Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Pyroptosis: a form of...
Mechanisms of Pyroptosis
Pyroptosis and Cardiovascular...
Targeting of NLRP3 inflammasome...
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(7):1345-1357. doi:10.7150/ijbs.33568 This issue Cite
Review
Role of Pyroptosis in Cardiovascular Diseases and its Therapeutic Implications
Cheng Zeng1, Renqing Wang1,2, Hongmei Tan1 ![]()
1. Department of Pathophysiology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou 510080, China;
2. Department of pathology, the Affiliated Drum Tower Hospital of Medical School of Nanjing University, Nanjing 21008, China
Received 2019-1-28; Accepted 2019-4-4; Published 2019-5-20
Abstract

Pyroptotic cell death or pyroptosis is characterized by caspase-1-dependent formation of plasma membrane pores, leading to the release of pro-inflammatory cytokines and cell lysis. Pyroptosis tightly controls the inflammatory responses and coordinates antimicrobial host defenses by releasing pro-inflammatory cellular contents, such as interleukin (IL)-1β and IL-18, and consequently expands or sustains inflammation. It is recognized as an important innate immune effector mechanism against intracellular pathogens. The induction of pyroptosis is closely associated with the activation of the NOD-like receptor 3 (NLRP3) inflammasome which has been linked to key cardiovascular risk factors including hyperlipidemia, diabetes, hypertension, obesity, and hyperhomocysteinemia. Emerging evidence has indicated pyroptosis as an important trigger and endogenous regulator of cardiovascular inflammation. Thus, pyroptosis may play an important role in the pathogenesis of cardiovascular diseases. Design of therapeutic strategies targeting the activation of NLRP3 inflammasome and pyroptosis holds promise for the treatment of cardiovascular diseases.
Keywords: pyroptosis, inflammasome, NLRP3, caspase-1, inflammation, cardiovascular diseases
Introduction
Cell death is one of the ancient processes with fundamental biological importance in both normal physiology and pathological states. Recently, caspase-1 in both human and mouse, caspase-4/5 in human, and caspase-11 in mouse, are recognized to mediate a novel form of programmed cell death termed as pyroptosis. Pyroptosis is characterized by the formation of membrane pores, cell lysis and the release of pro-inflammatory cytokines and intracellular content. This event is predicted to be inherently inflammatory and coincides with the activation of inflammasome, a molecular platform resulting in caspase-1 activation and interleukin (IL)-1β and IL-18 secretion upon cellular infection or stress. The active caspase-1 triggered by inflammasome activation cleaves gasdermin D (GSDMD) to generate an N-terminal GSDMD fragment (GSDMD-NT), which further induces the formation of membrane pores and subsequent inflammatory responses. As an innate immune effector mechanism, pyroptosis has the ability to defense against infection. Emerging evidence has indicated that pyroptosis and related inflammasome activation play important roles in the progression of vascular inflammation and cardiovascular diseases. In this review, we summarize the molecular pathways of pyroptosis and the implications in cardiovascular research.
Pyroptosis: a form of pro-inflammatory programmed cell death
In 1992, morphological features of pyroptosis were firstly observed in macrophages infected with the Gram-negative bacterial pathogen Shigella flexneri, which were mistakenly considered as morphological changes of apoptosis at that time[1]. A similar phenotype was observed in Salmonella-infected macrophages in 1999[2]. Further studies revealed that both selective caspase-1 inhibitor and caspase-1 knockdown efficiently blocked the shigella flexneri- induced cell death, whereas caspase-3 inhibitor did not, suggesting a novel form of cell death, depending on the activation of caspase-1, but not classical apoptotic caspase-3[3-5]. Until 2001, pyroptotic cell death (pyroptosis) was firstly defined as a novel form of caspase-1-dependent programmed cell death by Cookson BT and Brennan MA[6]. In addition to microbial signaling, endogenous contents released by cells in overwhelming stress were also identified to induce macrophage pyroptosis. Pyroptosis has been found not only in monocyte/macrophages, but also in other cells including dendritic cells[7], hepatic cells[8], endothelial cells[9] and myocardial cells[10].
Macrophages undergoing pyroptosis show several morphological features of apoptosis, and also exhibit some characters which are similar to necrosis. Indeed, pyroptosis is characterized by rapid formation of membrane pores with a diameter of 10-15 nm[11]. Cellular ionic gradients are dissipated by these pores, which allow water influx, cell swelling and osmotic lysis with the release of intracellular pro-inflammatory contents including IL-1β, IL-18, high mobility group box-1 protein (HMGB-1) and heat shock protein (HSP). These are similar to oncosis but remarkably opposite to apoptosis which is characterized with the formation and non- inflammatory phagocytic uptake of apoptotic bodies. Several features of pyroptotic cells seem to overlap with apoptotic cells. Both pyroptosis and apoptosis share the feature of chromatin condensation, but the nucleus remains intact and karyorrhexis does not occur in pyroptosis[12]. Another feature that is shared between pyroptosis and apoptosis is annexin V positive staining. During the early stages of apoptosis, phosphatidylserine is translocated to the outer leaflet, leading to positive cell surface staining with annexin V[13]. As cell membrane is ruptured during pyroptosis, annexin V is permited to enter the cell and stains the inner leaflet of the membrane. In contast, 7-aminoactinomycin or propidium iodide, as alive cell membrane impermeant dyes, can stain the nucleus of pyroptotic cell through the membrane pores, but not the early-stage apoptotic cell[14]. Thus, these dyes are applied to differentiate between apoptosis and pyroptosis. Moreover, pore formation causes cell swelling in pyroptotic cell, whereas cell shrinks in apoptosis[14]. Caspases play central role in initiating both apoptosis and pyroptosis. The characterized effectors of apoptosis are caspase-3, -5 and -7, whereas pyroptosis is induced by pro-inflammatory caspases (especially for caspase-1).
Mechanisms of Pyroptosis
The host can sense intracellular and extracellular 'danger' signals generated by invading microorganisms or by the host in response to tissue injury. The innate immune responses rely on specific host- receptors which are termed as pattern-recognition receptors (PRRs) to detect pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs)[15-18]. Based on their location, the PRRs can be differentiated into membrane-bound PRRs that recognize signals of infection in the cellular milieu or endosomes, such as Toll-like receptors (TLRs) and C-type lectin-like receptors, and cytoplasmic PRRs that recognize invasive pathogens, such as nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), retinoic acid-inducible gene I-like receptors, absent in melanoma 2 (AIM2)-like receptors (ALRs), cyclic GMP-AMP synthase, and stimulator of interferon gene[18-21]. Detection of PAMPs and DAMPs by NLRs and ALRs triggers the assembly of a caspase-1- activating complex that was firstly termed inflammasome by Tschopp et al[22]. Active caspase-1 further processes pro-inflammatory cytokines (IL-1β and IL-18), and initiates pyroptosis by disruption of plasma membrane integrity[18, 23]. In addition, mouse caspase-11 and human caspase-4 and -5 can directly bind to cytosolic lipopolysaccharide (LPS), leading to non-canonical inflammasome activation [18]. Non-canonical inflammasome can also induce pyroptosis, but cannot directly process pro-inflammatory IL-1β and IL-18. However, non-canonical inflammasome is capable of inducing the processing of pro-inflammatory cytokines indirectly by activating the canonical inflammasome through a not well-defined mechanism.
Pyroptotic pathway by caspase-1 dependent model
It is critical for pyroptosis to activate pro-inflammatory caspases. Thus, how inflammatory caspases are activated is a key question to elucidate the mechanism of pyroptosis. In 2002, inflammasome was firstly identified as the molecular platform triggering the activation of inflammatory caspases and consequently processing of pro-IL-1β and pro-IL-18 by Tschopp et al[22]. Inflammasome is multiprotein complex, composed of sensor, the adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD)) and caspase-1. To date, several types of inflammasomes have been identified, including NLRP1, NLRP3, NLRC4, NLRP6, and AIM2 inflammasomes. Among these different types of inflammasomes, the NLRP3 inflammasome has been extensively studied in a variety of mammalian cells, and has been linked with various autoimmune and inflammatory diseases. Therefore, the role of NLRP3 inflammasome and related pyroptosis in cardiovascular disease will be discussed in this review.
The NLR protein of NLRP3 inflammasome contains a conserved nucleotide-binding and oligomerization domain (NACHT domain), C-terminal leucine rich repeats and pyrin domain (PYD) for multimerization[24]. Upon the activation of NLRP3 inflammasome, the NLRs oligomerize via the NACHT domains, followed by the recruitment of adapter protein ASC through PYD-PYD interaction. ASC forms large speck-like structures and recruits pro-caspase-1 via CARD-CARD interaction. Pro-caspase-1 is subsequently autocatalytically cleaved into p10 and p20 subunits that form the active caspase-1 p10/p20 tetramer, mediating the maturation and secretion of IL-1β and IL-18[25, 26]. Caspase-1 can also cleave GSDMD to generate GSDMD-NT, which forms plasma membrane pores to induce pyroptosis (Figure 1)[11, 27-29].
Caspase-1-dependent canonical pyroptotic cell death induced by NLRP3 inflammasome activation. The NLRP3 oligomerization and ASC recruitment trigger pro-caspase-1 autocleavage, leading to autocatalytic activation of caspase-1, which in turn converts inactive pro-IL-1β and pro-IL-18 into their bioactive and secreted forms (IL-1β and IL-18). The active caspase-1 also cleaves GSDMD to generate GSDMD-NT, which forms plasma membrane pores to induce pyroptosis. Three models about the assembly of NLRP3 inflammasome have been proposed: (1) The NLRP3 inflammasome is activated by extracellular ATP through one of the following mechanisms: the P2X7-activated pannexin-1 pore allows cytoplasmic entry of extracellular factors that are direct NLRP3 ligands, or NLRP3 senses either K+ efflux or the loss of membrane integrity. (2) Crystalline or particulate agonists are phagocytosed, leading to the leakage of lysosomal cathepsins B and L, which are sensed by the NLRP3 inflammasome. (3) NLRP3 agonists including DAMPs and PAMPs trigger the production of ROS, which leads to the activation of the NLRP3 inflammasome. In general, the three models discussed above are not functionally independent but rather interactive. (4) Besides above three models, the activation of NLRP3 inflammasome also requires 'priming' with TLRs agonist (such as LPS).
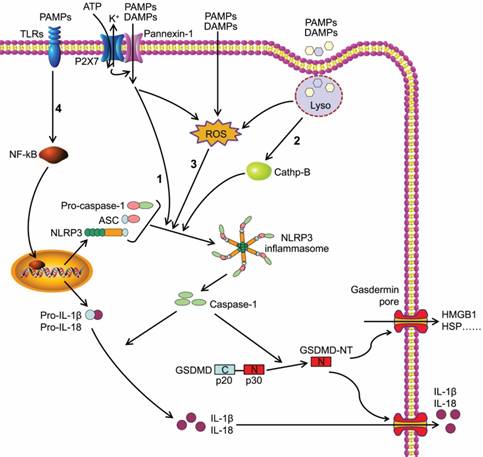
The canonical inflammasome activation has been proposed to be a two-step process involving priming followed by assembly[30]. The activation of NLRP3 inflammasome requires 'priming' with TLRs agonists (such as LPS) to initiate signaling cascades (primarily nuclear factor-κB (NF-κB)-dependent pathway) that ultimately promote a transcriptional response to up-regulate pro-inflammatory mediators (such as pro-IL-1β) (Figure 1). After this priming phase, inflammasome assembles in response to a diverse range of PAMPs or DAMPs to form a large molecular platform triggering activation of inflammatory caspases and processing of pro-IL-1β.
In addition, three models about the assembly of NLRP3 inflammasome have been proposed (Figure 1). Extracellular ATP stimulates the purinergic receptor P2X, ligand-gated ion channel 7 (P2X7) on cell membrane[31], triggering K+ efflux and inducing recruitment of pannexin 1 to form large membrane pores[32]. Therefore, extracellular agonists are able to penetrate the cell membrane and activate NLRP3 inflammasome. A second model was presented for particulate or crystalline agonists, such as cholesterol crystal, monosodium urate, silica and asbestos. Uptake of such substances leads to lysosomal damage, resulting in the leakage of lysosomal cathepsins B and L that are sensed by the NLRP3 inflammasomes[33]. Under the third model, agonists trigger the generation of reactive oxygen species (ROS), which is the common pathway engaging the NLRP3 inflammasome. Intriguingly, all three models are not functionally independent but rather interactive.
The agonist of NLRP3 inflammasome serves as potential inducer for cell pyroptosis. The detailed molecular basis for pyroptosis was unclear until very recently. Several studies simultaneously identified GSDMD as a critical executor of pyroptosis.
GSDMD belongs to a gasdermin family that shares the pore-forming domain[34]. Both caspase-1 and caspase-4/ 5/11 can cleave GSDMD to release GSDMD-NT that perforates the plasma membrane to induce cell swelling and osmotic lysis. Other gasdermin family members also show pore-forming activity, but they are not substrates of the inflammatory caspases[28]. GSDMD is cleaved at the conserved residue D276, which separates GSDMD into a GSDMD-NT domain (p30) and a GSDMD C-terminal domain (p20)[11]. GSDMD- NT can combine with lipid in the plasma membrane and form large oligomeric pores, leading to the disruption of cell membrane integrity, the increase of intracellular osmotic pressure, and the release of inflammatory intracellular contents, such as HMGB-1 and HSP (Figure 1)[11, 28, 35, 36]. This caspase-1- dependent GSDMD cleavage is defined as canonical pyroptotic pathway[37].
Pyroptotic pathway by caspase-1 independent model
The other inflammatory caspases, mouse caspase-11 and human caspase-4 and -5, are required for non-canonical inflammasome activation, thus are major mediators of non-canonical pyroptosis (Figure 2). In 2011, Kayagaki et al reported that gram-negative bacteria (Escherichia coli, Vibrio cholerae, and Citrobacter rodentium)-induced IL-1β maturation and secretion were blocked in caspase-11-/- macrophages rather than caspase-1-/- macrophages, whereas ATP and monosodium urate-induced canonical inflammasome activation was intact in caspase-11-/- macrophages, indicating caspase-11 but not caspase-1 was required for non-canonical inflammasome activation and related cell death[38]. Follow-up studies identified intracellular LPS as the trigger of TLR4-independent non-canonical inflammasome[39, 40]. Recent study demonstrated that the high-affinity interaction between the lipid A component of LPS and the CARD domain of cytoplasmic caspase-11/4/5 led to direct binding of them, and resulted in inflammatory caspase oligomerization and activation[41, 42]. Active caspase-4/5/11 ultimately cleaved GSDMD to induce cell pyroptosis similar to canonical pathway (Figure 2)[41]. In addition, non-canonical inflammasome activation led to secondary activation of the canonical inflammasome and caspase-1, and subsequent maturation and secretion of proinflammatory cytokines[38], which may be an indirect consequence of GSDMD-NT[37]. Although a study reported that caspase-4 could cleave pro-IL-1β directly[43], the inability of caspase-4/5/11 to cleave the precursors of proinflammatory cytokines was widely accepted and considered as a major difference between canonical and non-canonical pyroptotic pathways.
Caspase-11/4/5-dependent pyroptotic cell death. Cytosol LPS is sensed by proinflammatory caspases including caspase-11 in mouse and caspase-4/5 in human through a direct binding between LPS and the caspases, which induces the oligomerization and activation of the inflammatory caspases. The active caspase-11/4/5 subsequently cleaves GSDMD to generate GSDMD-NT and induces pyroptosis.
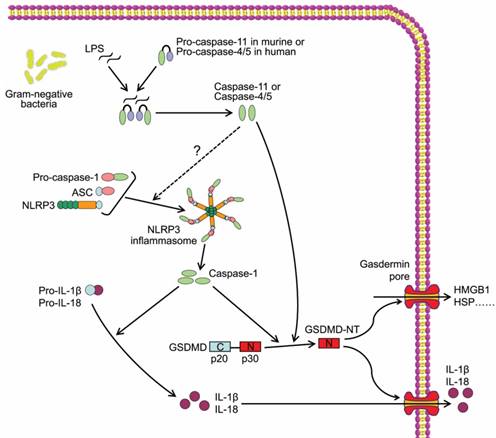
In addition, gasdermin E (GSMDE) can shift caspase-3-mediated classical apoptosis induced by tumor necrosis factor alpha (TNFα) or chemotherapy drug to pyroptosis. GSDME is specifically cleaved by non-proinflammatory caspase-3, producing a GSDME N-terminal cleavage product that perforates membrane and thereby induces cell pyroptosis[44]. These findings reveal novel insights into mechanisms underlying pyroptosis.
Pyroptosis and Cardiovascular Disease
Pyroptosis and Atherosclerosis
Atherosclerosis is complicated by a number of events: endothelial dysfunction; accumulation and oxidation of low-density lipoprotein (LDL); recruitment of monocytes and lymphocytes; migration and proliferation of smooth muscle cells; activation of proinflammatory cytokines; adherence of platelets. In these processes, inflammatory reaction was recognized in the atherosclerotic lesions from the early stage of fatty streak to culmination into an acute cardiovascular event caused by plaque erosion or rupture. However, how the inflammation contributes to atherosclerosis is still largely unclear.
More and more studies revealed that risk factors of atherosclerosis could activate NLRP3 inflammasomes in both endothelial cells and macrophages. In addition, NLRP3 inflammasome- mediated pyroptosis has been observed in the atherosclerotic plaques and is positively correlated with the plaque rupture and vascular inflammation, indicating that NLRP3 inflammasome and related pyroptosis play an important role in the progression of atherosclerosis.
Pyroptosis and Dyslipidemia
Cholesterol crystals have been identified as a potential biomarker for atherosclerosis and a major risk factor for plaque vulnerability[45]. Duewell et al firstly reported that cholesterol crystals activated NLRP3 inflammasomes by inducing lysosomal damage, and led to the release of IL-1β[46]. Combined confocal-reflection microscopy revealed that a large load of cholesterol crystals were deposited in the aortic wall and coincided with increased recruitment of macrophages in high cholesterol diet-fed low-density lipoprotein receptor-null (LDLR-/-) mice. In contrast, a significant decrease of atherosclerotic lesion size was also observed at the aortic sinus of high fat diet-fed LDLR-/- mice reconstituted with NLRP3-, ASC- or IL-1α/β-knockout bone marrow cells[46]. Consistent with these in vivo findings, cholesterol crystals activated NLRP3 inflammasomes in human macrophages, which was suppressed by silencing of NLRP3[47]. These studies confirmed that NLRP3 inflammasomes were activated by cholesterol crystals and required for atherogenesis. Given that caspase-1 activation induced by NLRP3 inflammasome is a major mediator for pyroptosis, these work further suggested that pyroptotic macrophages might take part in the progression of atheroslerosis.
In addition, oxidized LDL (ox-LDL), a major risk factor for atherosclerosis, triggered the assembly of NLRP3 inflammasome, and subsquently activated caspase-1 and processed IL-1β in macrophages[46, 48]. Immunohistochemistry results showed that caspase-1 was activated in advanced atherosclerotic lesion and co-located with macrophages and TUNEL positive area, suggesting that pyroptosis was implicated in advanced plaque. Another in vitro experiment further confirmed that ox-LDL induced pyroptosis in macrophages[49], which was inhibited by the depletion of mitochondrial DNA[50].
In addition to ox-LDL, NLRP3 inflammasome was also activated by high level of triglyceride, a marker for several types of atherogenic lipoproteins [51, 52]. Yin et al also reported that early hyperlipidemia induced caspase-1 activation along with enhanced expression of intercellular adhension molecule, vascular cell adhension molecule and E-selectin in the aorta, and promoted monocyte recruitment in ApoE-/- mice, whereas the knockout of caspase-1 obviously suppressed these effects and decreased the area of atheroclerotic plaque[53]. They further performed in vitro expriments and found that ox-LDL induced cell pyroptosis through ROS- dependent pathway in endothelial cells. The pyroptosis of vascular endothilal cells was firstly linked to atherosclerotic progression in this work.
As same as ox-LDL, palmitic acid, a saturated fatty acid, has been reported to induce pyroptosis in ROS-dependent manner in human umbilical venin endothelial cells (HUVECs), while pretreatment with dihydromyricetin inhibited palmitic acid-induced pyroptosis by activating Nrf2-antioxidant response element signaling pathway[54]. Another study also pointed out that cholesterol crystal obviously induced NLRP3 inflammasome-dependent pyroptosis in primary coronary endothelial cells[55], indicating that inflammasome-mediated pyroptosis might play an important role in cholesterol crystal-induced atherosclerosis. As endothelial cells cover the inner surface of vessel wall and are directly exposed to the atherosclerosis-related danger signals in the circulation, endothelial dysfunction is an initial event responsible for monocyte recruitment in atherogenesis[56]. It is very likely that cell pyroptosis can switch endothelial cells to the proinflammatory state which subsequently contributes to atherosclerosis.
Pyroptosis and Hyperhomocysteinemia
Hyperhomocysteinemia (HHcy) was defined as elevated plasma level of homocysteine (Hcy) (>15 μmol/L). HHcy is an important and independent risk factor involved in several disorders including atherosclerosis[57]. Earlier reports have shown that plasma levels of proinflammatory cytokines including IL-6 and TNFα were increased in HHcy individuals, which played a crucial role in endothelial damage and atherosclerosis[58]. Since Hcy can be autooxidized with anthor Hcy molecule to generate ROS and the disulfide[59], Hcy has the potential to activate NLRP3 inflammasome. Recently, a study from our laboratory demonstrated that HHcy-induced activation of NLRP3 inflammasome in macrophages contributed to vascular inflammation and atherosclerosis[60]. ROS scavenger N-acetyl-L-cysteine suppressed the activation of NLRP3 inflammasomes and alleviated atherosclerosis induced by HHcy[60]. In fact, there was an evidence showing that the conbination of Hcy and LPS synergistically induced endothelial cell pyroptosis via caspase-1-dependent inflammasome activation[61]. Further study found that intracellular ROS levels determined Hcy/LPS-induced death destiny in endothelial cells: relatively high ROS levels directly induced apoptosis, while moderate ROS generation preferred to trigger pyroptosis[61]. These phenomena indicate that HHcy-aggravated atheroclerosis may be closely associated with not only apoptosis of endothelial cells but also the pyroptosis-mediated inflammation.
Evidence shows that certain heavy metals may regulate NLRP3 inflammasome activation. Cadmium (Cd), a toxic and nonessential heavy metal, has been linked to cardiovascular diseases including atherosclerosis[62]. Chen et al recently found that Cd induced NLRP3 inflammasome activation and subsequent pyroptosis via mitochondrial ROS- dependent pathway in vascular endothelial cells[9]. Whether NLRP3 inflammasome and related pyroptosis contribute to Cd-induced cardiovascular diseases needs further exploration in animal models.
Pyroptosis in Obesity and Diabetes: Reminding of NLRP3 Inflammasome
Obesity and related metabolic syndrome pose huge threats to human health. Active inflammation in obese adipose tissue is increasingly recognized as a key event underlying metabolic diseases. Obesity provides a proinflammatory condition which contributes to the increase of circulating proinflammatory cytokines[63]. Studies showed that the expression of caspase-1, ASC and NLRP3 was upregulated in high fat diet-induced obesity mice, while the knockout of NLRP3 or caspase-1 suppressed obesity-induced inflammasome activation in fat depots and liver[64]. Interestingly, obese db/db mice exhibited ultrastructural abnormalities similar to pyroptotic cells with ROS increment and caspase-1 activation in the subcutaneous and visceral hypertrophic adipocytes [65]. In addition, Konene et al found that the expression of caspase-1 in the abdominal adipose tissue was markedly higher than in the subcutaneous fat, indicating that pyroptotic cell death was probably presented in the abdominal adipose tissue[66].
A study showed that peripheral blood-derived macrophages from drug-naïve patients with type 2 diabetes displayed enhanced expression of NLRP3 and ASC together with caspase-1 activation and IL-1β maturation[67]. It was reported that high glucose stimulated IL-1β secretion in pancreatic β cells[68]. Further study demostrated that high glucose induced NLRP3 inflammasomes activation and IL-1β secretion, leading to pancreatic β cell death[68]. In normal condition, the thioredoxin (TRX)-interacting protein (TXNIP) was bound to TRX, however, TXNIP was dissociated from TRX to interact with and activate NLRP3 inflammasome in various types of cells when incubated with high glucose[68]. In contrast, γ-tocotrienol, known to exert potent anti-inflammatory function in various cell types[69], was shown to inhibit NLRP3 inflammasome activation and the recruitment of macrophages to adipose tissues, thus ameliorated insulin resistance [70]. In fact, γ-tocotrienol not only inhibited the tumor necrosis factor receptor-associated factor 6/NF-κB pathway, but also activated AMP-activated protein kinase/autophagy axis, leading to reduced cleavage of caspase-1. These results suggested that NLRP3 inflammasome and related pyroptosis might be potentially promising targets for new drug research in diabetes.
Pyroptosis in Hypertension: Reminding of NLRP3 Inflammasome
Hypertension is a well-known risk factor for atherogenesis-related cardiovascular and cerebrovascular diseases. Recently, the role of inflammation in the pathologenesis of hypertension has attracted great interests. Although there was no direct proof that cell pyroptosis contributed to pathological process of hypertension, studies showed that circulating levels of IL-1β and IL-18 were elevated in hypertensive mice and rats[71, 72]. Dalekos et al found that serum level of IL-1β in patients with essential hypertension was higher than in normotensive controls[73]. Given that IL-1β maturation is dependent on caspase-1, there is a possibility that hypertension may be assiociated with the activation of caspase-1. Another study showed that mRNA expression of pro-caspase-1 in the aorta and renal artery was increased in spontaneously hypertensive rats, which indirectly supported this possibility[74]. Moreover, high salt increased the expression of NLRP3, ASC and pro-caspase-1, the activation of caspase-1, and the maturation of IL-1β in mouse kidney[71]. Interestingly, NF-κB knockdown was associated with reduced expression of NLRP3 inflammasome components in hypothalamic paraventricular nucleus, and thus delayed the progression of hypertension in Dahl salt-sensitive hypertensive rats[75]. Therefore, targeting NLRP3 inflammasome may be a therapeutic strategy to treat hypertension. Based on this hypothesis, Tang et al found that ellagic acid, a promising treatment candidate for pulmonary artery hypertension (PAH), inhibited NLRP3 inflammasomes activation primarily by exerting its antioxidant effect[76].
Pyroptosis and Myocardial Ischemia/Reperfusion Injury
Therapeutic strategies aimed at rescuing ischemic myocardium have been studied extensively. Reperfusion remains the definitive treatment for acute myocardial infarction (MI). However, restoring blood flow carries the potential to exacerbate lethal tissue injury, a process termed as “ischemia/reperfusion injury”. The major contributory factors of reperfusion injury include oxidative stress[34], calcium overload [41], opening of mitochondrial permeability transition pores[42], and in particular inflammation[18]. Myocardial ischemia/reperfusion injury (MIRI) may cause MI and cardiac arrhythmias as well as cardiac dysfunction[77]. MI is defined as myocardial cell death due to prolonged ischemia and usually caused by the sudden thrombotic occlusion of coronary artery [78]. Recent studies found that NLRP3 inflammasome and related pyroptosis were strongly associated with pathological process of MIRI and MI. As previously mentioned, NLRP3 inflammasome activation and pyroptosis contributed to plaque rupture, and the release of IL-1β was an early and prominent event of inflammatory responses in MI[49]. Investigators have also found that inflammatory responses and subsequent injuries, including myocardial dysfunction and fibrosis, were significantly alleviated in ASC or caspase-1 deficient mice[79]. Further studies showed that NLRP3 inflammasome was activated in the ischemic heart as evidenced by increased NLRP3 expression and IL-1β and IL-18 production with caspase-1 activation, whereas NLRP3 siRNA, treated via intramyocardial injection, inhibited these effects and decreased TUNEL positive areas. Another study showed that pyroptosis was induced by ROS-dependent NLRP3 inflammasome activation, and inhibition of NLRP3 inflammasome activation alleviated MIRI in diabetic rats, suggesting that NLRP3 inflammasome-mediated pyroptosis played a critical role in inflammatory responses and MIRI in diabetic rats[80]. In addition, silence of cryopyrin (also known as NLRP3) and P2X7 prevented the formation of NLRP3 inflammasome, limited myocardial infarct size and ameliorated cardiac remodeling after acute MI[81]. These results indicated that NLRP3 inflammasome activation and related pyroptosis are potential novel therapeutic targets for MIRI.
Pyroptosis and Diabetic Cardiomyopathy
Diabetic cardiomyopathy (DCM) is a diabetes- induced cardiomyopathy that occurs in diabetic subjects without coronary artery disease and hypertension[82, 83]. DCM is characterized by functional and structural abnormalities, including myocardial dysfunction[84], myocardial cell death[85], myocardial fibroblast activation[86], and metabolic deregulation[87]. Among them, myocardial cell death is a key factor in the progression of DCM, leading to a loss of contractile units, conduction disturbances, compensatory hypertrophy of myocardial cells, and myocardial fibrosis[88]. Apoptosis has been widely recognized in the pathogenesis of DCM. In addition, recent evidence showed pyroptosis could occur in DCM heart. Electron microscopy studies demonstrated that most of the dying cells had characteristics of swollen fibril and mitochondria similar to the features of pyroptosis in the myocardium of diabetic mice and rats[89-91]. Intriguingly, Luo et al reported that NLRP3 inflammasome-caspase-1- mediated pyroptosis was presented in myocardium of diabetic rats, while silencing of NLRP3 ameliorated cardiac inflammation and pyroptosis, and improved myocardial function[92].
Numerous studies have shown that microRNAs (miRNAs) are involved in the regulation of cardiomyocyte pyroptosis in DCM. For example, Li et al demonstrated that miR-30d was upregulated in high glucose-treated cardiomyocytes and streptozotocin (STZ)-induced diabetic rat hearts, which regulated cardiomyocyte pyroptosis in DCM[10]. In an effort to explore the pro-pyroptotic mechanisms of miR-30d, they found that miR-30d directly repressed the expression of Forkhead box O3 and its downstream factor, apoptosis repressor with caspase recruitment domain[10]. Moreover, miR-9 inhibited hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes[93], suggesting that miR-9 may have a protective effect on DCM.
Pyroptosis in Viral Myocarditis: Reminding of NLRP3 Inflammasome
About 20 common types of viruses have been related to myocarditis in human[94]. The dominant etiological agent of viral myocarditis is coxsackievirus B3 (CVB3)[95]. Wang et al firstly reported that the activation of NLRP3 inflammasome was involved in CVB3-induced myocarditis[96]. Both in vivo and in vitro results showed that CVB3 infection activated NLRP3 inflammasome. Conversely, once inflammasome activation was inhibited, CVB3-induced myocarditis manifested by weight loss, increased serological indexes of creatine kinase and CK-MB activities, was obviously suppressed. Caspase-1 inhibitor also improved left ventricular ejection fraction and left ventricular fractional shortening in CVB3-induced myocarditis[96]. Further study showed that ROS generation and K+ efflux were involved in the activation of NLRP3 inflammasome caused by CVB3 infection[96]. Recent study demonstrated that NOD2 mRNA expression was higher in endomyocardial biopsy tissue from CVB3-positive patients as compared with myocarditis patients without persistent CVB3 infection[97]. Interestingly, mesenchymal stromal cells suppressed NOD2 expression and NLRP3 inflammasome activation, which was associated with improved expression of markers for cardiac contractility and fibrosis in the heart of CVB3-infected mice, suggesting that NOD2 might coordinate NLRP3 inflammasome activation in CVB3-infected myocardium[98]. In addition, Wang et al. found that CVB3-induced myocarditis was aggravated by cathepsin B-induced activation of NLRP3 inflammasome and pyroptosis [99]. It is still not clear whether other types of viruses regulate NLRP3 inflammasome activation in viral myocarditis.
Pyroptosis and Heart Failure
Heart failure (HF) means that the heart is unable to pump sufficiently to sustain blood flow to meet the needs of the body[100]. HF is the end stage of many cardiovascular diseases. Common causes of HF include myocarditis, MI, cardiomyopathy, hypertension, atrial fibrillation (AF), valvular heart disease, alcohol abuse, and infection. Recently, elevated mRNA of NLRP3 and enhanced cleavage of caspase-1 were observed along with cardiac hypertrophy and ventricular dilatation in calcineurin transgene (CNTg) mice, an established mouse model for chronic HF[101]. As expected, they also discovered that genetic ablation of NLRP3 or administration of IL-1 receptor antagonist attenuated cardiac inflammation and systolic dysfunction[101]. Similarly, the expression of NLRP3 inflammasome components was significantly increased, and associated with elevated inflammatory mediators and profibrotic factors and impaired cardiac function in a mouse model of transverse aortic constriction (TAC)-induced ventricular hypertrophy[102] [103]. Interestingly, the inhibition of NLRP3 by triptolide[103] or pirfenidone[102] attenuated myocardial remodeling and improved cardiac function in TAC mice. In addition, the activation of NLRP3 inflammasome was observed in LPS- stimulated cardiac fibroblasts[104] and myofibroblasts[105], suggesting that NLRP3 inflammasome and related pyroptosis may contribute to myocardial dysfunction in sepsis.
AF, a common kind of arrhythmia, is associated with an increased risk of worsening HF[106, 107]. A strong inflammatory response is frequently associated with AF progression[108, 109]. Increased serum levels of IL-1β and IL-18 were positively correlated with AF progression from paroxysmal AF (pAF) to long-lasting persistent AF (perAF)[109, 110]. Recently, Yao et al firstly showed that NLRP3 inflammasome was activated in the atrial cardiomyocytes from patients with pAF and perAF[111]. Cardiomyocyte- specific activation of NLRP3 induced abnormal Ca2+ release from sarcoplasmic reticulum and promoted inducible AF, which was attenuated by MCC950, a specific NLRP3 inflammasome inhibitor, and genetic silencing of NLRP3[111]. Genetic silencing of NLRP3 also prevented AF development in cAMP-responsive element modulator (CREM)-transgenic mice, a spontaneous AF mouse model[111].
Targeting of NLRP3 inflammasome or cell pyroptosis as a therapeutic strategy
Based on experimental data, inhibiting NLRP3 inflammasome or cell pyroptosis may supply a new therapeutic strategy for cardiovascular disease. The complicated signaling of NLRP3 inflammasome and related pyroptosis offers diverse targets for inhibiting their activation, including suppression of upstream signals of NLRP3 inflammasome, prevention of NLRP3 inflammasome assembly, inhibition of caspase-1 activation and GSDMD cleavage, and neutralizing antibodies targeting inflammasome-derived inflammatory cytokines. Preventing the assembly of NLRP3 inflammasome could be established by inhibition of K+ efflux with glyburide[112] and β-hydroxybutyrate[113], blockage of P2X7 signaling with human cathelicidin LL-37[114], as well as the application of ROS scavenger[115]. It was reported that MCC950 disrupted the interaction between NLRP3 and ASC, and showed strong inhibitory effect on NLRP3 inflammasome and beneficial effect on inflammatory diseases[116]. In addition, parthenolide, BAY 11-7082, INF39, and 3,4- methylenedioxy-β-nitrostyrene (MNS) were reported to directly inhibit the ATPase activity of NLRP3, but these inhibitors have unspecific effects limiting their use[117-119]. Recently, Jiang et al. showed that CY-09 specifically blocked NLRP3 inflammasome activation by binding to the ATP-binding motif of NLRP3 NACHT domain and thus inhibiting the ATPase activity of NLRP3. Treatment with CY-09 showed therapeutic effects on cryopyrin-associated autoinflammatory syndrome and type 2 diabetes in mouse models[120]. The suppression of NLRP3 inflammasome components could be achieved by genetic silencing of NLRP3, ASC or caspase-1 using specific shRNAs[111] or clustered regularly interspaced short palindromic repeat (CRISPR)-associated system 9 (Cas9)[121]. Xu et al screened an optimized cationic lipid-assisted nanoparticle to deliver Cas9 mRNA and guide RNA targeting NLRP3 into macrophages, which improved insulin sensitivity and reduced adipose inflammation in high fat diet-induced diabetic mice[122]. Moreover, caspase-1 inhibitors are currently under development for inflammatory diseases[123, 124]. However, further studies are needed to determine whether these inhibitors are appropriate for cardiovascular diseases.
To date, most drug candidates targeting NLRP3 inflammasome have been studied only in animal models, and need to be further translated into clinical practices. The current available clinical treatments for NLRP3 inflammasome-related diseases are the agents that block IL-1β, including the recombinant IL-1 receptor antagonist anakinra, the IL-1β-neutralizing antibody canakinumab, and the soluble decoy IL-1β receptor rilonacept[125]. Interestingly, the recently completed large-scale clinical trial CANTOS showed that canakinumab targeting the IL-1β innate immunity pathway significantly decreased the rate of recurrent cardiovascular events[126], and reduced incident lung cancer and lung cancer mortality in patients with atherosclerosis[127]. However, IL-1β is not the only biological production of NLRP3 inflammasome and pyroptosis, other inflammatory cytokines, such as IL-18 and HMGB-1, also involved in the pathogenesis of cardiovascular diseases. Therefore, inhibition of IL-1β signaling may have limitations in clinical treatments.
Conclusion
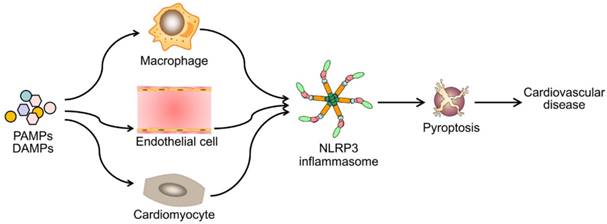
Emerging evidence has demonstrated that NLRP3 inflammasome activation is a critical regulator for pyroptosis involved in the progression of various cardiovascular diseases (Figure 3). Pyroptosis has been observed in different types of cells and tissues in in vivo and in vitro models of cardiovascular diseases (Table 1). Despite of growing studies focused on the role of pyroptosis in cardiovascular diseases, the detailed molecular mechanisms remain elusive. Although to what extent does NLRP3 inflammasome activation lead to pyroptosis remains unclear, NLRP3 inflammasome activation indeed mediates pyroptosis in most situations. Therefore, blocking the inflammasome pathway has been predicated to have clinical benefits in delaying the progression of cardiovascular diseases. Several types of NLRP3 inflammasome inhibitors have been developed and validated in cell culture studies and animal models of inflammation- related diseases. However, prospective clinical trials are needed for their potential translation into clinical practice. Also other potential candidates are required for further development, such as autophagy-inducer, antioxidant and miRNA agents. Future studies are needed to answer many unresolved questions. For example, can drugs or antibodies targeting GSDMD pyroptotic pathway enhance the therapeutic potential of single NLRP3 inflammasome inhibitor on cardiovascular diseases? Further studies exploring novel signaling pathways of pyroptosis will be important for the development of new therapeutic drugs.
Diverse PAMPs and DAMPs induce cardiovascular disease by NLRP3 inflammasome-mediated pyroptosis. PAMPs and DAMPs trigger the activation of NLRP3 inflammasome and induce pyroptosis, which promotes vascular inflammation and heart remodeling, and ultimately lead to cardiovascular diseases.
Types of cell and tissue injury subjected to cell pyroptosis in various cardiovascular diseases
| Diseases | Cell types/Organ | Injury types | Effects | References |
|---|---|---|---|---|
| Atherosclerosis | Macrophages | Cholesterol crystals | NLRP3 inflammasome activation | Duewell et al.[46] |
| Atherosclerosis | Macrophages | Ox-LDL | Pyroptosis | Lin et al.[49] |
| Atherosclerosis | Macrophages | Triglyceride | Pyroptosis | Son et al.[52] |
| Atherosclerosis | Macrophages | Hcy | NLRP3 inflammasome activation | Wang et al.[60] |
| Atherosclerosis | HUVECs | Palmitic acid | Pyroptosis | Hu et al.[54] |
| Atherosclerosis /MI/ Hypertension | HUVECs | Cd | Pyroptosis | Chen et al.[9] |
| Atherosclerosis | Mouse primary aortic endothelial cells | Ox-LDL | Pyroptosis | Yin et al.[53] |
| Atherosclerosis | Mouse primary coronary arterial endothelial cells | Cholesterol crystals | NLRP3 inflammasome activation | Zhang et al.[55] |
| Atherosclerosis | Mouse primary aortic endothelium cells/HUVECs | Hcy+LPS | Pyroptosis | Xi et al.[61] |
| Obesity | Subcutaneous and visceral hypertrophic adipocytes | High Fat | Ultrastructural abnormalities and caspase-1 activity | Giordano et al.[65] |
| Obesity | Human abdominal adipose tissue* | Caspase-1 expression | Koenen et al.[66] | |
| Diabetes | Mouse primary β cells | High glucose | NLRP3 inflammasome activation | Zhou et al.[68] |
| Type 2 diabetes | Human monocyte-derived macrophages* | NLRP3 inflammasome activation | Lee et al.[67] | |
| Hypertension | Rat aorta and renal artery | Spontaneity | mRNA expression of pro-caspase-1 | Chen et al.[74] |
| Hypertension | Mouse kidney | Deoxycorticosterone acetate and salt or angiotensin II | NLRP3 inflammasome activation | Krishnan et al.[71] |
| Hypertension | Mouse myocardium | TAC | NLRP3 inflammasome activation | Wang et al.[102] |
| Hypertension | Rat hypothalamic paraventricular nucleus | Dahl salt | NLRP3 inflammasome activation | Qi et al.[128] |
| Hypertension (PAH) | Rat lung tissue | Monocrotaline | NLRP3 inflammasome activation | Tang et al.[76] |
| Hypertension | Human serum* | Increased serum levels of IL-1β and IL-18 | Krishnan and Saito et al.[71, 72] | |
| MI | Mouse cardiac microvascular endothelial cells | Coronary artery ligation | Pyroptosis | Liu et al.[115] |
| MI | Mouse ischemic heart | Coronary artery ligation | Pyroptosis | Mezzaroma et al.[81] |
| DCM | Rat myocardium/H9c2 cardiomyocytes | High glucose and STZ | Pyroptosis | Luo and Li et al.[10, 92] |
| Viral myocarditis | Mouse cardiomyocyte/Myocardial tissue | CVB3 | NLRP3 inflammasome activation and pyroptosis | Wang et al.[96, 99] |
| Heart remodeling | Mouse myocardium | TAC | NLRP3 inflammasome activation | Wang et al.[102] |
| AF | Cardiomyocyte from human atrial biopsies* and myocardial tissue | CREM transgene | Pyroptosis | Yao et al.[111] |
| HF | Mouse myocardial tissue | CNTg | Cleaved caspase-1 expression | Bracey et al.[101] |
| Bacterial myocarditis | Mouse cardiac fibroblasts and myofibroblasts | LPS | NLRP3 inflammasome activation | Zhang et al.[104] |
* indicated this study with cells or tissue from human.
Abbreviations
AF: atrial fibrillation; AIM2: absent in melanoma 2; ALRs: absent in melanoma 2-like receptors; ASC: apoptosis-associated speck-like protein; CANTOS: The Canakinumab Anti-inflammatory Thrombosis Outcomes Study; CARD: a caspase recruitment domain; Cd: cadmium; CK-MB: creatine kinase-MB; CNTg: calcineurin transgene; CREM: cAMP-responsive element modulator; CRISPR/Cas9: clustered regularly interspaced short palindromic repeat-associated system 9; CVB3: coxsackievirus B3; DAMPs: danger-associated molecular patterns; DCM: diabetic cardiomyopathy; GSDMD: gasdermin D; GSDMD-NT: gasdermin D N-terminal domain; Hcy: homocysteine; HF: heart failure; HHcy: hyperhomocysteinemia; HMGB-1: high mobility group box-1 protein; HSP: heat shock protein; HUVECs: human umbilical venin endothelial cells; IL: interleukin; LDL: low-density lipoprotein; LPS: lipopolysaccharide; MI: myocardial infarction; MIRI: myocardial ischemia/ reperfusion injury; miRNAs: microRNAs; NF-κB: nuclear factor-κB; NLRP: NOD-like receptor protein; NLRs: NOD-like receptors; NOD: nucleotide-binding oligomerization domain; Ox-LDL: oxidized low density lipoprotein; P2X7: purinergic receptor P2X, ligand-gated ion channel 7; pAF: paroxysmal AF; PAH: pulmonary artery hypertension; PAMPs: pathogen-associated molecular patterns; perAF: persistent AF; PRRs: pattern-recognition receptors; ROS: reactive oxygen species; STZ: streptozotocin; TAC: transverse aortic constriction; TLRs: toll-like receptors; TNFα: tumor necrosis factor alpha; TRX: thioredoxin; TXNIP: thioredoxin-interacting protein.
Acknowledgements
This work was supported by National Natural Science Foundation of China (grant numbers 81570394, 81873514, and 81370371), Natural Science Foundation of Guangdong Province (grant numbers 2017A030311017 and 2014A030313066), and 111 Project from the Ministry of Education of the People's Republic of China (grant number B13037).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358:167-9
2. Hersh D, Monack DM, Smith MR, Ghori N, Falkow S, Zychlinsky A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc Natl Acad Sci U S A. 1999;96:2396-401
3. Koterski JF, Nahvi M, Venkatesan MM, Haimovich B. Virulent Shigella flexneri causes damage to mitochondria and triggers necrosis in infected human monocyte-derived macrophages. Infect Immun. 2005;73:504-13
4. Haimovich B, Venkatesan MM. Shigella and Salmonella: death as a means of survival. Microbes Infect. 2006;8:568-77
5. Hilbi H, Moss JE, Hersh D, Chen Y, Arondel J, Banerjee S. et al. Shigella-induced apoptosis is dependent on caspase-1 which binds to IpaB. J Biol Chem. 1998;273:32895-900
6. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113-4
7. Edgeworth JD, Spencer J, Phalipon A, Griffin GE, Sansonetti PJ. Cytotoxicity and interleukin-1beta processing following Shigella flexneri infection of human monocyte-derived dendritic cells. Eur J Immunol. 2002;32:1464-71
8. Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A. et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59:898-910
9. Chen H, Lu Y, Cao Z, Ma Q, Pi H, Fang Y. et al. Cadmium induces NLRP3 inflammasome-dependent pyroptosis in vascular endothelial cells. Toxicol Lett. 2016;246:7-16
10. Li X, Du N, Zhang Q, Li J, Chen X, Liu X. et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. 2014;5:e1479
11. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H. et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153-8
12. Sasaki M, Nakanuma Y. Stress-induced cellular responses and cell death mechanisms during inflammatory cholangiopathies. Clin Res Hepatol Gastroenterol. 2017;41:129-38
13. Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184:39-51
14. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73:1907-16
15. Kimbrell DA, Beutler B. The evolution and genetics of innate immunity. Nat Rev Genet. 2001;2:256-67
16. Imler JL, Hoffmann JA. Toll signaling: the TIReless quest for specificity. Nat Immunol. 2003;4:105-6
17. Pedra JH, Cassel SL, Sutterwala FS. Sensing pathogens and danger signals by the inflammasome. Curr Opin Immunol. 2009;21:10-6
18. Liu X, Lieberman J. A Mechanistic Understanding of Pyroptosis: The Fiery Death Triggered by Invasive Infection. Adv Immunol. 2017;135:81-117
19. Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M. et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515-8
20. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805-20
21. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786-91
22. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417-26
23. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013-22
24. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821-32
25. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519-50
26. Wilson KP, Black JA, Thomson JA, Kim EE, Griffith JP, Navia MA. et al. Structure and mechanism of interleukin-1 beta converting enzyme. Nature. 1994;370:270-5
27. Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N. et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci U S A. 2016;113:7858-63
28. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J. et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111-6
29. Liu X, Lieberman J. How ICE lights the pyroptosis fire. Cell Death Differ. 2017;24:197-9
30. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D. et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787-91
31. Kahlenberg JM, Dubyak GR. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am J Physiol Cell Physiol. 2004;286:C1100-8
32. Jin C, Flavell RA. Molecular mechanism of NLRP3 inflammasome activation. J Clin Immunol. 2010;30:628-31
33. Lima H Jr, Jacobson LS, Goldberg MF, Chandran K, Diaz-Griffero F, Lisanti MP. et al. Role of lysosome rupture in controlling Nlrp3 signaling and necrotic cell death. Cell Cycle. 2013;12:1868-78
34. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci. 2017;42:245-54
35. Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H. et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766-78
36. Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K. et al. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics. 2007;89:618-29
37. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S. et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666-71
38. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J. et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117-21
39. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S. et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246-9
40. Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250-3
41. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187-92
42. Yi YS. Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology. 2017;152:207-17
43. Kamens J, Paskind M, Hugunin M, Talanian RV, Allen H, Banach D. et al. Identification and characterization of ICH-2, a novel member of the interleukin-1 beta-converting enzyme family of cysteine proteases. J Biol Chem. 1995;270:15250-6
44. Wang Y, Gao W, Shi X, Ding J, Liu W, He H. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99-103
45. Abela GS, Aziz K. Cholesterol crystals rupture biological membranes and human plaques during acute cardiovascular events-a novel insight into plaque rupture by scanning electron microscopy. Scanning. 2006;28:1-10
46. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG. et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357-61
47. Zheng F, Xing S, Gong Z, Mu W, Xing Q. Silence of NLRP3 suppresses atherosclerosis and stabilizes plaques in apolipoprotein E-deficient mice. Mediators Inflamm. 2014;2014:507208
48. Jiang Y, Wang M, Huang K, Zhang Z, Shao N, Zhang Y. et al. Oxidized low-density lipoprotein induces secretion of interleukin-1beta by macrophages via reactive oxygen species-dependent NLRP3 inflammasome activation. Biochem Biophys Res Commun. 2012;425:121-6
49. Lin J, Shou X, Mao X, Dong J, Mohabeer N, Kushwaha KK. et al. Oxidized low density lipoprotein induced caspase-1 mediated pyroptotic cell death in macrophages: implication in lesion instability? PLoS One. 2013;8:e62148
50. Yan H, Li Y, Peng X, Huang D, Gui L, Huang B. Resistance of mitochondrial DNA-depleted cells against oxidized low-density lipoprotein-induced macrophage pyroptosis. Mol Med Rep. 2016;13:4393-9
51. Talayero BG, Sacks FM. The role of triglycerides in atherosclerosis. Curr Cardiol Rep. 2011;13:544-52
52. Son SJ, Rhee KJ, Lim J, Kim TU, Kim TJ, Kim YS. Triglyceride-induced macrophage cell death is triggered by caspase-1. Biol Pharm Bull. 2013;36:108-13
53. Yin Y, Li X, Sha X, Xi H, Li YF, Shao Y. et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler Thromb Vasc Biol. 2015;35:804-16
54. Hu Q, Zhang T, Yi L, Zhou X, Mi M. Dihydromyricetin inhibits NLRP3 inflammasome-dependent pyroptosis by activating the Nrf2 signaling pathway in vascular endothelial cells. Biofactors. 2018;44:123-36
55. Zhang Y, Li X, Pitzer AL, Chen Y, Wang L, Li PL. Coronary endothelial dysfunction induced by nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3 inflammasome activation during hypercholesterolemia: beyond inflammation. Antioxid Redox Signal. 2015;22:1084-96
56. Chowienczyk PJ, Watts GF, Cockcroft JR, Ritter JM. Impaired endothelium-dependent vasodilation of forearm resistance vessels in hypercholesterolaemia. Lancet. 1992;340:1430-2
57. Ueland PM, Refsum H, Stabler SP, Malinow MR, Andersson A, Allen RH. Total homocysteine in plasma or serum: methods and clinical applications. Clin Chem. 1993;39:1764-79
58. Cheung GT, Siow YL, O K. Homocysteine stimulates monocyte chemoattractant protein-1 expression in mesangial cells via NF-kappaB activation. Can J Physiol Pharmacol. 2008;86:88-96
59. Huang RF, Huang SM, Lin BS, Wei JS, Liu TZ. Homocysteine thiolactone induces apoptotic DNA damage mediated by increased intracellular hydrogen peroxide and caspase 3 activation in HL-60 cells. Life Sci. 2001;68:2799-811
60. Wang R, Wang Y, Mu N, Lou X, Li W, Chen Y. et al. Activation of NLRP3 inflammasomes contributes to hyperhomocysteinemia-aggravated inflammation and atherosclerosis in apoE-deficient mice. Lab Invest. 2017;97:922-34
61. Xi H, Zhang Y, Xu Y, Yang WY, Jiang X, Sha X. et al. Caspase-1 Inflammasome Activation Mediates Homocysteine-Induced Pyrop-Apoptosis in Endothelial Cells. Circ Res. 2016;118:1525-39
62. Borne Y, Fagerberg B, Persson M, Ostling G, Soderholm M, Hedblad B. et al. Cadmium, Carotid Atherosclerosis, and Incidence of Ischemic Stroke. J Am Heart Assoc. 2017;6:e006415
63. Makki K, Froguel P, Wolowczuk I. Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm. 2013;2013:139239
64. Stienstra R, van Diepen JA, Tack CJ, Zaki MH, van de Veerdonk FL, Perera D. et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci U S A. 2011;108:15324-9
65. Giordano A, Murano I, Mondini E, Perugini J, Smorlesi A, Severi I. et al. Obese adipocytes show ultrastructural features of stressed cells and die of pyroptosis. J Lipid Res. 2013;54:2423-36
66. Koenen TB, Stienstra R, van Tits LJ, Joosten LA, van Velzen JF, Hijmans A. et al. The inflammasome and caspase-1 activation: a new mechanism underlying increased inflammatory activity in human visceral adipose tissue. Endocrinology. 2011;152:3769-78
67. Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62:194-204
68. Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136-40
69. Jiang Q. Natural forms of vitamin E: metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic Biol Med. 2014;72:76-90
70. Kim Y, Wang W, Okla M, Kang I, Moreau R, Chung S. Suppression of NLRP3 inflammasome by gamma-tocotrienol ameliorates type 2 diabetes. J Lipid Res. 2016;57:66-76
71. Krishnan SM, Dowling JK, Ling YH, Diep H, Chan CT, Ferens D. et al. Inflammasome activity is essential for one kidney/deoxycorticosterone acetate/salt-induced hypertension in mice. Br J Pharmacol. 2016;173:752-65
72. Saito T, Miyagawa K, Chen SY, Tamosiuniene R, Wang L, Sharpe O. et al. Upregulation of Human Endogenous Retrovirus-K Is Linked to Immunity and Inflammation in Pulmonary Arterial Hypertension. Circulation. 2017;136:1920-35
73. Dalekos GN, Elisaf M, Bairaktari E, Tsolas O, Siamopoulos KC. Increased serum levels of interleukin-1beta in the systemic circulation of patients with essential hypertension: additional risk factor for atherogenesis in hypertensive patients? J Lab Clin Med. 1997;129:300-8
74. Chen H, Lu ZZ, Wei H, Han C. Induction of ICE and inhibition of c-fos, jun D and zif 268 in 12-month old spontaneously hypertensive rats. Life Sci. 1997;61:PL27-31
75. Qi J, Yu XJ, Shi XL, Gao HL, Yi QY, Tan H. et al. NF-kappaB Blockade in Hypothalamic Paraventricular Nucleus Inhibits High-Salt-Induced Hypertension Through NLRP3 and Caspase-1. Cardiovasc Toxicol. 2016;16:345-54
76. Tang B, Chen GX, Liang MY, Yao JP, Wu ZK. Ellagic acid prevents monocrotaline-induced pulmonary artery hypertension via inhibiting NLRP3 inflammasome activation in rats. Int J Cardiol. 2015;180:134-41
77. Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59:418-58
78. Little WC, Constantinescu M, Applegate RJ, Kutcher MA, Burrows MT, Kahl FR. et al. Can coronary angiography predict the site of a subsequent myocardial infarction in patients with mild-to-moderate coronary artery disease? Circulation. 1988;78:1157-66
79. Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H. et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;123:594-604
80. Qiu Z, Lei S, Zhao B, Wu Y, Su W, Liu M. et al. NLRP3 Inflammasome Activation-Mediated Pyroptosis Aggravates Myocardial Ischemia/Reperfusion Injury in Diabetic Rats. Oxid Med Cell Longev. 2017;2017:9743280
81. Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN. et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci U S A. 2011;108:19725-30
82. Braunwald E. Cardiomyopathies: An Overview. Circ Res. 2017;121:711-21
83. Timmis AD. Diabetic heart disease: clinical considerations. Heart. 2001;85:463-9
84. Poanta L, Fodor D, Albu A. Left ventricular function in patients with uncomplicated well-controlled diabetes mellitus. Med Ultrason. 2010;12:184-7
85. Cai L, Kang YJ. Cell death and diabetic cardiomyopathy. Cardiovasc Toxicol. 2003;3:219-28
86. Asbun J, Villarreal FJ. The pathogenesis of myocardial fibrosis in the setting of diabetic cardiomyopathy. J Am Coll Cardiol. 2006;47:693-700
87. An D, Rodrigues B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2006;291:H1489-506
88. Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A. et al. Myocardial cell death in human diabetes. Circ Res. 2000;87:1123-32
89. Shen X, Zheng S, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes. 2006;55:798-805
90. Van Linthout S, Spillmann F, Riad A, Trimpert C, Lievens J, Meloni M. et al. Human apolipoprotein A-I gene transfer reduces the development of experimental diabetic cardiomyopathy. Circulation. 2008;117:1563-73
91. Yu W, Wu J, Cai F, Xiang J, Zha W, Fan D. et al. Curcumin alleviates diabetic cardiomyopathy in experimental diabetic rats. PLoS One. 2012;7:e52013
92. Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C. et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One. 2014;9:e104771
93. Jeyabal P, Thandavarayan RA, Joladarashi D, Suresh Babu S, Krishnamurthy S, Bhimaraj A. et al. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem Biophys Res Commun. 2016;471:423-9
94. Grist NR, Reid D. Organisms in myocarditis/endocarditis viruses. J Infect. 1997;34:155
95. Esfandiarei M, McManus BM. Molecular biology and pathogenesis of viral myocarditis. Annu Rev Pathol. 2008;3:127-55
96. Wang Y, Gao B, Xiong S. Involvement of NLRP3 inflammasome in CVB3-induced viral myocarditis. Am J Physiol Heart Circ Physiol. 2014;307:H1438-47
97. Tschope C, Muller I, Xia Y, Savvatis K, Pappritz K, Pinkert S. et al. NOD2 (Nucleotide-Binding Oligomerization Domain 2) Is a Major Pathogenic Mediator of Coxsackievirus B3-Induced Myocarditis. Circ Heart Fail. 2017;10:e003870
98. Miteva K, Pappritz K, Sosnowski M, El-Shafeey M, Muller I, Dong F. et al. Mesenchymal stromal cells inhibit NLRP3 inflammasome activation in a model of Coxsackievirus B3-induced inflammatory cardiomyopathy. Sci Rep. 2018;8:2820
99. Wang Y, Jia L, Shen J, Wang Y, Fu Z, Su SA. et al. Cathepsin B aggravates coxsackievirus B3-induced myocarditis through activating the inflammasome and promoting pyroptosis. PLoS Pathog. 2018;14:e1006872
100. Sacco SJ, Park CL, Suresh DP, Bliss D. Living with heart failure: psychosocial resources, meaning, gratitude and well-being. Heart Lung. 2014;43:213-8
101. Bracey NA, Beck PL, Muruve DA, Hirota SA, Guo J, Jabagi H. et al. The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1beta. Exp Physiol. 2013;98:462-72
102. Wang Y, Wu Y, Chen J, Zhao S, Li H. Pirfenidone attenuates cardiac fibrosis in a mouse model of TAC-induced left ventricular remodeling by suppressing NLRP3 inflammasome formation. Cardiology. 2013;126:1-11
103. Li R, Lu K, Wang Y, Chen M, Zhang F, Shen H. et al. Triptolide attenuates pressure overload-induced myocardial remodeling in mice via the inhibition of NLRP3 inflammasome expression. Biochem Biophys Res Commun. 2017;485:69-75
104. Zhang W, Xu X, Kao R, Mele T, Kvietys P, Martin CM. et al. Cardiac fibroblasts contribute to myocardial dysfunction in mice with sepsis: the role of NLRP3 inflammasome activation. PLoS One. 2014;9:e107639
105. Boza P, Ayala P, Vivar R, Humeres C, Caceres FT, Munoz C. et al. Expression and function of toll-like receptor 4 and inflammasomes in cardiac fibroblasts and myofibroblasts: IL-1beta synthesis, secretion, and degradation. Mol Immunol. 2016;74:96-105
106. Freeman JV, Wang Y, Akar J, Desai N, Krumholz H. National Trends in Atrial Fibrillation Hospitalization, Readmission, and Mortality for Medicare Beneficiaries, 1999-2013. Circulation. 2017;135:1227-39
107. Andrade J, Khairy P, Dobrev D, Nattel S. The clinical profile and pathophysiology of atrial fibrillation: relationships among clinical features, epidemiology, and mechanisms. Circ Res. 2014;114:1453-68
108. Harada M, Van Wagoner DR, Nattel S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ J. 2015;79:495-502
109. Chen G, Chelu MG, Dobrev D, Li N. Cardiomyocyte Inflammasome Signaling in Cardiomyopathies and Atrial Fibrillation: Mechanisms and Potential Therapeutic Implications. Front Physiol. 2018;9:1115
110. Luan Y, Guo Y, Li S, Yu B, Zhu S, Li S. et al. Interleukin-18 among atrial fibrillation patients in the absence of structural heart disease. Europace. 2010;12:1713-8
111. Yao C, Veleva T, Scott L Jr, Cao S, Li L, Chen G. et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation. 2018;138:2227-42
112. Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K. et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol. 2009;187:61-70
113. Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D. et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21:263-9
114. Hu Z, Murakami T, Suzuki K, Tamura H, Kuwahara-Arai K, Iba T. et al. Antimicrobial cathelicidin peptide LL-37 inhibits the LPS/ATP-induced pyroptosis of macrophages by dual mechanism. PLoS One. 2014;9:e85765
115. Liu Y, Lian K, Zhang L, Wang R, Yi F, Gao C. et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res Cardiol. 2014;109:415
116. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC. et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248-55
117. Cocco M, Pellegrini C, Martinez-Banaclocha H, Giorgis M, Marini E, Costale A. et al. Development of an Acrylate Derivative Targeting the NLRP3 Inflammasome for the Treatment of Inflammatory Bowel Disease. J Med Chem. 2017;60:3656-71
118. He Y, Varadarajan S, Munoz-Planillo R, Burberry A, Nakamura Y, Nunez G. 3,4-methylenedioxy-beta-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J Biol Chem. 2014;289:1142-50
119. Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW. et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem. 2010;285:9792-802
120. Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J. et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med. 2017;214:3219-38
121. Schmid-Burgk JL, Gaidt MM, Schmidt T, Ebert TS, Bartok E, Hornung V. Caspase-4 mediates non-canonical activation of the NLRP3 inflammasome in human myeloid cells. Eur J Immunol. 2015;45:2911-7
122. Xu C, Lu Z, Luo Y, Liu Y, Cao Z, Shen S. et al. Targeting of NLRP3 inflammasome with gene editing for the amelioration of inflammatory diseases. Nat Commun. 2018;9:4092
123. Howley B, Fearnhead HO. Caspases as therapeutic targets. J Cell Mol Med. 2008;12:1502-16
124. MacKenzie SH, Schipper JL, Clark AC. The potential for caspases in drug discovery. Curr Opin Drug Discov Devel. 2010;13:568-76
125. Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633-52
126. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C. et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. 2017;377:1119-31
127. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ. et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1833-42
128. Qi J, Zhao XF, Yu XJ, Yi QY, Shi XL, Tan H. et al. Targeting Interleukin-1 beta to Suppress Sympathoexcitation in Hypothalamic Paraventricular Nucleus in Dahl Salt-Sensitive Hypertensive Rats. Cardiovasc Toxicol. 2016;16:298-306
Author contact
![]() Corresponding author: Hongmei Tan, MD, PhD, Professor, Department of Pathophysiology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou 510080, China, Tel: (8620) 87334055, E-mail: tanhmsysu.edu.cn.
Corresponding author: Hongmei Tan, MD, PhD, Professor, Department of Pathophysiology, Zhongshan School of Medicine, Sun Yat-sen University, Guangzhou 510080, China, Tel: (8620) 87334055, E-mail: tanhmsysu.edu.cn.