ISSN: 1449-2288International Journal of Biological Sciences

Int J Biol Sci 2019; 15(6):1240-1251. doi:10.7150/ijbs.33044 This issue Cite
Research Paper
Epigenetic Down-Regulation of Sirt 1 via DNA Methylation and Oxidative Stress Signaling Contributes to the Gestational Diabetes Mellitus-Induced Fetal Programming of Heart Ischemia-Sensitive Phenotype in Late Life
Zewen Chen1,2, Lei Gong1, Peng Zhang1, Yong Li1, Bailin Liu1, Lubo Zhang1, Jian Zhuang2 ![]() , Daliao Xiao1
, Daliao Xiao1 ![]()
1. Lawrence D. Longo, MD Center for Perinatal Biology, Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, California, USA
2. Department of Cardiac Surgery, Guangdong Cardiovascular Institute, Guangdong Provincial People's Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China
Abstract

Rationale: The incidence of gestational diabetes mellitus (GDM) is increasing worldwide. However, whether and how GDM exposure induces fetal programming of adult cardiac dysfunctional phenotype, especially the underlying epigenetic molecular mechanisms and theranostics remain unclear. To address this problem, we developed a late GDM rat model.
Methods: Pregnant rats were made diabetic on day 12 of gestation by streptozotocin (STZ). Experiments were conducted in 6 weeks old offspring.
Results: There were significant increases in ischemia-induced cardiac infarction and gender-dependent left ventricular (LV) dysfunction in male offspring in GDM group as compared to controls. Exposure to GDM enhanced ROS level and caused a global DNA methylation in offspring cardiomyocytes. GDM attenuated cardiac Sirt 1 protein and p-Akt/Akt levels, but enhanced autophagy-related proteins expression (Atg 5 and LC3 II/LC3 I) as compared to controls. Ex-vivo treatment of DNA methylation inhibitor, 5-Aza directly inhibited Dnmt3A and enhanced Sirt 1 protein expression in fetal hearts. Furthermore, treatment with antioxidant, N-acetyl-cysteine (NAC) in offspring reversed GDM-mediated DNA hypermethylation, Sirt1 repression and autophagy-related gene protein overexpression in the hearts, and rescued GDM-induced deterioration in heart ischemic injury and LV dysfunction.
Conclusion: Our data indicated that exposure to GDM induced offspring cardiac oxidative stress and DNA hypermethylation, resulting in an epigenetic down-regulation of Sirt1 gene and aberrant development of heart ischemia-sensitive phenotype, which suggests that Sirt 1-mediated signaling is the potential therapeutic target for the heart ischemic disease in offspring.
Keywords: GDM, ROS, Sirt 1, DNA methylation, heart ischemia-sensitive phenotype
Introduction
The prevalence of gestational diabetes mellitus (GDM) has been increasing all over the world and growing evidence suggests that GDM has adverse effects not only on mothers but also on their offspring [1]. Previous studies have reported that GDM could cause cardiac development defects and increase morbidity of congenital heart defects [2]. Furthermore, maternal GDM has been shown to act as an independent risk factor for development of cardiovascular disease in offspring late in life [3]. However, the molecular epigenetic mechanisms underlying the maternal GDM-induced development of heart dysfunctional phenotypes in offspring are not fully understood.
Several molecular epigenetic mechanisms including DNA methylation, histone modification, and small non-code RNAs may play a key role in epigenetic inheritance and aberrant development of cardiovascular disease in late life. DNA methylation is the major mechanism in modification of gene expression patterns, and occurs at cytosine of the dinucleotide sequence CpG [4, 5]. Methylation in gene promoter regions is generally associated with a long-term shutdown of the associated gene. Clinical studies have shown that prenatally exposed to adverse factors could alter the global and gene- specific DNA methylation profiles [6]. Treatment with a DNA methylation donor or inhibitor can restore the gene expression patterns and prevent perinatal programed disease late in life [7-9]. In addition, recently we have demonstrated that the alteration of DNA methylation plays a key role in paternal hyperglycermia-induced transgenerational inheritance of susceptibility to hepatic steatosis in animal model [10]. These studies lead to hypothesize that aberrant DNA methylation contributes to the GDM- mediated fetal programming of heart ischemia- sensitive phenotypes in postnatal life.
Reactive oxygen species (ROS) are well- recognized as important signaling molecules involving in diverse biological responses [4, 11]. There is increasing evidence that oxidative stress plays a key role in the fetal programming of cardiovascular disorders in adulthood [12-14]. It has been shown a significant role of oxidative stress in the pathophysiology of GDM. GDM is associated with a heightened level of ROS, which affects both the mother and the fetal developing [15]. Furthermore, recent studies have showed a tight relation between ROS and DNA methylation [16]. ROS can differentially regulate DNA methylation patterns in carcinogenesis. ROS can function as catalysts of DNA methylation and also can induce site-specific hypermethylation via either the up-regulation of DNA methyltransferases (DNMTs) expression or the formation of a new DNMT containing complex [16]. These studies suggest that GDM-induced oxidative stress may directly up-regulate DNA methylation patterns and target specific genes in offspring hearts.
Sirtuin 1 (Sirt1) may be one of those genes which are epigenetically regulated by ROS-mediated DNA methylation signaling mechanism. In the pathophysiology, Sirt1 acts as a protector when suffering from cardiovascular disease and other disorders [12, 17]. Of particular interest, previous studies have also demonstrated that activation of Sirt1 signaling mimics ischemic preconditioning and protects against myocardial I/R injury [18-20]. Of importance, previous studies have demonstrated that maternal pathologic status could induce offspring renal and liver disorders, which are associated with Sirt 1 repression [21, 22]. Furthermore, increased expression of Sirt 1 attenuated offspring metabolic and liver disorders [22]. In addition, deletion of cardiac Sirt1 gene sensitized myocardium to ischemic insults and impaired left ventricle functions, such as ejection fraction (EF) and fractional shortening (FS) [23]. These studies suggest that aberrant Sirt1 may contribute to the fetal programming of heart ischemia-sensitive phenotypes in postnatal life, and rescue of the impaired Sirt1 level may reduce the adverse effect.
The present study was designed to reveal the potential epigenetic molecular mechanistic signaling pathway and therapeutic target for GDM-induced aberrant development of heart ischemia-sensitive phenotype in offspring. First, we established a GDM pregnant rat model to investigate whether GDM induces fetal programming of heart ischemia- sensitive phenotype late in life. Then we investigated the underlying epigenetic mechanisms and potential therapeutic molecular targets for reversing the heart ischemia-sensitive phenotype. In present study we examined cardiac ROS production, DNA methyltransferases and DNA methylation profile, Sirt 1 protein and its down-stream autophagy-related gene protein levels in the offspring. Furthermore, we determined whether antioxidant and DNA methylation inhibitor rescued Sirt 1 and its down-stream proteins expression, consequently rescued the heart ischemia-sensitive phenotype.
Methods
Experimental animals
All the procedures and protocols involved in this study were approved by the Institutional Animal Care and Use Committee of Loma Linda University and followed the NIH (National Institutes of Health, USA) Guide for the Care and Use of Laboratory Animals. Pregnant Sprague-Dawley rats (day 10 of gestation) were purchased from Charles River Laboratories (Portage, MI) and housed individually in cages which located in air-conditioned rooms (room temperature 22 °C, relative humidity 60%; lights on from 8:00 a.m. to 8:00 p.m.). Pellet food and tap water were available ad libitum. On day 12 of gestation, rats were divided into two groups randomly: 1) saline control (n=6) and 2) streptozotocin (STZ) (n=8) group. The pregnant rats were received a subcutaneous injection of saline or STZ (50 mg/kg) (Sigma, USA) as described in previous studies [24-26]. STZ has been widely used as a tool for inducing experimental diabetes because of its relatively specific pancreatic β cell cytotoxic effect [18, 24, 26]. The experimental rationale for use of STZ to develop an animal model of late gestational diabetes is that STZ can induce maternal hyperglycemia without directly significantly affecting the fetuses [26, 27]. Because STZ is imported by β cells via the GLUT2 transporter followed by induction of β cell necrosis, whereas, fetal β cell has lower levels of GLUT2 compared to adults [27]. The blood glucose levels in the dams and offspring were measured via tail-nicking using the Germaine Laboratories AimStrip Plus Blood Glucose Testing System (Fisher Scientific, Pittsburgh PA. Cat# 23-111-275) following manufacturer's instructions. A total of 64 pups from saline controls and 93 pups from STZ- treated dams were born. There was no difference of the litter sizes between the controls (10.7 ± 2.2 pups/ litter) and STZ-treated dams (11.6 ± 0.4 pups/litter).
After weaning (3 weeks after birth), the offspring from both groups were divided randomly into four groups: 1) control - ROS inhibitor, N-acetyl-cysteine (NAC); 2) control + NAC; 3) STZ - NAC; and 4) STZ +NAC. Antioxidant NAC (500 mg/kg/day) was placed in the drinking water which was concurrently started at weaning day (3-week-old) until 6-week-old. The rationale for using this dosage of NAC is that it can inhibit ROS production without significant side effect in rats as previously described [14, 28, 29]. After treatment, the male and female rat offspring were used for following cardiac functional and molecular biologic studies.
Heart ischemic model and measurement of myocardial infarct size
At the age of 6 weeks-old, the offspring rats were subjected to induce heart ischemia procedure in vivo as described previously [30]. Briefly, rats were anesthetized with isoflurane (5% for induction, 3% for maintenance) (Vet ONE, USA) by inhalation and placed on the RoVent Jr. Small Animal Ventilator (Kent Scientific). The rat body weight was used to adjust respiratory parameters on the mechanical ventilator. The ischemia was induced by an occlusion on left anterior descending artery (LAD) with a 6-0 PROLENE® suture (Ethicon, USA). After surgery, offspring were recuperated for 20 min in a single cage and then returned to their house cages. The rationale for use of 6-week old LAD ligation model in this study is to identify the early biomarkers of heart ischemic dysfunction and to provide an early time point to assess young offspring hearts' susceptibility to ischemic injury.
For measurement of cardiac infarct size, one set of rats from each group were sacrificed after 24 hours of ischemia. The whole hearts were collected and cut into five slices on the ice. Then, heart slices were incubated in 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma-Aldrich, USA) for 10 minutes at room temperature and immersed in formalin (Thermo Scientific, USA) over night. Slices were photographed and the areas of myocardial infraction and LV were analyzed by Image J (National Institutes of Health, USA), which is a java-based image processing program. The size of myocardial infarct was expressed as the ratio of myocardial infarct size to whole LV area, as previously described [9, 31].
Measurement of cardiac function
Another set of rats from each group were used for cardiac functional studies. The cardiac function was assessed by echocardiography (GE Healthcare, USA) before and 7 days after heart ischemia as previously described [32]. Briefly, offspring were anesthetized with inhalation of 3.5% isoflurane and placed on a pre-warmed (37 ℃) work surface. Then the rats were placed in the left lateral decubitus position. M-mode recording of the LV was obtained at the level of the mitral valve in the parasternal view using two-dimensional (2D) echocardiographic guidance in both short and long axis views. Cardiac function and heart dimensions were evaluated by 2D echocardiography on the anesthetized (2% isoflurane) rat. M-mode tracing was used to measure functional parameters such as LV end-diastolic dimension (LVEDD), LV end-systolic dimension (LVESD), LV end-diastolic volume (LVEDV) and LV end-systolic volume (LVESV) were calculated using the above primary measurements and accompanying software. The percentage of LV ejection fraction (EF) was calculated as (LVEDV-LVESV)/LVEDV x 100% and the percentage of LV fractional shortening (FS) was calculated as (LVEDD-LVESD)/LVEDD x 100%. Echocardiography data were recorded and analyzed blindly to the different treatments.
Western blot analysis
The total protein samples were isolated from left ventricles (LVs) tissues from the offspring and were homogenized in lysis buffer containing 150 mM NaCl (Bio-Rad, USA), 50 mM Tris·HCl (Bio-Rad, USA), 10 mM EDTA (Bio-Rad, USA), 0.1% Tween (Fisher, USA), 0.1% β-mercaptoethanol (Bio-Rad, USA), 0.1 mM phenylmethylsulfonyl fluoride (Bio-Rad, USA), 5 ug/mL leupeptin (Bio-Rad, USA), and 5 ug/mL aprotinin (Bio-Rad, USA), pH 7.4. Then, the homogenates were centrifuged at 4°C for 15 min at 12,000 g and the supernatants were collected. Samples with equal amount of total protein were loaded in the 8% or 10% polyacrylamide gel with 0.1% SDS (Bio-Rad, USA) and were separated by electrophoresis at 100 V for 80 min. Proteins were then transferred onto nitrocellulose membranes (Bio-rad, USA) and blocked for 3 h at room temperature. The membranes were incubated at 4 ℃ with primary antibodies against Sirtuin 1 (SIRT1) (Cell Signaling Technology, USA), DNA methyltransferase (DNMT) 1, 3a, 3b (Cell Signaling Technology, USA), NADPH oxidase (NOX) 1, 2, 4 (Boster Biological Technology, USA), p-Akt (Cell Signaling Technology, USA), Akt (Cell Signaling Technology, USA), Beclin1 (Cell Signaling Technology, USA), Atg5 (Cell Signaling Technology, USA), LC3B (Cell Signaling Technology, USA) and GAPDH (MilliporeSigma, USA), respectively. After washing and incubating with secondary antibodies, protein bands were visualized with enhanced chemiluminescence reagents and captured by photographic films (MidSci, USA). The films were analyzed with the document imaging scanner (HP, USA) with gray mode. Band intensities were normalized to GAPDH.
Measurement of ROS level
Tissues from LVs were homogenized on the ice at 30 mg/mL in Phosphate Buffered Saline (PBS) and centrifuged at 10000 for 5 min at 4 ℃. The total ROS levels in the LV tissue samples were measured with the OxiselectTM in vitro ROS/RNS assay kit (Cell Biolabs, Inc. San Diego, CA), following the manufacturer's instruction. Briefly, 50 µL of the samples or standard were added to a 96-well plate and mixed with 50 µL of catalyst and 100 µL of 2',7'-dichlorodihydrofluorescein diacetate (DCF). After incubation at room temperature for 30 min, the fluorescence (Ex480 nm/Em530 nm) was measured using a Synergy HT Multi-Mode Microplate Reader (Bio-Tek Instruments, Inc., Winooski, VT, USA).
5-mC DNA ELISA
Total DNA were isolated from LVs with A260/280 >1.6. The global DNA methylation levels in each sample of LV were determined by the detection of global 5-methylcytosine (5-mC) in genomic DNA that was isolated from the LV tissues using a 5-mC DNA ELISA kit (Zymo Research, USA) following the manufacturer's instructions. Briefly, 100 ng of each DNA sample and Negative/Positive Controls were mixed respectively with 5-mC coating Buffer, denatured at 98 ℃ for 5 min and cooled on ice for 10 min. After incubation at 37 ℃ for 1 h, each well of the 96 well plate was washed 3 times with 5-mC ELISA Buffer and incubated again at 37 ℃ for 30 min. Antibody mix consisting of Anti-5-Methylcytosine and Secondary Antibody in 5-mC ELISA Buffer were added into each well and incubated at 37 ℃ for 1 h. Then wells were washed with 5-mC ELISA buffer 3 times and a HRP developer was added to each well and incubated at room temperature for 30 min. The absorbance at 405 nm was measured using an ELISA plate reader. The 5-mC percentage for DNA samples was calculated using the logarithmic second-order regression equation at the standard curve that was constructed with negative and positive controls in the same experiment.
Treatment with DNA methylation inhibitor, 5-aza-2'-deoxycytodine (5-Aza) in ex vivo cultured fetal heart
Fetal rat hearts were isolated on the gestational day 19 and were cultured in M199 media (Hyclone) supplemented with 10% FBS and 1% penicillin/ streptomycin at 37 °C in 95% air/ 5% CO2 as previously described [33, 34]. After 24 h incubation in M199 culture media. The DNA methylation inhibitor, 5-aza-2'-deoxycytodine (Sigma), was added to the media at a final concentration of 10 μM. The control group was given the same volume of vehicle. Media was changed at 24 h intervals during the treatment. After 48 h of treatment, fetal hearts were harvested for further western blotting analysis.
Statistical analysis
All data are expressed as the mean ± SEM obtained from the number (n) of experimental animals given. Difference between the groups was compared by Student's t-test or analysis of variance (ANOVA) using the Graph-Pad Prism software (GraphPad Software Version 5, San Diego, CA, USA), where appropriate. For all comparisons, P-values less than 0.05 indicated statistical significance.
Results
Effect of GDM on body weight and blood glucose in pregnant rats and their offspring
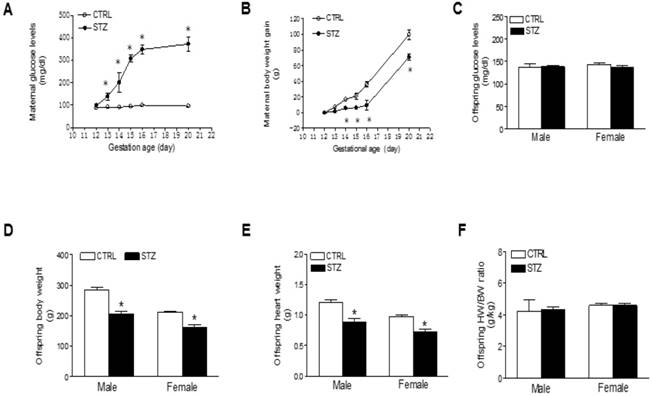
To develop a GDM model, we treated the pregnant rats on gestational day 12 with STZ as previously described [24, 25, 35, 36]. As shown in Figure 1, treatment with STZ showed a time-dependent increase in blood glucose level and constantly remained higher blood glucose level throughout period of pregnancy (Figure 1A), which was associated a decrease in maternal body weight gain (Figure 1B). As shown in figure 1C and Table 1, prenatal STZ exposure had no effect on blood glucose in both male and female offspring. However, prenatal STZ exposure significantly decreased the body weight (Figure 1D) and heart weight (Figure 1E) in both male and female offspring, while the heart to body weight ratio did not have difference between STZ group and control group (Figure 1F).
Effect of GDM on baseline heart function and ischemia-induced heart dysfunction in offspring
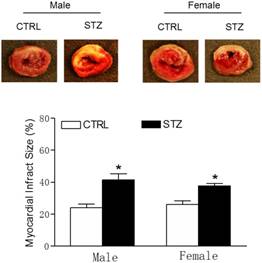
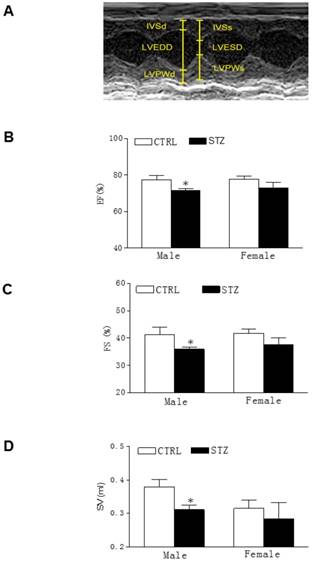
As shown in the Table 2, there were no significant differences of the baseline heart function measured by echocardiography between the GDM exposed group and the control group of both male and female offspring at the age of 6-week-old. However, 24 h ligation ischemia produced higher cardiac infarction size in the GDM exposed group than in the control group for both male and female offspring (Figure 2). In addition, as shown in the Table 2 and figure 3, GDM exposure significantly affected heart function 7 days after ischemia and attenuated the post-ischemic recovery of EF (Figure 3B), FS (Figure 3C) and SV (Figure 3D) only in male but not in female offspring as compared with control group.
GDM induced oxidative stress in offspring hearts
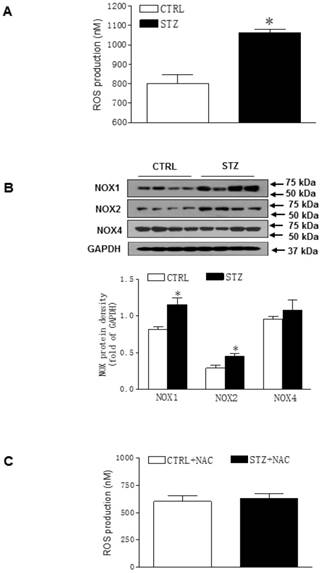
Because there was no difference of cardiac function between STZ and control group after heart ischemia in the female offspring (Figures 3 and Table 2), all of the following molecular biologic studies were only performed in male offspring. As shown in Figure 4A, the levels of ROS in LV tissues were significantly increased in STZ-treated group as compared with the control group in male offspring. In addition, the protein levels of NOX1 and NOX2 but not NOX4 were also up-regulated in LV tissues of male offspring in STZ-treated group as compared with the control group (Figure 4B). However, chronic treatment with the antioxidant, NAC reversed GDM-mediated ROS production in the male offspring (Figure 4C). Furthermore, treatment with NAC rescued GDM-mediated ischemia-induced infarction (data not shown) and cardiac dysfunction (Table 3) in the male offspring.
Effect of prenatal STZ on blood glucose, HW, BW and HW/BW in offspring 7 days after heart ischemia in the absence and presence of NAC
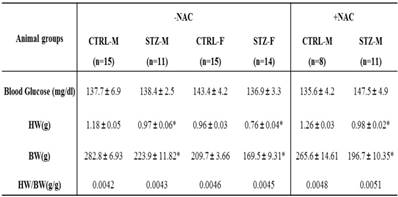
Note: STZ, streptozotocin; CTRL, control; HW, heart weight; BW, body weight; NAC, N-acetyl-cysteine; -NAC, in the absence of NAC; +NAC, in the presence of NAC; M, male; F, female. *P < 0.05
Effect of GDM on body weight and blood glucose in pregnant rats and their offspring. Pregnant rats were administered with either saline or STZ with 50 mg/kg on 12th day of gestation. A) Showing the daily blood glucose values in pregnant dams with diabetes induced by injection of STZ (●) on day 12 of gestation and sham control (○). B) Showing the maternal body weight gain values after STZ treatment (●) and sham control (○). C) Showing the blood glucose levels in both male and female offspring of STZ-treated (■) and control (□) groups at 6 weeks-old. D) Showing the body weights in both male and female offspring of STZ-treated (■) and control (□) groups at 6 weeks-old. E) Showing the heart weights in both male and female offspring of STZ-treated (■) and control (□) groups at 6 weeks-old. F) Showing the heart weight (HW)/body weight (BW) ratio in both male and female offspring of STZ-treated (■) and control (□) groups at 6 weeks-old. Values are means ± SE, *p < 0.05 compared to control.
Effect of prenatal STZ on LV parameters in offspring before heart ischemia and 7 days after heart ischemia
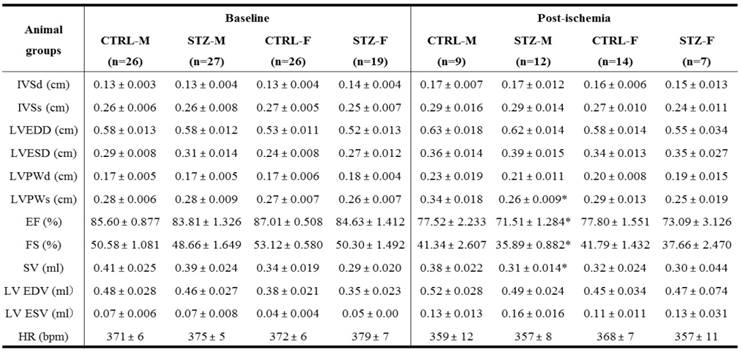
Note: CTRL, control; STZ, streptozotocin; IVSd/ IVSs, interventricular septal end diastolic/systolic dimension; LVEDD/ LVESD, left ventricular end-diastolic/ systolic dimension; LVPWd/ LVPWs, left ventricular posterior wall thickness at end diastole/systole; EF, ejection fraction; FS, fractional shortening; SV, stroke volume; LV EDV/ LV ESV, left ventricular end-diastolic/ systolic volume; HR, heart rate; M, male; F: female. *P < 0.05 vs. control.
Effect of NAC treatment of prenatal STZ-induced changes of LV parameters in male offspring before heart ischemia and 7 days after heart ischemia
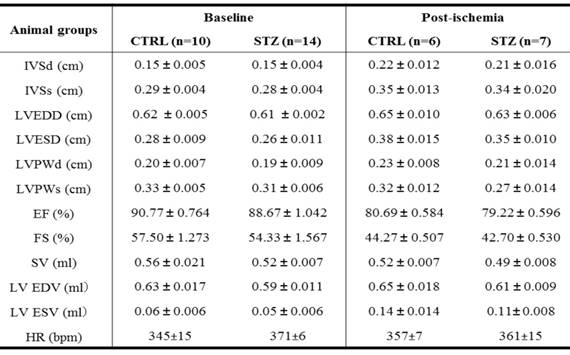
Note: NAC, N-acetyl-cysteine; CTRL, control; STZ, streptozotocin; IVSd/ IVSs, interventricular septal end diastolic/systolic dimension; LVEDD/ LVESD, left ventricular end-diastolic/ systolic dimension; LVPWd/ LVPWs, left ventricular posterior wall thickness at end diastole/systole; EF, ejection fraction; FS, fractional shortening; SV, stroke volume; LV EDV/ LV ESV, left ventricular end-diastolic/ systolic volume; HR, heart rate.
Effect of GDM on Sirt1 and autophagy-related protein abundances in male offspring hearts
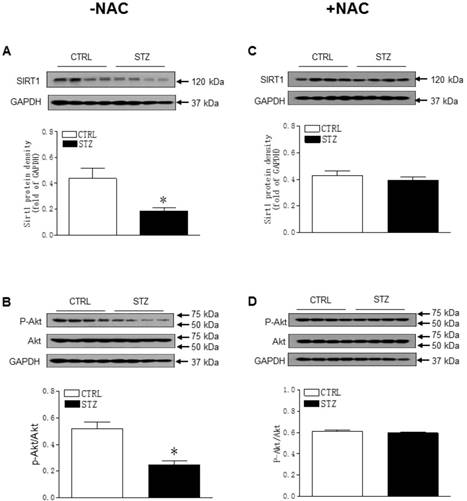
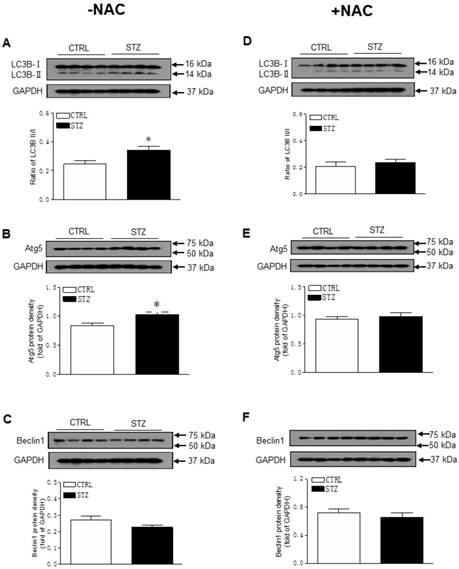
As shown in Figure 5A-B, GDM exposure significantly decreased the offspring cardiac protein expressions of Sirt1 and its downstream target p-Akt/Akt as compared with control group in the absence of NAC treatment. However, there were no significant differences of the protein expressions of Sirt1 and p-Akt/Akt ratio between the GDM exposed and control offspring after the treatment of NAC (Figure 5C and 5D). Furthermore, prenatal STZ exposure promoted autophagy activity in offspring heart tissues. As shown in Figure 6, there were significant increases in autophagy-related protein LC3B II/I ratio (Figure 6A) and Atg 5 (Figure 6B) but not Beclin 1 (Figure 6C) levels of cardiac tissues in STZ exposed offspring as compared to the controls. However, NAC treatment reversed the effects of STZ on LC3B II/I ratio (Figure 6C), Atg 5 (Figure 6D) and Beclin 1 (Figure 6E) protein expression levels in the offspring heart tissues.
Effect of GDM exposure on ischemia-induced myocardial infarction in offspring. Offspring from each group were subjected heart ischemia as indicated in the Methods Section. 24 hours after heart ischemia, the hearts were isolated and their infarct sizes were determined with 2% TTC staining (A). The bar graph (B) showing percent of left ventricle infarct size (infarct area/risk area x100%) in each offspring group. Data are means ± SEM of animals from each group (n=6-5 in male offspring, n=5-7 in female offspring) were analyzed by 2-way ANOVA. *P < 0.05 versus.
Effect of GDM on heart function in offspring. The echocardiographic data of both the control and STZ exposed groups were examined at 7 days after heart ischemia, as described under Methods Section. (A) A representative echocardiography shows the measurement of LVSd, LVEDd, LVPWd, LVSs, and LVPWs. (B) Percent of ejection fraction (EF), (C) Percent of fractional shortening (FS), and (D) Stroke volume (SV) in both male and female offspring of STZ-treated (■) and control (□) groups at 7th day after heart ischemia. Data are means ± SEM of animals from each group (n=9-12 in male offspring, n=14-7 in female offspring) were analyzed by 2-way ANOVA. *P < 0.05 versus control.
Effect of GDM on DNA methylation and the role of DNA methylation in regulation of Sirt 1 gene expression
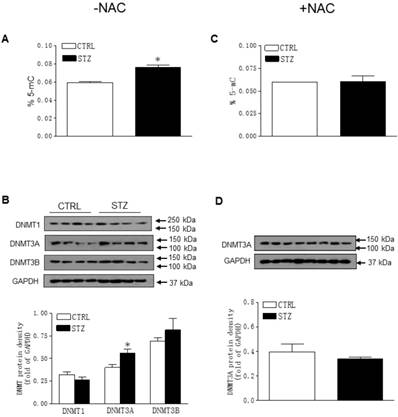
As shown in Figure 7A, the global DNA methylation levels in the LV tissues were significantly higher in the STZ-treated offspring than in the control group. The changes of DNA methylation were associated with a selective increase in DNMT3A but not DNMT1 and DNMT3B protein expression (Figure 7B). As shown in Figure 7C and 7D, treatment with NAC reversed the effects of STZ on global DNA methylation and DNMT3A expression in the offspring heart tissues.
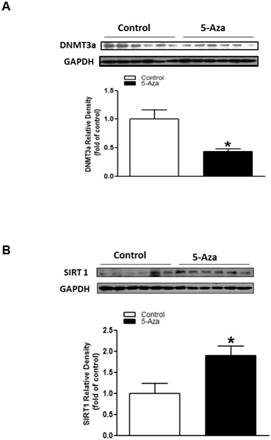
To see whether DNA methylation plays a key role in regulation of Sirt 1 gene, we treated the fetal hearts with DNA methylation inhibitor, 5-Aza ex vivo. As shown in Figure 8, treatment with 5-Aza significantly decreased cardiac DNMT3A protein expression (Figure 8A), but increased the expression of Sirt 1 (Figure 8B) in the fetal hearts.
Discussion
GDM is a worldwide disease which affects up to 16% of pregnant women globally, and it has adverse effects on both mother and child [37, 38]. Exposure to diabetes in utero is one of the most important risk factors for development of cardiovascular dysfunction [39-41]. Most of the previous studies have been mainly focusing on the effects of pre-gestational diabetes. In present study, we have developed a rat model of diabetes with maternal hyperglycemia limited to the last third of pregnancy (gestational day 16 ~ 21), which is similar to that occurring with gestational diabetes in clinic. Our present data indicated that late maternal gestational hyperglycemia significantly decreased the body and heart weights of both male and female offspring as compared with the control group. However, the effects of maternal diabetes on offspring body size are not consistent. Pre-gestational maternal diabetes and later gestation maternal diabetes have produced either large [26, 42] or small [26, 43] offspring. The reasons for this discrepancy are unclear. However, previous studies suggest that the heavier offspring tended to come from mothers with mild hyperglycemia, whereas the underweight offspring were most likely from mothers with more severe hyperglycemia [26].
In human and animal studies, in utero exposure to diabetes is associated with aberrant development of fetal heart and fetal heart dysfunction [39-41, 44]. The present study further demonstrated that exposure to a hyperglycemia in utero environment leads to impairment of cardiac function that persist into late life. Specifically, although the bassline cardiac functions measured by echocardiography were no difference between the control and GDM-exposed offspring at 6 weeks of age, we found that ischemic insult-induced cardiac infarction sizes were significantly increased in GDM-exposed offspring as compared to controls. In addition, we also found that compared to controls, GDM-exposed offspring had diminished cardiac function in response to ischemic challenge. These findings suggest that GDM exposure may not impair offspring heart function at resting condition but enhance the heart susceptibility to ischemic insult and development of heart ischemia- sensitive phenotype in young offspring. Similar findings have been reported in different animal models where in utero exposure to adverse environmental stimuli have had no effect on cardiac function at resting condition while enhances the cardiac ischemic infarction and dysfunction after ischemia stimulation [14, 32]. The present finding that GDM caused a gender-dependent attenuation of cardiac functional recovery after 7 days of ischemic stimulation in male but not female offspring, suggests a gender difference in response to GDM exposure. The gender differences found in this study are not surprised and have been reported in different models of fetal programming of cardiovascular disease [31, 45, 46]. The majority of these studies have shown that male offspring are more affected by the adverse in utero environment than female offspring. Furthermore, in present study, we found that GDM exposure enhanced ischemia-induce cardiac infarction in both male and female offspring after 24 h of ischemic stimulation. However, the enhanced cardiac dysfunction was only observed in male but not female offspring after 7 days of ischemic stimulation. These findings suggest that female offspring heart has much higher compensated mechanism than male offspring heart in response to ischemic insult. The underlying mechanisms by which biological sex contributes to these developmental programed cardiovascular dysfunctions remains unclear at present; nevertheless, it has been proposed that some of the intriguing mechanistic molecules and proteins such as sex hormones and recently reported sexual dimorphism in the transcriptome of a variety of tissues many play a key role in the gender difference in the development of cardiovascular disease late in life. Therefore, in our future studies, we will consider the underlying mechanisms of gender differences as further investigation into the developmental origins of cardiovascular dysfunction.
Effect of GDM on the ROS level in offspring. Heart tissues were isolated from male offspring. (A) ROS levels in the left ventricle (LV) tissues isolated from both control (□) and STZ-treated (■) groups were measured using in vitro ROS/RNS assay kit. (B) NOX1, 2, and 4 protein abundances in the LV tissues isolated from both control (□) and STZ-treated (■) groups were determined by Western blot analysis. Their protein densities were normalized to internal control (GAPDH). (C) After NAC pretreatment, ROS levels in the LV tissues isolated from both control (□) and STZ-treated (■) groups were measured using in vitro ROS/RNS assay kit. Data are means ± SEM (n=4 animals/group). *P < 0.05 vs. control, as determined by Student's t-test.
Although the underlying mechanisms that may lead to enhanced susceptibility to ischemic insult in the GDM-exposed male offspring are unknown, exaggerated ROS or enhanced sensitivity to oxidative stress may be one of the key mechanisms contributing to the heart dysfunction. Recent studies have demonstrated that an adverse uterine environment, including maternal diabetes and obesity, alters oxidative pathways and induces oxidative stress in offspring [47, 48]. Consistent with the previous studies, the present study also showed increases in cardiac ROS productions and ROS-associated NADPH oxidases (NOX1 and NOX2) in the GDM- exposed offspring. In addition, previous studies have suggested that ROS is a hallmark of cardiovascular disease and participates the pathophysiological change in cardiac ischemia/reperfusion injury [49, 50]. Our current data that pretreatment of the offspring with an antioxidant N-acetylcysteine (NAC) blocked the enhanced ROS and improved heart functional recovery following the ischemic insult, suggests that the enhanced ROS is one of the important molecular mechanisms underlying GDM- induced fetal programming of cardiac dysfunction late in life. Furthermore, the present study also provides novel therapeutic approaches for improvement of the programmed cardiac function with supplemental antioxidant.
Effect of GDM on the protein expression of Sirt1 and p-Akt/Akt in male offspring. The hearts were isolated from both control (□) and STZ-treated (■) groups of male offspring. The protein abundance in the LV tissue was determined by Western blot analysis. The Sirt1 protein density (A) and P-Akt/Akt protein density (B) in the LV tissues of the groups which had not treated with NAC. The Sirt1 protein density (C) and P-Akt/Akt protein density (D) in the LV tissues of the groups which had treated with NAC. Data are means ± SEM (n=4 animals/group). *P < 0.05 vs. control, as determined by Student's t-test.
A decrease in Sirt1 protein expression in male offspring heart tissues was observed in GDM-exposed group as compared to the controls. This finding suggests that Sirt1 may be one of the molecular mechanisms in maternal GDM-induced aberrant development of heart ischemia-sensitive phenotype in offspring. Indeed, previous studies have demonstrated that Sirt 1 expression is reduced in diabetic heart and overexpression of Sirt 1 by adenoviral vectors encoding Sirt 1 could reduce diabetes-exacerbated myocardial infarct size and improves cardiac function in diabetic rats [18]. Sirt1 functions as an acetylation regulator to maintain cardiac mitochondrial integrity and normal postnatal myocardium development [51]. Moreover, Sirt1-mediated deacetylation of lysines and PDK1 locates in the PH domain of Akt, resulting in direct upregulation of Akt signaling [52, 53]. Similar to the reduced Sirt 1 expression, our present study has shown that Akt phosphorylation levels were significantly attenuated in GDM-exposed offspring hearts as compared to the controls. Regulation of Akt signaling by Sirt 1 has significant implication in the development of cardiac disease via control of a variety of cellular processes, such as apoptosis and autophagy [54]. Akt is one of the key signaling pathways in negative regulation of autophagy in myocardial ischemic injury [55].
Autophagy is well documented to be involved in diverse pathological processes [56]. In present study, we examined the status of autophagy-related biomarkers including Atg5, Beclin1 and LC3B II/I in the offspring heart tissues. Our data indicated that Atg5 and LC3B-II/I was overexpressed in GDM- exposed hearts as compared to the controls. These findings suggest that the exaggerated autophagy signaling may be one of the molecular mechanisms underlying GDM-mediated development of heart ischemia-sensitive phenotype in rat offspring. Furthermore, our present finding that pretreatment of the offspring with antioxidant NAC reversed GDN- mediated changes of Sirt 1, Akt phosphorylation and autophagy-related protein expressions in the heart tissues, suggests that GDM-induced ROS is the up-stream regulator.
DNA methylation is one of the key epigenetic mechanisms in modification of gene expression patterns and occurs at CpG sites in promoter region, leading to a long-term shutdown of the associated gene [4, 5]. Previous studies have shown that GDM exposure could alter global and gene-specific DNA methylation profile in offspring [57, 58]. Similarly, in present study, our data showed that global DNA methylation level was increased in GDM-exposed offspring as compared to the controls, which was associated with a selective increase in Dnmt 3A protein expression in the offspring heart tissues. Furthermore, our present data showed that pre-treatment of fetal hearts ex vivo with DNA methylation inhibitor 5-Aza significantly attenuated Dnmt 3A protein expression and increased Sirt 1 protein expression. These findings suggest that GDM-mediated Sirt 1 repression is epigenetically regulated by DNA hypermethylation mechanism in the offspring heart. However, it is unknown at present whether this is a direct or indirect regulation. In our future study we will investigate the effect of DNA methylation inhibitor on Sirt 1 gene promoter activity to confirm whether this is a direct regulation. In addition, the present findings of enhanced global cardiac DNA methylation suggests that altered methylation does not only regulate Sirt 1 expression but also could regulate other target genes expressions.
Effect of GDM on autophagy-related protein expression in male offspring. The hearts were isolated from both control (□) and STZ-treated (■) groups of male offspring. The protein abundance in the LV tissue was determined by Western blot analysis. The LC3B-I/II protein density (A), Atg5 protein density (B), and Beclin 1 protein density (C) in the LV tissues of the groups which had not treated with NAC. The LC3B-I/II protein density (D), Atg5 protein density (E), and Beclin 1 protein density (F) in the LV tissues of the groups which had treated with NAC. Data are means ± SEM (n=4 animals/group). *P < 0.05 vs. control, as determined by Student's t-test.
Enhanced levels of both ROS and DNA methylation are one of the most common characteristics in human programmed disease. However, the relation between these two is not well understood. Previous studies have reported that ROS-induced oxidative stress can function as catalysts of DNA methylation and induce site-specific hypermethylation via either the up-regulation of Dnmts expression or the production of a new Dnmt containing complex [16, 54]. In present study, our findings that pre-treatment of offspring with ROS inhibitor NAC reversed GDM-mediated Dnmt 3A over-expression and DNA hypermethylation in cardiac tissues, suggest a cause-consequence relationship between GDM-induced ROS and DNA methylation. The ROS-induced DNA methylation pattern alteration may play an important role in epigenetic down-regulation of Sirt 1 gene in the GDM -exposed offspring hearts.
Study limitations: 1) In our present study, we found that GDM exposure caused offspring growth restriction. However, the molecular mechanisms underlying GDM-mediated offspring growth restriction are not fully understood. Therefore, in our future studies, we need to investigate whether GDM exposure epigenetically alters development and growth-associated hormones levels and genes expressions. 2) In present study we investigated the effect of fetal GDM exposure on the postnatal cardiovascular function. Although the offspring from each dam are randomly divided into each experimental group, one of the limitations for this study is that we used the number of offspring as experimental number (n). 3) Our present results indicated that GDM exposure had no effect on cardiac function in female offspring, however, this does not mean that the heart ischemia-sensitive target proteins are not altered by GDM exposure in females, which opens a wide door for our future studies.
In conclusion, our present study provides a novel evidence that GDM exposure is one of the important risk factors in the aberrant development of heart ischemia-sensitive phenotype in offspring in gender-dependent manner. Specifically, our data indicated that GDM exposure caused an increase in ROS production and induced oxidative stress in offspring heart tissues. The enhanced ROS selectively activated and increased DNA methyltransferase (Dnmt 3A) expression, leading to DNA hypermethylation. GDM exposure attenuated cardiac Sirt1 protein expression and decreased Akt phosphorylation, which was regulated by DNA methylation. The GDM-mediated down-regulation of Sirt 1/Akt signaling promoted the progress of autophagy, resulting in aberrant development of heart ischemia- sensitive phenotype in offspring. Furthermore, antioxidant treatment could reverse the GDM- mediated DNA hypermethylation, Sirt 1 repression, and autophagy signaling, consequently rescue GDM- induced heart ischemia-sensitive phenotype. With improved understanding of the epigenetic molecular mechanisms underlying gestational diabetes milieu-induced fetal programming of cardiovascular dysfunction, our findings may provide novel therapeutic strategies to prevent or rescue the development of cardiovascular disease later in life.
Effect of GDM on the global DNA methylation and DNA methyltransferase (DNMT) protein expression in male offspring. The hearts were isolated from both control (□) and STZ-treated (■) groups of male offspring. Global DNA methylation levels in LV tissues of male offspring, which were without (A) or with (C) NAC treatment, were measured using in 5-mC DNA ELISA kit. The protein abundances of DNMTs in the left ventricle tissues of male offspring, which were without (B) or with (D) NAC treatment, were determined by Western blot analysis. Data are means ± SEM (n=4 animals/group). *P < 0.05 vs. control, as determined by Student's t-test.
Effect of DNA methylation inhibitor on Sirt 1 protein expression in fetal hearts ex vivo. Fetal rat hearts were isolated from gestational day 19 and were cultured in an incubator. After 48 hours of treatment with DNA methylation inhibitor, 5-aza-2'-deoxycytodine (5-Aza, 10 μM) (■) or control vehicle (□). The protein levels of DNMT3a (A) and Sirt 1 (B) in the fetal hearts were determined by Western blot analysis. Data are means ± SEM (n=6 hearts/group). *P < 0.05 vs. control, as determined by Student's t-test.
Abbreviations
GDM: gestational diabetes mellitus; ROS: reactive oxygen species; STZ: streptozotocin; NAC: N-acetyl-cysteine; DNMT: DNA methyltransferase; I/R: ischemia/reperfusion; TTC: 2,3,5-triphenyltetrazolium chloride; LAD: left anterior descending artery; IVSd/IVSs: interventricular septal end diastolic/ systolic dimension; LVEDD/LVESD: left ventricular end-diastolic/systolic dimension; LVPWd/LVPWs: left ventricular posterior wall thickness at end diastole/systole; EF: ejection fraction; FS: fractional shortening; SV: stroke volume; LV EDV/ LV ESV: left ventricular end-diastolic/systolic volume; HR: heart rate; NOX: NADPH oxidase; Akt: protein kinas B; DCF: 2',7'-dichlorodihydrofluorescein diacetate; 5-mc: 5-methylcytosine; 5-Aza: 5-aza-2'-deoxycytodine.
Acknowledgements
This work was supported by National Institutes of Health Grants HL135623 (DX), DA041492 (DX), HD088039 (DX), and HL118861 (LZ). The author Zewen Chen thanks Guangdong Cardiovascular Institute and Loma Linda University for their support. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Russo LM, Nobles C, Ertel KA, Chasan-Taber L, Whitcomb BW. Physical activity interventions in pregnancy and risk of gestational diabetes mellitus: a systematic review and meta-analysis. Obstet Gynecol. 2015;125:576-82
2. Dervisoglu P, Kosecik M, Kumbasar S. Effects of gestational and pregestational diabetes mellitus on the foetal heart: a cross-sectional study. J Obstet Gynaecol. 2018;38:408-12
3. Leybovitz-Haleluya N, Wainstock T, Landau D, Sheiner E. Maternal gestational diabetes mellitus and the risk of subsequent pediatric cardiovascular diseases of the offspring: a population-based cohort study with up to 18 years of follow up. Acta Diabetol. 2018
4. Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597-610
5. Martinez SR, Gay MS, Zhang L. Epigenetic mechanisms in heart development and disease. Drug Discov Today. 2015;20:799-811
6. Breton CV, Byun HM, Wenten M, Pan F, Yang A, Gilliland FD. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am J Respir Crit Care Med. 2009;180:462-7
7. Bakker R, Timmermans S, Steegers EA, Hofman A, Jaddoe VW. Folic acid supplements modify the adverse effects of maternal smoking on fetal growth and neonatal complications. J Nutr. 2011;141:2172-9
8. Steegers-Theunissen RP, Obermann-Borst SA, Kremer D, Lindemans J, Siebel C, Steegers EA, Slagboom PE, Heijmans BT. Periconceptional maternal folic acid use of 400 microg per day is related to increased methylation of the IGF2 gene in the very young child. PLoS One. 2009;4:e7845
9. Ke J, Dong N, Wang L, Li Y, Dasgupta C, Zhang L, Xiao D. Role of DNA methylation in perinatal nicotine-induced development of heart ischemia-sensitive phenotype in rat offspring. Oncotarget. 2017;8:76865-80
10. Li X, Shi X, Hou Y, Cao X, Gong L, Wang H, Li J, Wu C, Xiao D, Qi H, Xiao X. Paternal hyperglycemia induces transgenerational inheritance of susceptibility to hepatic steatosis in rats involving altered methylation on Pparalpha promoter. Biochim Biophys Acta Mol Basis Dis. 2019;1865:147-60
11. Hamanaka RB, Chandel NS. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci. 2010;35:505-13
12. Cambonie G, Comte B, Yzydorczyk C, Ntimbane T, Germain N, Le NL, Pladys P, Gauthier C, Lahaie I, Abran D, Lavoie JC, Nuyt AM. Antenatal antioxidant prevents adult hypertension, vascular dysfunction, and microvascular rarefaction associated with in utero exposure to a low-protein diet. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1236-45
13. Roghair RD, Wemmie JA, Volk KA, Scholz TD, Lamb FS, Segar JL. Maternal antioxidant blocks programmed cardiovascular and behavioural stress responses in adult mice. Clin Sci (Lond). 2011;121:427-36
14. Xiao D, Wang L, Huang X, Li Y, Dasgupta C, Zhang L. Protective Effect of Antenatal Antioxidant on Nicotine-Induced Heart Ischemia-Sensitive Phenotype in Rat Offspring. PLoS One. 2016;11:e0150557
15. Lappas M, Hiden U, Desoye G, Froehlich J, Hauguel-de Mouzon S, Jawerbaum A. The role of oxidative stress in the pathophysiology of gestational diabetes mellitus. Antioxid Redox Signal. 2011;15:3061-100
16. Wu Q, Ni X. ROS-mediated DNA methylation pattern alterations in carcinogenesis. Curr Drug Targets. 2015;16:13-9
17. Guarente L. Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med. 2011;364:2235-44
18. Ding M, Lei J, Han H, Li W, Qu Y, Fu E, Fu F, Wang X. SIRT1 protects against myocardial ischemia-reperfusion injury via activating eNOS in diabetic rats. Cardiovasc Diabetol. 2015;14:143
19. Yamamoto T, Tamaki K, Shirakawa K, Ito K, Yan X, Katsumata Y, Anzai A, Matsuhashi T, Endo J, Inaba T, Tsubota K, Sano M, Fukuda K, Shinmura K. Cardiac Sirt1 mediates the cardioprotective effect of caloric restriction by suppressing local complement system activation after ischemia-reperfusion. Am J Physiol Heart Circ Physiol. 2016;310:H1003-14
20. Wang YH, Li SA, Huang CH, Su HH, Chen YH, Chang JT, Huang SS. Sirt1 Activation by Post-ischemic Treatment With Lumbrokinase Protects Against Myocardial Ischemia-Reperfusion Injury. Front Pharmacol. 2018;9:636
21. Nguyen LT, Chen H, Pollock C, Saad S. SIRT1 reduction is associated with sex-specific dysregulation of renal lipid metabolism and stress responses in offspring by maternal high-fat diet. Sci Rep. 2017;7:8982
22. Nguyen LT, Chen H, Zaky A, Pollock C, Saad S. SIRT1 overexpression attenuates offspring metabolic and liver disorders as a result of maternal high-fat feeding. J Physiol. 2018
23. Wang L, Quan N, Sun W, Chen X, Cates C, Rousselle T, Zhou X, Zhao X, Li J. Cardiomyocyte-specific deletion of Sirt1 gene sensitizes myocardium to ischaemia and reperfusion injury. Cardiovasc Res. 2018;114:805-21
24. Sutariya B, Saraf M. Betanin, isolated from fruits of Opuntia elatior Mill attenuates renal fibrosis in diabetic rats through regulating oxidative stress and TGF-beta pathway. J Ethnopharmacol. 2017;198:432-43
25. Domouky AM, Hegab AS, Al-Shahat A, Raafat N. Mesenchymal stem cells and differentiated insulin producing cells are new horizons for pancreatic regeneration in type I diabetes mellitus. Int J Biochem Cell Biol. 2017;87:77-85
26. Segar EM, Norris AW, Yao JR, Hu S, Koppenhafer SL, Roghair RD, Segar JL, Scholz TD. Programming of growth, insulin resistance and vascular dysfunction in offspring of late gestation diabetic rats. Clin Sci (Lond). 2009;117:129-38
27. Hathout EH, Kumagai AK, Sangkharat A, Geffner ME, Mullen Y. Absence of GLUT2 protein in near-term fetal rat pancreatic islets. Pancreas. 1997;14:318-21
28. Xiao D, Huang X, Li Y, Dasgupta C, Wang L, Zhang L. Antenatal Antioxidant Prevents Nicotine-Mediated Hypertensive Response in Rat Adult Offspring. Biol Reprod. 2015;93:66
29. Zhu R, Huang X, Hu XQ, Xiao D, Zhang L. Gestational hypoxia increases reactive oxygen species and inhibits steroid hormone-mediated upregulation of Ca(2+)-activated K(+) channel function in uterine arteries. Hypertension. 2014;64:415-22
30. Wu DM, Wang YJ, Han XR, Wen X, Li L, Xu L, Lu J, Zheng YL. Tanshinone IIA prevents left ventricular remodelling via the TLR4/MyD88/NF-kappaB signalling pathway in rats with myocardial infarction. J Cell Mol Med. 2018;22:3058-72
31. Lawrence J, Xiao D, Xue Q, Rejali M, Yang S, Zhang L. Prenatal nicotine exposure increases heart susceptibility to ischemia/reperfusion injury in adult offspring. J Pharmacol Exp Ther. 2008;324:331-41
32. Zhang P, Ke J, Li Y, Huang L, Chen Z, Huang X, Zhang L, Xiao D. Long-term exposure to high altitude hypoxia during pregnancy increases fetal heart susceptibility to ischemia/reperfusion injury and cardiac dysfunction. Int J Cardiol. 2019;274:7-15
33. Meyer K, Zhang H, Zhang L. Direct effect of cocaine on epigenetic regulation of PKCepsilon gene repression in the fetal rat heart. J Mol Cell Cardiol. 2009;47:504-11
34. Wildenthal K. Long-term maintenance of spontaneously beating mouse hearts in organ culture. J Appl Physiol. 1971;30:153-7
35. Moazzen H, Lu X, Ma NL, Velenosi TJ, Urguhart BL, Wisse LJ, Gittenberger-de Groot AC, Feng Q. N-Acetylcysteine prevents congenital heart defects induced by pregestational diabetes. Cardiovasc Diabetol. 2014;13:46
36. Bequer L, Gomez T, Molina JL, Alvarez A, Chaviano, Clapes S. Experimental diabetes impairs maternal reproductive performance in pregnant Wistar rats and their offspring. Syst Biol Reprod Med. 2018;64:60-70
37. Coustan DR, Lowe LP, Metzger BE, Dyer AR. The Hyperglycemia and Adverse Pregnancy Outcome (HAPO) study: paving the way for new diagnostic criteria for gestational diabetes mellitus. Am J Obstet Gynecol. 2010;202:654 e1-6
38. Marcinkevage JA, Narayan KM. Gestational diabetes mellitus: taking it to heart. Prim Care Diabetes. 2011;5:81-8
39. Gardiner HM, Pasquini L, Wolfenden J, Kulinskaya E, Li W, Henein M. Increased periconceptual maternal glycated haemoglobin in diabetic mothers reduces fetal long axis cardiac function. Heart. 2006;92:1125-30
40. Veille JC, Sivakoff M, Hanson R, Fanaroff AA. Interventricular septal thickness in fetuses of diabetic mothers. Obstet Gynecol. 1992;79:51-4
41. Gandhi JA, Zhang XY, Maidman JE. Fetal cardiac hypertrophy and cardiac function in diabetic pregnancies. Am J Obstet Gynecol. 1995;173:1132-6
42. Soulimane-Mokhtari NA, Guermouche B, Yessoufou A, Saker M, Moutairou K, Hichami A, Merzouk H, Khan NA. Modulation of lipid metabolism by n-3 polyunsaturated fatty acids in gestational diabetic rats and their macrosomic offspring. Clin Sci (Lond). 2005;109:287-95
43. Eriksson UJ, Siman CM. Pregnant diabetic rats fed the antioxidant butylated hydroxytoluene show decreased occurrence of malformations in offspring. Diabetes. 1996;45:1497-502
44. Chu C, Gui YH, Ren YY, Shi LY. The impacts of maternal gestational diabetes mellitus (GDM) on fetal hearts. Biomed Environ Sci. 2012;25:15-22
45. Bellinger L, Langley-Evans SC. Fetal programming of appetite by exposure to a maternal low-protein diet in the rat. Clin Sci (Lond). 2005;109:413-20
46. Katkhuda R, Peterson ES, Roghair RD, Norris AW, Scholz TD, Segar JL. Sex-specific programming of hypertension in offspring of late-gestation diabetic rats. Pediatr Res. 2012;72:352-61
47. He F, Peng Y, Yang Z, Ge Z, Tian Y, Ma T, Li H. Activated ClC-2 Inhibits p-Akt to Repress Myelination in GDM Newborn Rats. Int J Biol Sci. 2017;13:179-88
48. Kinalski M, Sledziewski A, Telejko B, Zarzycki W, Kinalska I. Antioxidant therapy and streptozotocin-induced diabetes in pregnant rats. Acta Diabetol. 1999;36:113-7
49. Sugamura K, Keaney JF Jr. Reactive oxygen species in cardiovascular disease. Free Radic Biol Med. 2011;51:978-92
50. Munzel T, Gori T, Bruno RM, Taddei S. Is oxidative stress a therapeutic target in cardiovascular disease?. Eur Heart J. 2010;31:2741-8
51. Planavila A, Dominguez E, Navarro M, Vinciguerra M, Iglesias R, Giralt M, Lope-Piedrafita S, Ruberte J, Villarroya F. Dilated cardiomyopathy and mitochondrial dysfunction in Sirt1-deficient mice: a role for Sirt1-Mef2 in adult heart. J Mol Cell Cardiol. 2012;53:521-31
52. Sundaresan NR, Pillai VB, Wolfgeher D, Samant S, Vasudevan P, Parekh V, Raghuraman H, Cunningham JM, Gupta M, Gupta MP. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci Signal. 2011;4:ra46
53. Zhang J. The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J Biol Chem. 2007;282:34356-64
54. Pillai VB, Sundaresan NR, Gupta MP. Regulation of Akt signaling by sirtuins: its implication in cardiac hypertrophy and aging. Circ Res. 2014;114:368-78
55. Tang H, Song X, Ling Y, Wang X, Yang P, Luo T, Chen A. Puerarin attenuates myocardial hypoxia/reoxygenation injury by inhibiting autophagy via the Akt signaling pathway. Mol Med Rep. 2017;15:3747-54
56. Ghavami S, Gupta S, Ambrose E, Hnatowich M, Freed DH, Dixon IM. Autophagy and heart disease: implications for cardiac ischemia-reperfusion damage. Curr Mol Med. 2014;14:616-29
57. Weng X, Liu F, Zhang H, Kan M, Wang T, Dong M, Liu Y. Genome-wide DNA methylation profiling in infants born to gestational diabetes mellitus. Diabetes Res Clin Pract. 2018;142:10-8
58. Ren J, Cheng Y, Ming ZH, Dong XY, Zhou YZ, Ding GL, Pang HY, Rahman TU, Akbar R, Huang HF, Sheng JZ. Intrauterine hyperglycemia exposure results in intergenerational inheritance via DNA methylation reprogramming on F1 PGCs. Epigenetics Chromatin. 2018;11:20
Author contact
![]() Corresponding authors: Daliao Xiao, PhD, Center for Perinatal Biology, Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350. Tel: 909-558-4325; Fax: 909-558-4029; E-mail: dxiaoedu or Jian Zhuang, MD, Department of Cardiac Surgery, Guangdong Cardiovascular Institute, Guangdong Provincial People's Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China 510000. Tel: 86 020 83827812; E-mail: zhuangj5413com
Corresponding authors: Daliao Xiao, PhD, Center for Perinatal Biology, Department of Basic Sciences, Loma Linda University School of Medicine, Loma Linda, CA 92350. Tel: 909-558-4325; Fax: 909-558-4029; E-mail: dxiaoedu or Jian Zhuang, MD, Department of Cardiac Surgery, Guangdong Cardiovascular Institute, Guangdong Provincial People's Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China 510000. Tel: 86 020 83827812; E-mail: zhuangj5413com
Received 2019-1-23
Accepted 2019-3-1
Published 2019-5-11