified in breast cancer and its expression is regulated by estrogen/ER signaling(20-22). SGK3 has been reported to play a key role in mediating AKT-independent oncogenic signaling in breast cancer harboring PIK3CA mutations(20, 23). Recent studies also revealed the role of SGK3 in conferring anti-tumor drug resistance, including AKT inhibition and endocrine therapy(24, 25). However, whether SGK3 is involved in rapamycin resistance is still unclear.
In the present study, we demonstrate the critical role of SGK3 in mediating rapamycin resistance in breast cancer cells. We found that the feedback activation of both SGK3 and AKT synergistically resulted in the limited inhibition of rapamycin on 4EBP1 phosphorylation and cap-dependent translation. Blockage of the feedback activation of SGK3 and AKT markedly increased the ability of rapamycin to inhibit cancer growth in vitro and in vivo. Mechanistic studies demonstrated that hVps34- and mTORC2-mediated SGK3 activation led to the transient suppression of 4EBP1 phosphorylation by re-activating mTORC1 via TSC2. Our data uncovers a new mechanism of rapamycin resistance, which might facilitate the development of novel strategies by targeting both SGK3 and AKT for overcoming rapamycin resistance in human breast cancer therapy.
Materials and Methods
Cell lines and cell culture
Human breast cancer MCF-7, T47D cells were obtained from the ATCC. All cell lines were authenticated by short tandem repeat (STR) analysis and confirmed to be free of mycoplasma within the past year. The cell lines MCF7, T47D were maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum (Gibco) and 10 mM HEPES in a humidified incubator at 37°C with 5% CO2. All cell lines were passaged less than 2 months.
Reagents
LY294002 was obtained from Cell Signaling Technology (Danvers, MA). RAD001, MK-2206 and VPS34-IN1 were obtained from Selleck (Shanghai, China).
Antibodies
The following antibodies were used in this study: 4EBP1 (catalog #9644, 1:4000 dilution), Phospho-4EBP1-Thr37/46 (#2855,1:1000), Phospho-4EBP1 -Ser65 (#9451,1:1000), p70S6 Kinase (#2708,1:1000), Phospho-p70S6 Kinase-Thr389 (#9234,1:1000), Phospho-S6-Ser240/244(#5364,1:2000), AKT(#4691,1:2000), Phospho-AKT-Ser473 (#4060,1:1000), EIF4E (#9742,1: 2000), EIF4G (#2498,1:1000), NDRG1(#5196,1:2000), Phospho-NDRG1-Thr 346(#5482,1:2000), Phospho-Tuberin/TSC2-Ser939 (#3615,1:1000), SGK3 (#85731:1000), Phospho-SGK3-Thr320 (#5642,1:500), Survivin (#2808, 1:1000) and Rictor (#2114,1:1000), and were purchased from Cell Signaling Technology (Danvers, MA). In addition, γ-Tubulin (sc-7396,1:500) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Tuberin (ab32554,1:1000) and INPP4B (ab81269, 1:1000) were purchased from Abcam (Cambridge, UK).
Western blot assay
Cell lysates were harvested and 20-100 ug of protein were resolved by 12% SDS-PAGE gel after measuring protein concentration using the BCA protein reagent (Pierce Chemical, Rockford, IL, USA) and then transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat milk for 1 h at room temperature and then incubated with primary antibodies overnight at 4°C, followed by incubation with HRP-conjugated anti-rabbit/ mouse/ goat IgG for 1 h at room temperature. Detection was performed using enhanced chemiluminescence (ECL) detection reagent (Thermo).
m7GTP pull down assay
The cells were treated with inhibitors for 24 h. Lysates were harvested and 1 ml aliquots were subjected to 30 min, 4°C high-speed centrifugation. Next, 15 μL m7GTP Sepharose beads (#GE4710566 GE USA) were added at 4°C and allowed to conjugate to the lysates for 2 h. The samples were centrifuged and the m7GTP Sepharose beads were collected and washed thrice for 10 min each. After centrifugation at 3000 rpm, 20 μL 1x SDS protein sample buffer solution was added to the samples, boiled for 10 min, and resolved by SDS-PAGE gel. Western blot analysis was performed to detect the expression of EIF4E, 4EBP1, and EIF4G. The dynamic changes of EIF4F protein complex activity were analyzed by the changes of 4EBP1 and EIF4G protein levels.
Cap-dependent Renilla/Polio IRES-dependent firefly luciferase assay
Cells were seeded in 10 cm culture dishes and cultured in DMEM containing 10% FBS. Once attached, MCF7 and ZR-75-1 breast cancer cells were transfected with pcDNA3.1-rLuc-PolioIRES-fLuc, which directs cap-dependent translation of the Renilla luciferase gene and cap-independent Polio IRES-mediated translation of the firefly luciferase gene. After 24 h, the transferred cells were inoculated into 24-well plates, the cells were treated with different inhibitors. The Renilla ratio of control cells to treated cells was detected by double fluorescence reporting kit (Promega) after treatment to detect the dynamic change of the functional activity of eIF4F protein complex after treatment.
CCK-8 assay
Cells were seeded in 96-well plates at a density of 1500 cells per well and cultured in DMEM containing 10% FBS. Once attached, the cells were treated with the indicated agents and cultured for the indicated times. After the indicated treatments, cell proliferation was determined with the Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Tokyo, Japan) assay according to the manufacturer's instructions. The absorbance of individual wells was determined at 450 nm. The OD value of the treatment group was normalized to the values from the untreated control group. All reactions were repeated at least 3 times.
siRNA transfections
siRNA for Rictor (HSS153834), Raptor (HSS126373), 4EBP1(HSS141934) and TSC2(HSS111011) were purchased from Invitrogen. MCF7 and T47D breast cancer cells were transfected with 0.2 nM siRNA or negative control using Lipofectamine RNAimax Reagent (#13788 Invitrogen) according to the manufacturer's instructions.
Immunoprecipitation and Western blot analysis
The cell lysates (250 µg protein) were immunoprecipitated with 1 µg of SGK3 antibody overnight followed by incubation with a 50% slurry of protein G Sepharose beads for 3 h at 4°C. The beads were washed three times with the lysis buffer and the immunoprecipitated protein complexes were resuspended in 5× SDS lording buffer followed by Western blot analysis using equivalent total protein loadings as described above.
Clinical samples and immunohistochemistry
All patients' samples were collected after informed consent in accordance with the Declaration of Helsinki, and the research protocol was reviewed and approved by the Ethics Committee of Beijing Institute of Biotechnology. A total of 67 primary breast carcinomas tissue microarray (CGt No. XT14-023, Lot No. HBre-Duc070CS-01) samples with the informed consent of patients were purchased from Shanghai Outdo Biotech Company. All cases were females of age 30-89 years (mean age: 55.7 years) with breast cancer. The samples were deparaffinized, rehydrated, and pretreated with 3% hydrogen peroxide for 20 min to quench endogenous peroxidase activity. The antibody-binding epitopes of the antigens were retrieved by microwave treatment, and the samples were then preincubated with 10% normal serum to block nonspecific binding. Rabbit anti- Phospho-4EBP1-Thr37/46 (#2855, Cell Signaling Technology), rabbit anti- Phospho-NDRG1-Thr 346(#5482, Cell Signaling Technology) were used at dilutions of 1:100, respectively, as the primary antibodies for IHC in breast cancer. The specimens were incubated with primary antibodies for 1 h at room temperature, followed by incubation with biotinylated anti-rabbit secondary antibody and streptavidin-horseradish peroxidase. 3, 3'-Diaminobenzidine was used as a chromogen and hematoxylin was used for counterstaining. The Phospho-4EBP1-Thr37/46 and Phospho- NDRG1-Thr 346 score was generated by multiplying the percentage of stained cells (0-100%) by the intensity of the staining (negative, 0; low, 1-4; medium, 5-8; strong, 9-12). The correlation of Phospho-4EBP1- Thr37/46 with Phospho-NDRG1-Thr 346 was determined using the Spearman's rank correlation test.
Mass spectrometry-based phosphoproteomics analysis
MCF7 cells were treated with DMSO or 2 nM RAD001 for 24 h. Mass spectrometry-based phosphoproteomics analysis was performed by Novogene Biotech Company.
Generation of knockout cell lines via CRISPR/Cas9 technology
The sgRNA oligonucleotide pairs against SGK3 (SGK3 5′-AGATCACACCATGGACTACA-3′, 5′-AGCTGGGAATGCTTACACTT-3′ and 5′ -CTCTCTGTGTTCATCGGAGC-3′) was phosphorylated, annealed, and cloned into LentiCRISPRv2-puro. Lentiviruses were produced in HEK293T cells by transfection to 80% confluency in 100 mm culture dish with 6 ug DNA (3 ug SGK3-KO, 2.25 ug psPAX2, and 0.75 ug pMD2.G). DNA was mixed with 40 μL of lipofectamine 3000 in 300 μL Opti-MEM media (Invitrogen, US). Media containing viruses was collected and filtered 48 h after transfection. MCF7 and T47D cells were cultured in virus-containing media with polybrene (6 μg/mL) for 24 h. Cells were allowed to recover in fresh media for 48 h before the media was replaced with the selection media containing puromycin (1 ug/mL). The cells were kept in selection media for 3 days before use in experiments, unless stated otherwise. SGK3 knockouts in MCF7 and T47D cells were single-cell sorted into wells of a 96-well plate using a BD FACS Aria cell sorter (BD Biosciences, US) to establish clonal knockout cell lines. Cells were grown for 2 weeks, and the resultant colonies were cultured.
Animal Studies
Animal studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee at Beijing Institute of Biotechnology. Six-week-old female BALB/c nude mice were obtained from Vital River Laboratory (Beijing, China). Implanted with estrogen pellets (E2, 0.36 mg/pellet, 60-day release) (Innovative Research of America, Sarasota, FL, USA). Two days after implantation, 5×107 MCF7 and MCF7-SGK3-KO cells in serum-free medium were injected into the abdominal mammary fat pad of nude mice. When tumors reached an approximate size of 100 mm3, the mice were randomly allocated to 6 groups (n=6/group) according to body weights and tumor volumes for the following treatments: vehicle control, RAD001(3mg/kg/2d, intraperitoneal injection) and the combination of RAD001+MK2206(100 mg/kg/2 d, i.p. injection). For combination treatment, both drugs were given concurrently. Control mice received vehicle alone for both drugs. The average tumor diameter (two perpendicular axes of the tumor were measured) was measured in control and treatment groups using a caliper. The data are expressed as the increase or decrease in tumor volume in mm3 (mm3 = π/6 × [larger diameter] × [smaller diameter]2). Unpaired, two-tailed Student's t test was used to assess statistical significance. The tumor volumes of mice were monitored every 7 d. Four weeks after initiation of the treatment, all mice were euthanized. Xenograft primary tumors were harvested and proteins were extracted for Western blot analysis.
Statistical analysis
Statistical analyses for each experiment were performed as described in the corresponding figure legends. Analyses were carried out using GraphPad Prism 6 for method validation. All data are presented as mean±SEM, if not stated otherwise. Statistical significance was determined using paired Student t test; * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001. Data presented is representative of two or more independent experiments, unless indicated otherwise.
Results
The transient inhibition of RAD001 on 4EBP1 phosphorylation and cap-dependent translation contributes to its weak inhibitory effect on breast cancer cell growth
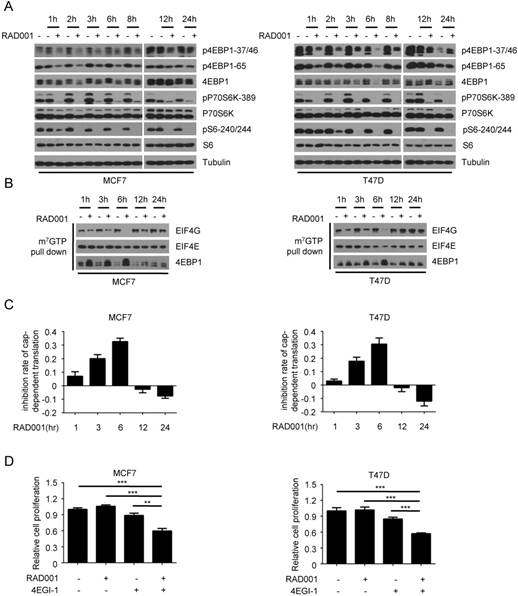
To explore the potential mechanism underlying rapamycin resistance in breast cancer, we first measured the effect of RAD001 treatment on the phosphorylation of p70S6K and 4EBP1, two downstream effectors of mTORC1. In both MCF7 and T47D cells, the abrogation of 4EBP1 phosphorylation (p-4EBP1-37/46 and p-4EBP1-65) by RAD001 was observed at 1 h and troughed after 6 h of treatment, while this inhibitory effect began to weaken at 8 h and was completely lost at 24 h (Fig 1A). However, the suppression of p70S6K phosphorylation and activity by RAD001 was evident at 1 h and persisted throughout the drug treatment process, as assessed by the phosphorylation of p70S6K (p-p70S6K-389) and its substrate S6 (pS6-240/244). To investigate the functional consequences of 4EBP1 re-phosphorylation after RAD001 treatment, we determined the dynamic changes of eIF4F translational initiation complex during the drug treatment process. Consistent with the changes of 4EBP1 phosphorylation, the formation of eIF4F translational initiation complex, as determined by the interaction of EFI4E and EIG4G, decreased gradually during 6 h of RAD001 treatment, but started to increase at 8 h and returned to pre-treatment levels at 24 h (Fig 1B). Furthermore, we measured the effect of RAD001 treatment on cap-dependent translation using a dual luciferase reporter system. As shown in Fig 1C, the inhibitory effect of RAD001 on cap-dependent translation peaked at 6 h and then gradually disappeared. These data in combination with the results that RAD001 treatment had minimal effect on the cell proliferation led us to speculate that the weak inhibitory effect of RAD001 on breast cancer cell growth might be attributed to its slight suppression on cap-dependent translation. To test this hypothesis, we added the 4EGI-1, an inhibitor of eIF4E/eIF4G interaction, to inhibit cap-dependent translation during RAD001 treatment. As expected, 4EGI-1 significantly enhanced the inhibitory effects of RAD001 on cell proliferation in both MCF7 and T47D cells (Fig 1 D). Together, these data suggested that the transient inhibition of RAD001 on 4EBP1 phosphorylation and cap-dependent translation contributed to its weak inhibitory effect on breast cancer cell growth.
The re-phosphorylation of 4EBP1 induced by RAD001 is dependent on PI3K, but only partially dependent on AKT
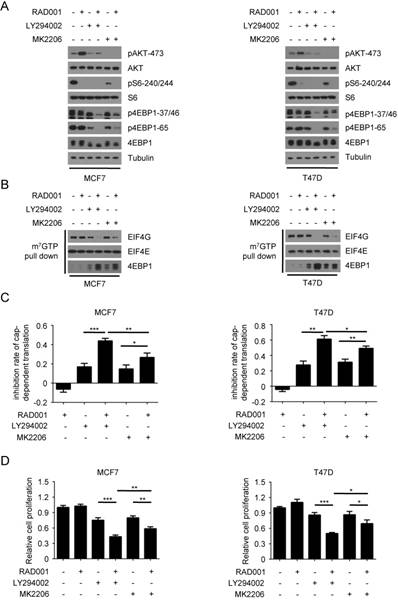
Previous studies have demonstrated that the feedback activation of PI3K/AKT contributes to the resistance to rapamycin in breast cancer cells(21, 26-28). Based on these previous observations and our data above, we hypothesized that rapamycin-induced PI3K/AKT activation may result in its transient inhibition on 4EBP1 phosphorylation and cap-dependent translation. To test this speculation, we first examined the effects of RAD001 and the PI3K inhibitor LY294002 or the AKT inhibitor MK2206, alone and in combination, on the phosphorylation of 4EBP1. In accordance with previous reports, RAD001 treatment profoundly activated AKT in both MCF7 and T47D cells, as indicated by phosphorylation on the Ser473 of AKT (Fig 2A). When combined with LY294002, RAD001 almost completely blocked the phosphorylation of 4EBP1 in both MCF7 and T47D cells. Although MK2206 treatment enhanced the inhibitory effect of RAD001 on the phosphorylation of 4EBP1, the degree of enhancement by MK2206 was much lower than that by LY294002 in both MCF7 and T47D cells. This effect was not due to more effective suppression of LY294002 on AKT activity, since LY294002 only partially inhibited the feedback activation of AKT while MK2206 almost blocked the phosphorylation of AKT in both MCF7 and T47D cells (Fig 2A). Consistent with the results of 4EBP1 phosphorylation, LY294002 exhibited the more potent inhibitory role than MK2206 in enhancing the inhibitory effect of RAD001 on the eIF4F translational initiation complex formation and cap-dependent translation in both MCF7 and T47D cells (Fig 2B and 2C). In addition, LY294002 more effectively increased the inhibitory effect of RAD001 on cancer cell proliferation than MK2206 (Fig 2D). Furthermore, LY294002 showed the more potent role than MK2206 in enhancing the effect of RAD001 on cell apoptosis in MCF7 cells (Fig. S2A and S2B), as assessed by apoptosis detection and Survivin expression. Collectively, these data demonstrated that the re-phosphorylation of 4EBP1 induced by RAD001 was dependent on PI3K, but only partially dependent on AKT, suggesting the possibility of other kinases that act downstream of PI3K that may be involved in the re-phosphorylation of 4EBP1 induced by RAD001.
The dynamic effects of rapamycin treatment on the 4EBP1 phosphorylation and cap-dependent translation in breast cancer cells. A. MCF7 and T47D cells were treated with DMSO or 2 nM RAD001 and collected at various time-points. Phosphorylation status of downstream mTORC1 effectors was analyzed using Western blot with indicated antibodies. B. MCF7 and T47D cells were treated with 2 nM RAD001 and collected at various time-points. Cell lysates were precipitated with m7GTP Sepharose beads followed by immunoblotting of eIF4G, eIF4E and 4EBP1. C. MCF7 and T47D cells were transfected with a bicistronic luciferase reporter plasmid that detects cap-dependent translation of the Renilla luciferase gene and cap-independent Polio IRES-mediated translation of the firefly luciferase gene. The transfected cells were treated with 2 nM RAD001 and collected at various time-points. Luciferase activities were measured by a dual-luciferase assay, and the Renilla/firefly luciferase luminescence ratio was calculated for cap-dependent translational activity. The results are expressed as the inhibition of cap-dependent translation relative to the DMSO-treated controls at each time and presented as means ± SEM (n = 3). D. MCF7 and T47D cells were treated with 2 nM RAD001 and 40 µM 4EGI-1, alone or in combination for 48 h, cell number was determined using the CCK-8 assay. Statistical significance was determined using paired Student t test; ** P < 0.01, *** P < 0.001. Values represent means ± SEM (n = 3).
SGK3 as a potent kinase involved in the re-phosphorylation of 4EBP1 induced by RAD001
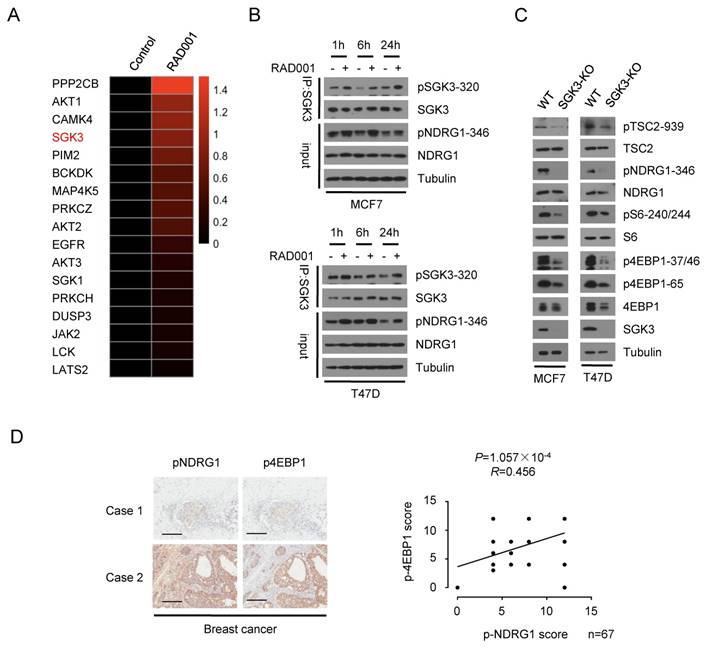
To identify the potential kinase that mediated the re-phosphorylation of 4EBP1 induced by RAD001, we collected MCF7 cells that were subjected to RAD001 treatment for 24 h and performed mass spectrometry-based phosphoproteomic analysis. Kinase-Substrate Enrichment Analysis (KSEA) revealed the feedback activation of AKT caused by RAD001, validating our screening strategy. In the kinases that were feedback-activated by RAD001, SGK3 attracted our attention because SGK3 has been reported to mediate the survival of certain oncogenic PIK3CA mutant breast cancer cells in which AKT is dispensable(20, 23) (Fig 3A). It was recently reported that SGK3 counteracts the inhibition of PI3K/AKT signaling and promotes tumor growth by re-activating the mTORC1 pathway(24), suggesting the possibility that SGK3 acts upstream of mTORC1 pathway similar to AKT. Thus, we speculated that SGK3 might be involved in the re-phosphorylation of 4EBP1 induced by RAD001. To test this hypothesis, we first confirmed the feedback activation of SGK3 induced by RAD001, as indicated by the increased phosphorylation of SGK3 and its substrate NDRG1 after RAD001 treatment (Fig 3B). To address the potential role of SGK3 in activating mTORC1 pathway in breast cancer cells, we created a targeted deletion for the SGK3 gene in MCF7 and T47D cells using the CRISPR/ Cas9 technology. In SGK3-deleted cells, the expression of SGK3 protein was completely lost and phosphorylation of NDRG1 was almost absent, confirming the functional deletion of SGK3 (Fig 3C). Furthermore, SGK3 deletion suppressed the phosphorylation of 4EBP1 and S6, suggesting the inhibition of the mTORC1 pathway (Fig 3C). To assess the clinical relevance of SGK3 activity in the mTORC1 pathway, we analyzed the expression of phosphorylated NDRG1 and 4EBP1 by immunohistochemical staining (IHC) in 67 human breast cancer samples. Consistent with SGK3 promotion of mTORC1 pathway in cultured cells, the expression of phosphorylated NDRG1 was positively associated with phosphorylated 4EBP1 (P = 1.057 × 10-4, R = 0.456) (Fig 3D). These data suggested that SGK3 acts as a potent kinase involved in the re-phosphorylation of 4EBP1 induced by RAD001.
Feedback activation of SGK3 and AKT contributes to rapamycin resistance by re-phosphorylating 4EBP1
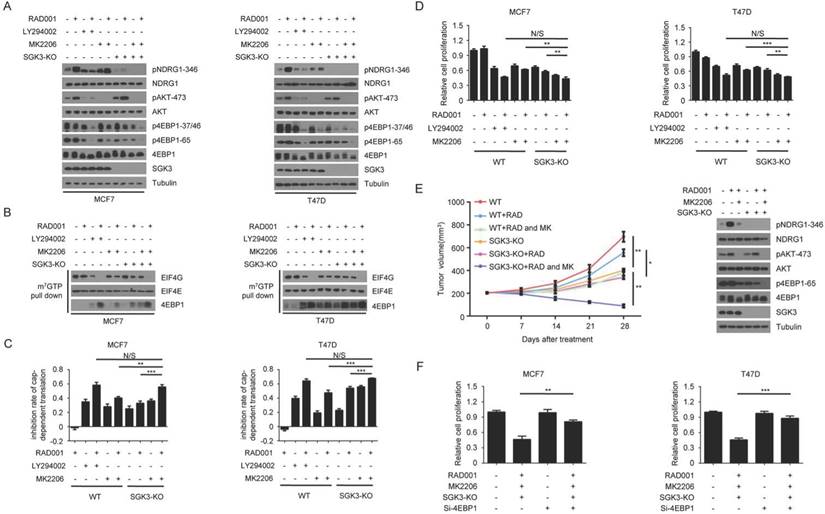
To explore whether SGK3 was involved in the re-phosphorylation of 4EBP1 induced by RAD001, we measured the phosphorylated 4EBP1 in wild type and SGK3-deleted MCF7 cells with or without RAD001 treatment. As shown in Fig. 4A, SGK3 deletion enhanced the inhibitory effect of RAD001 on 4EBP1 phosphorylation, but the degree of enhancement was much lower than that by LY294002, suggesting that SGK3 alone only partially contributed to the re-phosphorylation of 4EBP1 induced by RAD001. However, when combined with MK2206, SGK3 deletion almost blocked the re-phosphorylation of 4EBP1 induced by RAD001, and the degree of inhibition was comparable with that of LY294002 (Fig. 4A). Moreover, the combination of SGK3 deletion and AKT inhibition showed greater effect than SGK3 deletion or AKT inhibition alone in preventing the formation of the eIF4F translational initiation complex and cap-dependent translation in RAD001-treated MCF7 cells (Fig. 4B and 4C, respectively). Functionally, the combination of SGK3 deletion and AKT inhibition dramatically enhanced the sensitivity of tumor cells to RAD001 when compared with SGK3 deletion and AKT inhibition alone, reflected by CCK8 assay in vitro and tumor xenografts in vivo (Fig. 4D and 4E, respectively). To further determine whether the re-phosphorylation of 4EBP1 caused by SGK3 and AKT feedback activation resulted in the moderate inhibition of RAD001 on cell growth, 4EBP1 gene expression was knocked down using a specific siRNA target sequence in MCF7 cells with and without SGK3 deletion. As shown in Fig. 4F, silencing 4EBP1 expression markedly reversed the inhibitory effects of combination of SGK3 deletion and AKT inhibition on the proliferation of both MCF7 and T47D cells treated with RAD001. Collectively, these data suggested that the feedback activation of SGK3 and AKT induced by RAD001 re-phosphorylated 4EBP1, which counteracted the inhibitory effect of RAD001 on cap-dependent translation and cell growth in breast cancer cells.
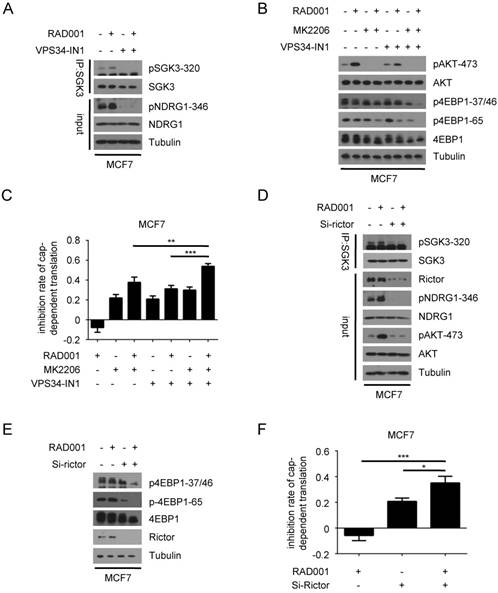
RAD001-mediated SGK3 phosphorylation is hVps34- and mTORC2-dependent
The above data have showed that the feedback activation of SGK3 and AKT induced by RAD001 resulted in the re-phosphorylation of 4EBP1 and counteracted the inhibitory effect of RAD001 on cell growth in breast cancer cells. In contrast to the well characterized mechanism by which RAD001 induced the feedback activation of AKT, the molecular basis underlying RAD001-mediated SGK3 phosphorylation remains unclear. Recently, hVps34 has been reported to regulate SGK3 activity through generating PtdIns(3)P, which recruits SGK3 to endosomal membrane and promotes its phosphorylation and activation by PDK1(24, 29). Thus, we first explored the role of hVps34 in mediating RAD001-mediated SGK3 phosphorylation by utilizing VPS34-IN1, a selective hVps34 inhibitor. As shown in Fig. 5A, elevated SGK3 phosphorylation as well as its kinase activity induced by RAD001 was profoundly suppressed by VPS34-IN1 treatment. Moreover, the combination of VPS34-IN1 and MK2206 exhibited greater effect than VPS34-IN1 or MK2206 alone in enhancing the inhibitory role of RAD001 on 4EBP1 phosphorylation, cap-dependent translation, and cell proliferation, functionally validating that hVps34 was required for RAD001-mediated SGK3 phosphorylation (Fig. 5B and C).
AKT partially contributes to Rapamycin-mediated re-phosphorylation of 4EBP1. A. MCF7 and T47D cells were treated with 2 nM RAD001, 2 µM LY294002 or 0.5 µM MK2206, alone or in combination for 24 h. Phosphorylation status of AKT and downstream mTORC1 effectors was analyzed using Western blot with indicated antibodies. B. MCF7 and T47D cells were treated with 2 nM RAD001, 2 µM LY294002 or 0.5 µM MK2206, alone or in combination for 24 h. Cell lysates were precipitated with m7GTP Sepharose beads followed by immunoblotting of eIF4G, eIF4E, and 4EBP1. C. MCF7 and T47D cells were treated with 2 nM RAD001, 2 µM LY294002 or 0.5 µM MK2206, alone or in combination for 24 h. The inhibition of cap-dependent translation was determined as in Figure 1C. Statistical significance was determined using paired Student t test; * P < 0.05, ** P < 0.01, *** P < 0.001. Values represent means ± SEM (n = 3). D. MCF7 and T47D cells were treated with 2 nM RAD001, 2 µM LY294002 and 0.2 µM MK2206, alone or in combination for 48 h, cell number was determined using the CCK-8 assay. Statistical significance was determined using paired Student t test; * P < 0.05, ** P < 0.01, *** P < 0.001. Values represent means ± SEM (n = 3).
Involvement of SGK3 in rapamycin-mediated re-phosphorylation of 4EBP1. A. Heat-map of rapamycin-mediated phosphorylation of protein kinase identified by the mass spectrometry-based phosphoproteomics analysis using MCF7 cells were treated with RAD001 or DMSO. B. MCF7 and T47D cells were treated with DMSO or 2 nM RAD001 and collected at various time-points. SGK3 was immunoprecipitated from lysates and analyzed by immunoblotting with the indicated antibodies. C. SGK3 was knocked out in MCF7 and T47D cells by using CRISPR/Cas9 and compared to the wild type cells. The cell lysates were subjected to immunoblot analysis with the indicated antibodies. D. Representative immunohistochemical staining of pNDRG1(T346) and p4EBP1(T37/46) in 67 human breast cancer samples. Case 1 and Case 2 refer to two representative samples categorized by low and high expression of pNDRG1(T346) and p4EBP1(T37/46). Scale bars, 50 μm. The correlation of pNDRG1(T346) with p4EBP1(T37/46) is shown. The P value was generated using the Spearman's rank correlation test.
Like AKT, hydrophobic motif phosphorylation of SGK3 is also controlled by mTORC2(30). We next investigated the role of mTORC2 in RAD001-mediated SGK3 phosphorylation. To specifically suppress the activity of mTORC2, we silenced the expression of Rictor, the unique component of mTORC2 complex compared with mTORC1 complex. As shown in Fig. 5D, the disruption of mTORC2 complex prevented SGK3 feedback activation induced by RAD001, indicating that RAD001-mediated SGK3 phosphorylation was dependent on mTORC2 activity. Given that mTORC2 activity is also required for RAD001-induced AKT feedback activation, we hypothesized that inhibiting mTORC2 activity alone could replace both SGK3 and AKT inhibition and prevent the re-phosphorylation of 4EBP1 induced by RAD001. As expected, Rictor silencing in combination with RAD001 treatment almost completely blocked phosphorylation of 4EBP1 in MCF7 cells (Fig. 5E). Accordingly, the disruption of mTORC2 complex significantly enhanced the inhibitory role of RAD001 on cap-dependent translation and cell proliferation in MCF7 cells (Fig. 5F). Collectively, these results suggested that RAD001-mediated SGK3 phosphorylation was dependent on hVps34 and mTORC2.
Feedback-activated SGK3 and AKT counteracts RAD001 treatment by phosphorylating TSC2 and reactivating mTORC1
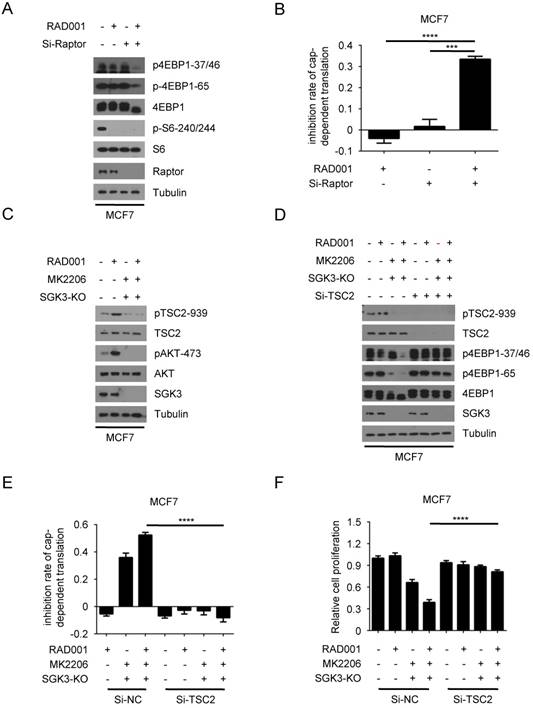
To explore whether the re-phosphorylation of 4EBP1 induced by RAD001 was still dependent on mTORC1 activity, we specifically inhibited the activity of mTORC1 by knocking down the expression of Raptor, the unique component of mTORC1 complex compared with mTORC2 complex. Although RAD001 or Raptor silencing alone only had a marginal effect on phosphorylated 4EBP1, the combination of RAD001 and Raptor silencing profoundly suppressed the phosphorylation of 4EBP1 (Fig. 6A). As a result, cap-dependent translation was significantly repressed by combination of RAD001 and Raptor silencing compared with either treatment alone in MCF7 cells (Fig. 6B). These data demonstrated that mTORC1 re-activation was required for the re-phosphorylation of 4EBP1 induced by RAD001.
We next investigated the mechanism by which mTORC1 was reactivated. The tubular sclerosis complex (TSC) is a key regulator of mTORC1 activity by integrating many upstream signals. Phosphorylated TSC2 releases its inhibitory role on Rheb GTPase, leading to mTORC1 activation(31, 32). Thus, we hypothesized that mTORC1 might be re-activated by the highly phosphorylated TSC2 caused by SGK3 and AKT feedback activation after RAD001 treatment. To test this hypothesis, we measured the effect of RAD001 treatment on phosphorylated TSC2. As expected, RAD001 enhanced the phosphorylation of TSC2 (Fig. 6C). Moreover, this enhancement was prevented by the combination of SGK3 deletion and MK2006 treatment, suggesting that the feedback activation of SGK3 and AKT promoted TSC2 phosphorylation induced by RAD001. We next tested whether TSC2 the high phosphorylation induced by RAD001 led to mTORC1 re-activation. As shown in Fig. 6D, TSC2 silencing significantly reversed the inhibitory effect of combined SGK3 deletion and MK2006 treatment on re-phosphorylation of 4EBP1 induced by RAD001. As a result, the inhibitory effect of combined SGK3 deletion and MK2006 treatment on cap-dependent translation and cell proliferation were profoundly repressed by TSC2 silencing, confirming that TSC2 high phosphorylation induced by feedback-activated SGK3 and AKT mediates mTORC1 re-activation (Fig. 6E and 6F). These data suggested that feedback-activated SGK3 and AKT counteracted RAD001 treatment by phosphorylating TSC2 and reactivating mTORC1 (Fig. 7).
Feedback activation of SGK3 and AKT confer rapamycin resistance by re-phosphorylating 4EBP1. A. SGK3-KO and WT cells were treated with 2 nM RAD001, 2 µM LY294002 and 0.5 µM MK2206, alone or in combination for 24 h. Phosphorylation status of AKT, TSC2 and 4EBP1 was analyzed using Western blot. B. SGK3-KO and WT cells were treated with 2 nM RAD001, 2 µM LY294002, and 0.5 µM MK2206, alone or in combination for 24 h. Cell lysates were precipitated with m7GTP Sepharose beads followed by immunoblotting of eIF4G, eIF4E, and 4EBP1. C. SGK3-KO and WT cells were treated with 2 nM RAD001, 2 µM LY294002, or 0.5 µM MK2206, alone or in combination for 24 h. The inhibition of cap-dependent translation was determined as in Figure 1C. Statistical significance was determined using paired Student t test; ** P < 0.01, *** P < 0.001. Values represent means ± SEM (n = 3). D. SGK3-KO and WT cells were treated with 2 nM RAD001, 2 µM LY294002 and 0.5 µM MK2206, alone or in combination for 48 h. Cell number was determined using the CCK-8 assay. Statistical significance was determined using paired Student t test; ** P < 0.01, *** P < 0.001. Values represent means ± SEM (n = 3). E. Volume of xenograft tumors derived from SGK3-KO and WT MCF7 cells treated with RAD001 (3 mg/kg) and MK2206 (100 mg/kg), alone or in combination. The tumor volume was measured once a week. Data are shown as mean ± s.d. (n = 6) (*P ˂ 0.05, **P ˂ 0.01 at 28 d). Tumor lysates were immunoblotted with the indicated antibodies. F. SGK3-KO and WT cells were treated with 2 nM RAD001, 10 µM LY294002, 0.5 µM MK2206 and (0.2 nM) 4EBP1 siRNA, alone or in combination for 48 h. Cell number was determined using the CCK-8 assay. Statistical significance was determined using paired Student t test; ** P < 0.01, *** P < 0.001. Values represent means ± SEM (n = 3).
RAD001-induced SGK3 phosphorylation is regulated by hVps34 and mTORC2. A. MCF7 cells were treated with 2 nM RAD001 and 1 µM VPS34-IN1, alone or in combination for 24 h. SGK3 was immunoprecipitated from lysates and analyzed by immunoblot with the indicated antibodies. B. MCF7 cells were treated with 2 nM RAD001, 0.5 µM MK2206, and 1 µM VPS34-IN1, alone or in combination for 24 h. Phosphorylation status of AKT and 4EBP1 were analyzed using Western blot. C. MCF7 cells were treated with 2 nM RAD001, 0.5 µM MK2206 and 1 µM VPS34-IN1, alone or in combination for 24 h. The inhibition of cap-dependent translation was determined as in Figure 1C. Statistical significance was determined using paired Student t test; ** P < 0.01, *** P < 0.001. Values represent means ± SEM (n = 3). D. MCF7 cells were transfected with 0.2 nM Rictor siRNA or negative control for 24 h, then treated with DMSO or 2 nM RAD001 for 24 h. SGK3 was immunoprecipitated from lysates were analyzed by immunoblot with the indicated antibodies. E. MCF7 cells were transfected with 0.2 nM Rictor siRNA or negative control for 24 hr, then treated with DMSO or 2 nM RAD001 for 24 h. Phosphorylation status of 4EBP1 was analyzed using Western blot. F. MCF7 cells were transfected with 0.2 nM Rictor siRNA or negative control for 24 h, then treated with DMSO or 2 nM RAD001 for 24 h. The inhibition of cap-dependent translation was determined as in Figure 1C. Statistical significance was determined using paired Student t test; * P < 0.05, *** P < 0.001. Values represent means ± SEM (n = 3).
Discussion
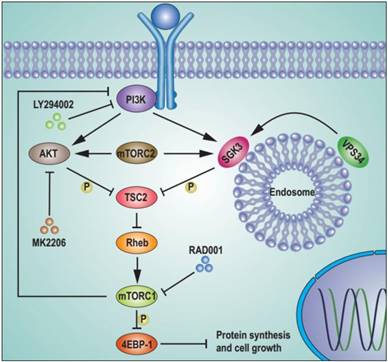
Although RAD001 has shown promising therapeutic effect in breast cancer treatment, the development of drug resistance limits it use in clinical therapies. In this study, we explored the molecular mechanism underlying RAD001 resistance in breast cancer. Our findings reveal that the transient inhibition of 4EBP1 phosphorylation and cap-dependent translation contributes to the modest anti-tumor activity of RAD001. Furthermore, we found that the feedback activation of SGK3 and AKT induced by RAD001 reactivates mTORC1 through phosphorylating TSC2, leading to the re-phosphorylation of 4EBP1 and limited repression of RAD001 on cap-dependent translation and tumor growth (Fig. 7). Our study provides new insight into the molecular basis mediating rapamycin resistance and may facilitate the development of novel therapeutic strategy against breast cancer.
As a major substrate of mTORC1, 4EBP1 plays an essential role in the regulation of cancer cell proliferation and survival by controlling translation initiation of oncogenic mRNAs. The weak inhibition of 4EBP1 phosphorylation has been reported to be involved in rapamycin resistance in certain cancer cells(33). Choo et al. first reported that rapamycin differentially inhibits S6Ks and 4EBP1; specifically, rapamycin persistently represses p70S6K activity but only transiently attenuates 4EBP1 phosphorylation (34). In acute myeloid leukemia (AML), PIM2-mediated 4EBP1 phosphorylation contributes to the limited repression of rapamycin on cell survival(35). In renal cell carcinoma (RCC), overexpressed GSK-3β leads to weak inhibition of 4EBP1 phosphorylation by rapamycin and combination of GSK-3β inhibitor and rapamycin remarkably suppressed 4EBP1 phosphorylation and cell proliferation(36). In breast cancer, the feedback activation of AKT is responsible for the limited inhibition of rapamycin on 4EBP1 phosphorylation and AKT inhibition dramatically enhances the inhibitory effect of rapamycin on 4EBP1 phosphorylation and cancer cell proliferation(16). In contrast, we found that AKT only partially contributes to the re-phosphorylation of 4EBP1, since LY294002 the inhibitor of PI3K, upstream of AKT, exhibited the more potent role than MK2206 the AKT inhibitor in enhancing the repression of RAD001 on 4EBP1 phosphorylation. This observation suggested that there were other kinases acting as downstream of PI3K that mediated the re-phosphorylation of 4EBP1 induced by RAD001 in coordination with AKT. By taking advantage of phosphoproteomic analysis, we identified SGK3 as a potent kinase involved in the re-phosphorylation of 4EBP1 induced by RAD001.
Feedback-activated SGK3 and AKT counteracts RAD001 treatment by phosphorylating TSC2 and reactivating mTORC1. A. MCF7 cells were transfected with 0.2 nM Raptor siRNA or negative control for 24 h, then treated with DMSO or 2 nM RAD001 for 24 h. Phosphorylation status of 4EBP1 was analyzed using western blot. B. MCF7 cells were transfected with 0.2 nM Raptor siRNA or negative control for 24 h, then treated with DMSO or 2 nM RAD001 for 24 h. The inhibition of cap-dependent translation was determined as in Figure 1C. Statistical significance was determined using paired Student t test; *** P < 0.001, **** P < 0.0001. Values represent means ± SEM (n = 3). C. SGK3-KO and WT MCF7 cells were treated with 2 nM RAD001 and 0.5 µM MK2206, alone or in combination for 24 h. Phosphorylation status of AKT and TSC2 was analyzed using Western blot. D. SGK3-KO and WT MCF7 cells were treated with 2 nM RAD001, 0.5 µM MK2206 and (0.2 nM) TSC2 siRNA, alone or in combination for 24 h. Phosphorylation status of 4EBP1was analyzed using Western blot. E. SGK3-KO and WT MCF7 cells were transfected with 0.2 nM TSC2 siRNA or negative control for 24 h, then treated with 2 nM RAD001, and 0.5 µM MK2206, alone or in combination for 24 h. The inhibition of cap-dependent translation was determined as in Figure 1C. Statistical significance was determined using paired Student t test; **** P < 0.0001. Values represent means ± SEM (n = 3). F. SGK3-KO and WT MCF7 cells were transfected with 0.2 nM TSC2 siRNA or negative control for 24 h, then treated with 2 nM RAD001 and 0.2 µM MK2206, alone or in combination for 24 h. Cell number was determined using the CCK-8 assay. Statistical significance was determined using paired Student t test; **** P < 0.0001. Values represent means ± SEM (n = 3).
Schematic representation of feedback activation of SGK3 and AKT contributes to rapamycin resistance by reactivating mTORC1/4EBP1 axis via TSC2. RAD001 treatment activated PI3K, which in combination with mTORC2 feedback activated AKT and SGK3. The feedback activation of SGK3 and AKT phosphorylates TSC2 and releases its inhibitory role on Rheb GTPase, leading to mTORC1 reactivation. The reactivated mTORC1 re-phosphorylates 4EBP1 and limits the repression of RAD001 on cap-dependent translation and tumor growth.
SGK3, with similar substrate specificity to AKT, is transcriptionally regulated by estrogen signaling and required for AKT-independent viability in breast cancer cells harboring PIK3CA mutations(20). However, the molecular mechanism by which SGK3 controls the development of breast cancer remains largely unknown. Our findings showed that SGK3 deletion profoundly suppressed the phosphorylation of 4EBP1. Additionally, the expression of phosphorylated NDRG1, the indicator of SGK3 activity, was positively associated with phosphorylated 4E-BP in human breast cancer tissues. These results indicated that SGK3 might exert its oncogenic role in breast cancer by activating mTORC1 signaling and enhancing cap-dependent transition. In line with this hypothesis, SGK3 has been shown to promote prostate cancer cell proliferation through activation of mTORC1 signaling(37). Furthermore, we found that the feedback-activated SGK3 in coordination with AKT, was responsible for the limited inhibition of 4EBP1 phosphorylation by RAD001. These data demonstrated that combined inhibition of mTORC1, SGK3, and AKT might be a promising therapeutic strategy for breast cancer. In addition, the phosphorylation of SGK3, like AKT and 4EBP1, might serve as a potential biomarker for predicting rapamycin resistance in clinic. We further found that RAD001-induced SGK3 activation was hVps34- and mTORC2-dependent. The observation that mTORC2 activity was essential for RAD001-induced SGK3 activation may explain why the ATP-competitive mTOR kinase inhibitors, which have capacity to inhibit both mTORC1 and mTORC2, exhibit much greater repression on 4EBP1 phosphorylation than rapamycin or its analogs(38).
Besides mTORC1, 4EBP1 can be phosphorylated by several other kinases, including PIM2 and GSK-3β, which are responsible for rapamycin resistance in AML or RCC respectively(35, 36). In contrast to these findings, we showed that the mTORC1 activity was required for the re-phosphorylation of 4EBP1, suggesting that the kinases involved in 4EBP1 phosphorylation after rapamycin exposure may be dependent on the tumor type. Furthermore, we showed that TSC2 high phosphorylation induced by feedback-activated SGK3 and AKT contributes to mTORC1 re-activation. In conclusion, our findings suggest that TSC2 functions downstream of SGK3 to activate mTORC1 in breast cancer cells.
Abbreviations
SGK3: Serum- and glucocorticoid-inducible kinase 3; AKT: Serine/threonine Kinase; mTORC1: Mammalian target of rapamycin complex 1; 4EBP1: 4E (eIF4E)-binding protein 1; eIF4E: Eukaryotic translation initiation factor 4E; eIF4F: Eukaryotic translation initiation factor; TSC2: Tubular sclerosis complex 2; KSEA: Kinase-Substrate Enrichment Analysis; NDRG1: N-Myc Downstream Regulated 1; PI3K: Phosphatidyl-inositol-3-kinase; p70S6K: 70 kDa ribosomal protein S6 kinase; PTEN: Phosphatase and tensin homolog; RCC: Renal cell carcinoma; AML: Acute myeloid leukemia; hVps34: Human type III phosphatidylinositol 3-kinase; ER: Estrogen receptor.
Supplementary Material
Supplementary figures and methods.
Acknowledgements
The authors thank Dr. Angelique Bordey, Departments of Neurosurgery and Cellular and Molecular Physiology, Yale University School of Medicine, for pcDNA3-rLuc-PolioIRES-fLuc plasmid. This study was supported by grants from the National Natural and Science Foundation of China (81672640 and 81772750), the National Key Subject of Drug Innovation of China (2018ZX09101001-003), the National Key Research and Development Program of China (2016YFC1202402).
Authors' contributions
YW and SL conceived and designed the study. HW, FH, ZZ, PW, NL and JW performed the experiments. HW, HL and JZ performed the data analysis. LY and HL helped to perform the partial experiments and review the statistical analysis. HW, YW and SL wrote the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;169:361-71
2. Kim LC, Cook RS, Chen J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene. 2017;36:2191-201
3. Chiarini F, Evangelisti C, McCubrey JA, Martelli AM. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol Sci. 2015;36:124-35
4. Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Targeting the translation machinery in cancer. Nat Rev Drug Discov. 2015;14:261-78
5. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
6. Clarke R, Tyson JJ, Dixon JM. Endocrine resistance in breast cancer-An overview and update. Mol Cell Endocrinol. 2015:418 Pt 3:220-34
7. Vicier C, Dieci MV, Arnedos M, Delaloge S, Viens P, Andre F. Clinical development of mTOR inhibitors in breast cancer. Breast Cancer Res. 2014;16:203
8. Lamming DW, Ye L, Sabatini DM, Baur JA. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J Clin Invest. 2013;123:980-9
9. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E. et al. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med. 2011;364:514-23
10. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S. et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449-56
11. Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R. et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381:125-32
12. Bachelot T, Bourgier C, Cropet C, Ray-Coquard I, Ferrero JM, Freyer G. et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. 2012;30:2718-24
13. Yardley DA, Noguchi S, Pritchard KI, Burris HA 3rd, Baselga J, Gnant M. et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv Ther. 2013;30:870-84
14. Steelman LS, Martelli AM, Cocco L, Libra M, Nicoletti F, Abrams SL. et al. The therapeutic potential of mTOR inhibitors in breast cancer. Br J Clin Pharmacol. 2016;82:1189-212
15. Vicier C, Dieci MV, Andre F. New strategies to overcome resistance to mammalian target of rapamycin inhibitors in breast cancer. Curr Opin Oncol. 2013;25:587-93
16. Mi W, Ye Q, Liu S, She QB. AKT inhibition overcomes rapamycin resistance by enhancing the repressive function of PRAS40 on mTORC1/4E-BP1 axis. Oncotarget. 2015;6:13962-77
17. Firestone GL, Giampaolo JR, O'Keeffe BA. Stimulus-dependent regulation of serum and glucocorticoid inducible protein kinase (SGK) transcription, subcellular localization and enzymatic activity. Cell Physiol Biochem. 2003;13:1-12
18. Castel P, Scaltriti M. The emerging role of serum/glucocorticoid-regulated kinases in cancer. Cell Cycle. 2017;16:5-6
19. Bruhn MA, Pearson RB, Hannan RD, Sheppard KE. AKT-independent PI3-K signaling in cancer - emerging role for SGK3. Cancer Manag Res. 2013;5:281-92
20. Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ. et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21-32
21. Xu J, Wan M, He Q, Bassett RL Jr, Fu X, Chen AC. et al. SGK3 is associated with estrogen receptor expression in breast cancer. Breast Cancer Res Treat. 2012;134:531-41
22. Wang Y, Zhou D, Phung S, Masri S, Smith D, Chen S. SGK3 is an estrogen-inducible kinase promoting estrogen-mediated survival of breast cancer cells. Mol Endocrinol. 2011;25:72-82
23. Gasser JA, Inuzuka H, Lau AW, Wei W, Beroukhim R, Toker A. SGK3 mediates INPP4B-dependent PI3K signaling in breast cancer. Mol Cell. 2014;56:595-607
24. Bago R, Sommer E, Castel P, Crafter C, Bailey FP, Shpiro N. et al. The hVps34-SGK3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC1 and tumour growth. EMBO J. 2016;35:2263
25. Wang Y, Zhou D, Phung S, Warden C, Rashid R, Chan N. et al. SGK3 sustains ERalpha signaling and drives acquired aromatase inhibitor resistance through maintaining endoplasmic reticulum homeostasis. Proc Natl Acad Sci U S A. 2017;114:E1500-e8
26. Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H. et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052-8
27. O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D. et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500-8
28. Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007;26:1932-40
29. Malik N, Macartney T, Hornberger A, Anderson KE, Tovell H, Prescott AR. et al. Mechanism of activation of SGK3 by growth factors via the Class 1 and Class 3 PI3Ks. Biochem J. 2018;475:117-35
30. Lien EC, Dibble CC, Toker A. PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol. 2017;45:62-71
31. Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10:151-62
32. Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H. et al. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell. 2014;156:771-85
33. Qin X, Jiang B, Zhang Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle. 2016;15:781-6
34. Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414-9
35. Kapelko-Slowik K, Owczarek TB, Grzymajlo K, Urbaniak-Kujda D, Jazwiec B, Slowik M. et al. Elevated PIM2 gene expression is associated with poor survival of patients with acute myeloid leukemia. Leuk Lymphoma. 2016;57:2140-9
36. Shin S, Wolgamott L, Tcherkezian J, Vallabhapurapu S, Yu Y, Roux PP. et al. Glycogen synthase kinase-3beta positively regulates protein synthesis and cell proliferation through the regulation of translation initiation factor 4E-binding protein 1. Oncogene. 2014;33:1690-9
37. Wang Y, Zhou D, Chen S. SGK3 is an androgen-inducible kinase promoting prostate cancer cell proliferation through activation of p70 S6 kinase and up-regulation of cyclin D1. Mol Endocrinol. 2014;28:935-48
38. Xie J, Wang X, Proud CG. mTOR inhibitors in cancer therapy. F1000Res. 2016:5
Author contact
![]() Corresponding authors: Shanhu Li, Department of Cell Engineering, Beijing Institute of Biotechnology, 27 Taiping Road, Beijing 100850, China. Email: lishanhu6com; Yongan Wang, State Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology and Toxicology, 27 Taiping Road, Beijing 100850, China. Email: yonganwcom.
Corresponding authors: Shanhu Li, Department of Cell Engineering, Beijing Institute of Biotechnology, 27 Taiping Road, Beijing 100850, China. Email: lishanhu6com; Yongan Wang, State Key Laboratory of Toxicology and Medical Countermeasures, Beijing Institute of Pharmacology and Toxicology, 27 Taiping Road, Beijing 100850, China. Email: yonganwcom.
Received 2018-12-23
Accepted 2019-2-14
Published 2019-3-9