Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2019; 15(7):1488-1499. doi:10.7150/ijbs.32718 This issue Cite
Research Paper
Increased expression of TRIP13 drives the tumorigenesis of bladder cancer in association with the EGFR signaling pathway
Yanjun Gao1#, Shanhui Liu1#, Qi Guo1, Su Zhang1, Youli Zhao1, Hanzhang Wang2, Tianbao Li3, Yuwen Gong1, Yuhan Wang1, Tao Zhang1, Zhilong Dong1 ![]() , Dean Bacich2, Wasim H. Chowdhury2, Ronald Rodriguez2, Zhiping Wang1
, Dean Bacich2, Wasim H. Chowdhury2, Ronald Rodriguez2, Zhiping Wang1 ![]()
1. Department of Urology, The Second Hospital of Lanzhou University, Key Laboratory of Urological Diseases in Gansu Province, Gansu Nephro-Urological Clinical Center, Lanzhou 730000, China
2. Department of Urology, University of Texas Health Science Center at San Antonio, San Antonio, TX 78229, USA
3. Department of Molecular Medicine, University of Texas Health Science Center at San Antonio, San Antonio, Texas 78229, USA
#These authors contributed to the work equally.
Received 2019-1-1; Accepted 2019-4-4; Published 2019-6-2
Abstract

Thyroid hormone receptor interactor 13 (TRIP13) is a crucial regulator of the spindle apparatus checkpoint and double-stranded break repair. The abnormal expression of TRIP13 was recently found in several human cancers, whereas the role of TRIP13 in the development of bladder cancer (BCa) has not been fully elucidated. Here, we reported that TRIP13 expression was elevated in BCa tissues compared with normal bladder tissues. Notably, the increased expression of TRIP13 was correlated with advanced tumor stage, lymph node metastasis, distant metastasis and reduced survival in BCa patients. Knockdown of TRIP13 in bladder cancer cells suppressed proliferation, induced cell cycle arrest, promoted apoptosis, and impaired cell motility, ultimately inhibiting tumor xenograft growth. Mechanistic investigations revealed that TRIP13 directly bound to epidermal growth factor receptor (EGFR), modulating the EGFR signaling pathway. Furthermore, TRIP13 expression was positively correlated with EGFR expression in BCa specimens, and the high expression of both TRIP13 and EGFR predicted poor survival. Overall, our results underscore the crucial role of TRIP13 in the tumorigenesis of BCa and provide a novel biomarker and therapeutic target for BCa treatment.
Keywords: TRIP13, Tumorigenesis, Bladder cancer, EGFR
Introduction
Bladder cancer (BCa) is one of the most common malignancies in the world and the most expensive malignancy to treat from diagnosis to death [1]. Similar to other carcinomas, BCa is associated with multiple genetic feature alterations, such as epigenetic regulation, copy number variation and mutations [2], which lead to the high molecular heterogeneity of particular biomarkers. Although new discoveries have improved our understanding of tumorigenesis and unveiled numerous potentially targetable genomic alterations in BCa, approximately one-third of BCa patients develop metastasis or progression and eventually die from the disease despite aggressive therapy [3]. Therefore, an improved understanding of the molecular mechanisms underlying the tumorigenesis of BCa and its progression is indispensable for identifying new biomarkers and developing novel treatment options for BCa.
Thyroid hormone receptor interactor 13 (TRIP13, also known as 16E1BP and pachytene checkpoint 2) is a member of the AAA+ protein family (ATPases associated with diverse cellular activities) [4]. Generally, TRIP13 is required for meiotic DNA break formation and recombination, chromosome synapsis and mitotic checkpoint regulation [5, 6]. Recently, evidence has indicated that TRIP13 may possess functions other than meiosis and mitosis, particularly in the regulation of tumorigenicity. Banerjee and colleagues [7] reported that in head and neck squamous cell carcinoma, TRIP13 overexpression resulted in aggressive, treatment-resistant tumors via enhanced double-strand break DNA repair through promoting nonhomologous end joining. The authors identified that TRIP13 had frequent copy number gains and concordant upregulation in lung cancer, suggesting its role as a potential oncogene [8]. Additionally, an increasing body of evidence suggests that high TRIP13 expression is positively correlated with a poor prognosis in multiple cancers, including breast, liver, and gastric cancers and multiple myeloma [9, 10]. By contrast, through exome sequencing, Yost and colleagues [11] found that individuals with biallelic loss-of-function mutations in TRIP13 had a high rate of chromosome missegregation, leading to a high risk of embryonal tumors, such as Wilms tumor. Therefore, TRIP13 may have multiple functions in cancer. Nevertheless, its specific role in BCa has not been elucidated thus far.
In the current study, we demonstrated that the increased expression of TRIP13 was a characteristic molecular change in BCa. Our data showed that TRIP13 overexpression was correlated with aggressive characteristics, such as advanced tumor stage, nodal metastasis, distant metastasis, and poor survival. Additionally, the loss of TRIP13 in bladder cancer cells inhibited the oncogenic phenotypes in vitro and subcutaneous tumor growth in vivo. Importantly, TRIP13 mechanistically promoted the tumorigenesis of BCa, partially by modulating the EGFR signaling pathway via direct binding to EGFR.
Results
Higher TRIP13 expression in human BCa tissues
We first examined the expression level of TRIP13 in a tissue microarray consisting of human BCa and paired adjacent normal bladder tissues (n=46 cases). TRIP13 expression was high in 10 (21.7%) of the tumor tissues but only in 1 (2.2%) of the adjacent normal tissues (Fig. 1a, P<0.01). Figure 1b shows representative images of TRIP13 staining in tumor and normal tissues. To further validate our TMA findings, we investigated the expression of TRIP13 in BCa cohorts from the Oncomine (Lee [12] and Sanchez-Carbayo[13], https://www.oncomine.org/) and The Cancer Genome Atlas (TCGA, https://cancergenome.nih.gov/) databases. TRIP13 expression was higher in tumor tissues than in normal bladder tissues (Fig. 1c and 1d, **, P<0.01), confirming our TMA results.
Increased expression of TRIP13 in human BCa tissues. (a) TRIP13 protein expression in the TMA consisting of BCa tissues and matched adjacent normal bladder tissues (n=46 cases). (b) Representative immunohistochemical staining of TRIP13 expression in human BCa tissues (T) and adjacent normal tissues (N). (c) TRIP13 gene expression in human BCa samples based on two independent studies (Lee et al., n=157 and Sanchez-Carbayo et al., n=256) from the Oncomine database (https://www.oncomine.org/). Box plots are shown for each study and include normal urothelium (NU), superficial cancer (SUP) and invasive cancer (INV; **, P < 0.01). (d) The mRNA level of TRIP13 in a published bladder cancer dataset from TCGA database (https://cancergenome.nih.gov/) with cancer tissues and paired adjacent normal bladder tissues (n = 19, P<0.01) is shown. (e) The association between TRIP13 expression and the overall survival rate in BCa patients. Patients with high TRIP13 expression (+, n =83) had significantly worse overall survival than those with low TRIP13 expression (-, n =82, P<0.01). (f) The association between TRIP13 expression and the disease-specific survival rate in BCa patients (P<0.001). (g) The association between TRIP13 expression and the overall survival rate in NMIBC patients (P<0.05). (h) The association between TRIP13 expression and the overall survival rate in MIBC patients (P=0.36).
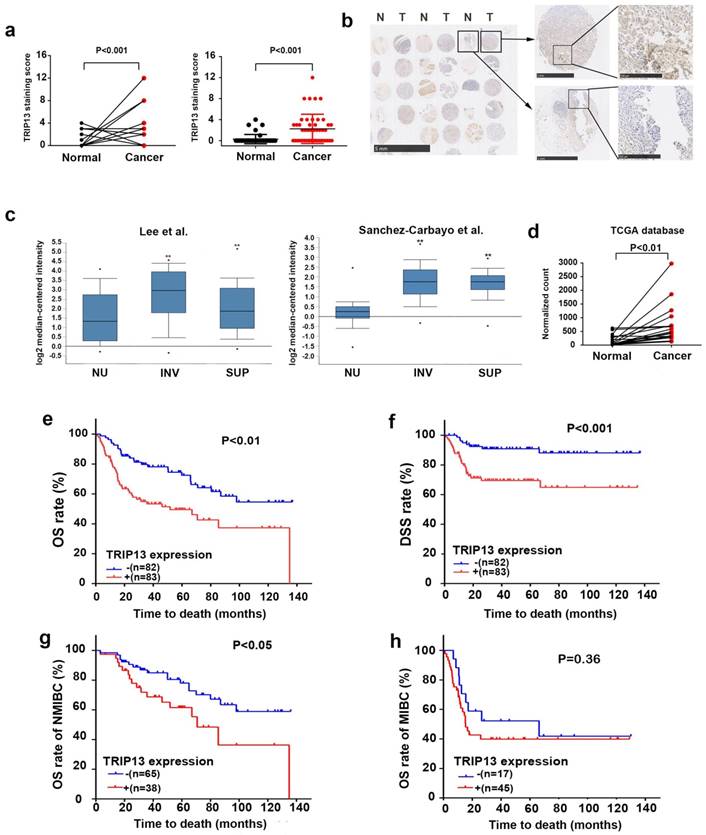
Elevated expression of TRIP13 in BCa is associated with stage progression, metastasis, and poor survival
To investigate the potential relationship between increased TRIP13 expression and the clinical features of BCa, we evaluated the expression of TRIP13 in 342 paraffin-embedded BCa tissue samples using immunohistochemical staining. We did not find a correlation of TRIP13 expression with age, sex or tumor size in BCa patients. Notably, the expression of TRIP13 was positively correlated with advanced American Joint Committee on Cancer (AJCC) stage, lymph node metastasis and distant metastasis (Table 1). Furthermore, Kaplan-Meier analysis of data in GSE13507 [12] indicated that patients with elevated TRIP13 expression displayed significantly reduced overall survival (OS) (Fig. 1e, P<0.01) and disease-specific survival (DSS) (Fig. 1f, P<0.001). Moreover, increased TRIP13 expression predicted poor overall survival in patients with NMIBC (non-muscle-invasive bladder cancer) (Fig. 1g, P<0.001) but not in patients with MIBC (muscle-invasive bladder cancer) (Fig. 1h, P=0.36).
Correlation between TRIP13 expression and clinicopathological features in BCa
| Number of patients | TRIP13 expression | χ2 a | P | ||
|---|---|---|---|---|---|
| Low | High | ||||
| All patients | 342 | 203(59.4%) | 139(40.6%) | ||
| Gender | 0.711 | 0.399 | |||
| Male | 291 | 170(58.4%) | 121(41.6%) | ||
| Female | 51 | 33(64.7%) | 18(35.3%) | ||
| Age (years) | 2.530 | 0.112 | |||
| <65 | 189 | 105(55.6%) | 84(44.4%) | ||
| ≥65 | 153 | 98(64.1%) | 55(35.9%) | ||
| Size of tumor (cm) | 0.275 | 0.600 | |||
| <3 | 183 | 111(60.7%) | 72(39.3%) | ||
| ≥3 | 159 | 92(57.9%) | 67(42.1%) | ||
| Grade | 1.102 | 0.294 | |||
| Low | 201 | 124(61.7%) | 77(38.3%) | ||
| High | 141 | 79(56.0%) | 62(44.0%) | ||
| AJCC stage | 12.050 | 0.017* | |||
| 0 | 163 | 109(66.9%) | 54(33.1%) | ||
| I | 86 | 48(55.8%) | 38(44.2%) | ||
| II | 30 | 18(60.0%) | 12(40.0%) | ||
| III | 15 | 9(60.0%) | 6(40.0%) | ||
| IV | 48 | 19(39.6%) | 29(60.4%) | ||
| Nodal metastasis | 10.536 | 0.001* | |||
| N0 | 313 | 194(62.0%) | 119(38.0%) | ||
| N1-N3 | 29 | 9(31.0%) | 20(69.0%) | ||
| Distant metastasis | 6.099 | 0.014* | |||
| M0 | 317 | 194(61.2%) | 123(38.8%) | ||
| M1 | 25 | 9(36.0%) | 16(64.0%) | ||
aPearson's chi-square test was used for the assay. Significant at P<0.05.
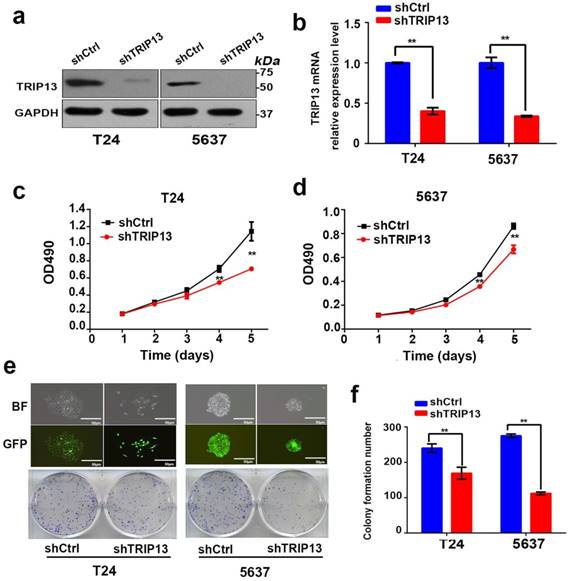
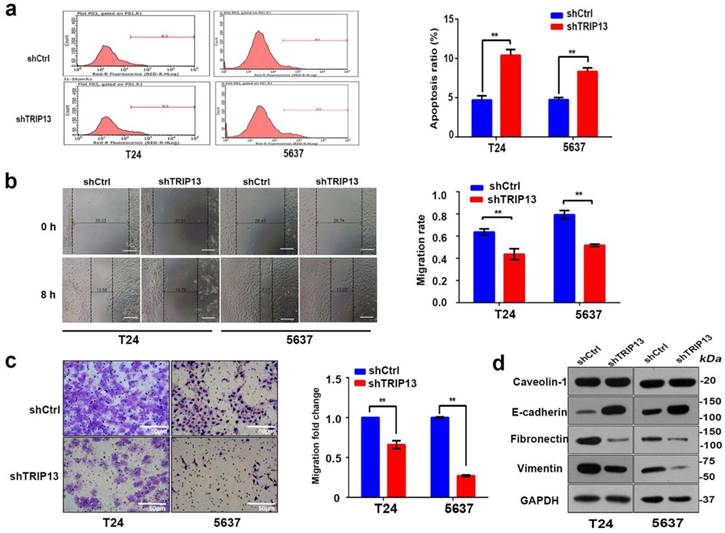
Knockdown of TRIP13 in bladder cancer cells suppresses proliferation, promotes apoptosis, and impairs cell motility
To further explore whether inhibition of TRIP13 affected the biologic activities of bladder cancer cells, TRIP13-silenced T24 and 5637 cell lines were established using lentiviruses encoding the specific shTRIP13. As shown in Fig. 2, the TRIP13 transcript (Fig. 2b, **, P<0.01) and protein expression (Fig. 2a) were consistently knocked down in both cell lines. We first tested whether the modulation of TRIP13 expression altered cell proliferation. An MTT assay demonstrated that the depletion of TRIP13 attenuated proliferation in both cell lines relative to the control (Fig. 2c and 2d, **, P<0.01). Additionally, we observed that the colony-forming ability of T24 and 5637 cells was significantly suppressed after the knockdown of TRIP13 by 29.6% and 59.3%, respectively (Fig. 2e and 2f, **, P<0.01). To evaluate the impact of TRIP13 on cell survival, an apoptosis assay was performed using flow cytometry in cells stained with FITC-annexin V. A significant difference between TRIP13-silenced and control cells was detected (Fig. 3a, **, P<0.01). In addition, wound-healing (Fig. 3b, **, P<0.01) and transwell migration assays (Fig. 3c, **, P<0.01) confirmed that silencing TRIP13 impeded the motility of T24 and 5637 cells. Then, we assessed whether TRIP13 expression was correlated with the epithelial-mesenchymal transition (EMT) in bladder cancer cells. Western blot analysis showed that after the depletion of TRIP13, the expression of the epithelial marker E-cadherin increased, whereas the expression of the mesenchymal markers vimentin and fibronectin decreased (Fig. 3d). Similar results were obtained for the other shRNA targeting TRIP13 (Fig. S1).
Knockdown of TRIP13 in bladder cancer cells suppresses proliferation in vitro. (a) The efficiency of shTRIP13 knockdown in T24 and 5637 cells was confirmed by western blot and (b) qPCR analyses. (c) and (d) The proliferation of T24 and 5637 cells transduced by lentiviruses expressing shCtrl or shTRIP13 was determined by the MTT assay. (e) and (f) Colony formation assay of T24 and 5637 cells transduced with lentiviruses expressing shCtrl or shTRIP13. BF: bright field; GFP: green fluorescent protein. Scale bar, 50 μm. The data are presented as the mean ± SD from three independent experiments. **, P<0.01.
Inhibition of TRIP13 in bladder cancer cells promotes apoptosis and impairs cell motility in vivo. (a) Cell apoptosis was measured using flow cytometry in T24 and 5637 cells treated with lentiviruses expressing shCtrl or shTRIP13. The motility of T24 and 5637 cells transduced with lentiviruses expressing shCtrl or shTRIP13 was determined by (b) wound-healing and (c) transwell assays. Scale bar, 50 μm. (d) The expression of caveolin-1, E-cadherin, vimentin, and fibronectin was evaluated by western blot in T24 and 5637 cells after treatment with lentiviruses expressing shCtrl or shTRIP13. The data are presented as the mean ± SD from three independent experiments. *, P<0.05, **, P<0.01.
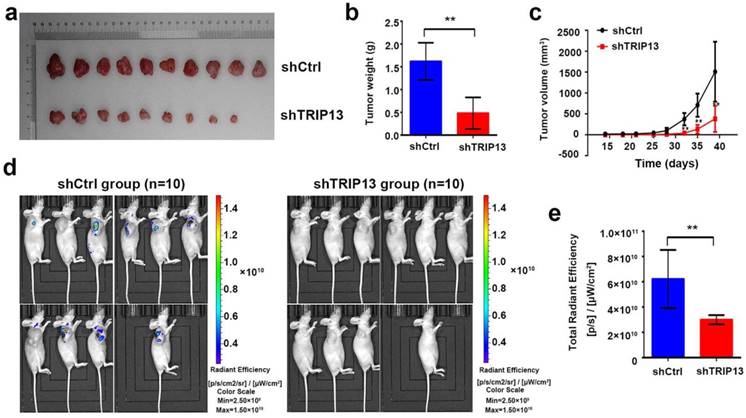
Loss of TRIP13 inhibits BCa carcinogenesis in vivo
Next, we evaluated the effect of TRIP13 depletion on BCa carcinogenesis in vivo. For this aim, T24 cells transduced with lentiviruses encoding shTRIP13 or shCtrl were subcutaneously inoculated into nude mice. The tumor sizes were measured twice each week for four weeks and then compared between the two groups. In the control group, all mice grew tumors four weeks after injection, whereas only 9 of 10 mice injected with TRIP13-silenced cells grew tumors (Fig. 4a). As shown in Figure 4c, tumors formed by TRIP13-silenced cells grew significantly slower than those in the control group at different time points. At the end of the experiment, bioluminescent signals in the TRIP13-knockdown group were lower than those in the control group (Fig. 4d and 4e, **, P<0.01), and the net weight of tumors formed by TRIP13-silenced cells was significantly reduced compared with that of the controls (Fig. 4b, **, P<0.01).
Loss of TRIP13 inhibits tumor growth in subcutaneous xenograft models. (a) Gross appearance of tumors from nude mice injected with T24/shCtrl (upper) or T24/shTRIP13 (lower) cells. Weight (b) and volume (c) of tumors from nude mice transplanted with T24/shCtrl (upper) or T24/shTRIP13 (lower) cells. **, P<0.01 (d) Bioluminescent imaging of the control and TRIP13-knockdown groups at the end of the experiment. (e) Total radiant efficiency of the control and TRIP13-knockdown groups at the end of the experiment. **, P<0.01.
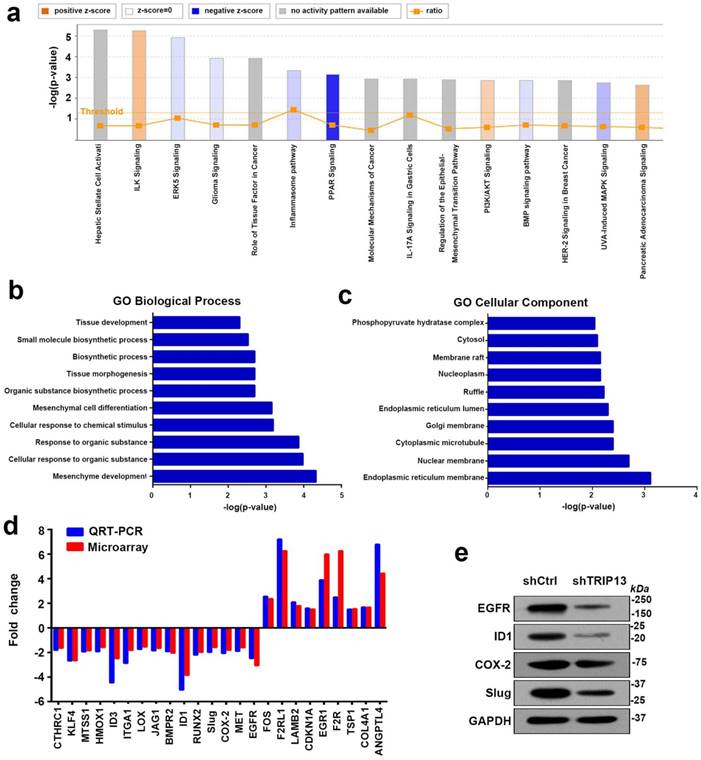
Inhibition of TRIP13 induces the differential expression of genes involved in mesenchyme development and suppresses the EGFR signaling pathway
To further study the underlying mechanism of TRIP13-mediated carcinogenesis in BCa, we performed a whole-genome expression microarray in T24/shCtrl and T24/shTRIP13 cells. Specifically, the Affymetrix Human Gene Expression Array was used and led to the identification of 1175 differentially expressed genes, of which 558 were upregulatedand 587 were downregulated. Gene Ontology enrichment analysis was performed using iPathwayGuide (https://www.advaitabio.com/). Among the enriched biological processes, mesenchyme development was at the top of the list (Fig. 5b). The cellular components involved included the endoplasmic reticulum membrane, the nuclear membrane and cytoplasmic microtubules (Fig. 5c). In addition, pathway analysis showed that among the top 15 significant pathways, seven pathways were associated with EGFR signaling: ERK5 signaling, glioma signaling, the role of tissue factor in cancer pathway, IL-17A signaling, regulation of the EMT pathway, HER-2 signaling and UVA-induced MAPK signaling (Fig. 5a). To validate the array-based findings, we measured the expression of several genes involved in cell proliferation and migration. Interestingly, the mRNA and protein expression of several key components in the EGFR signaling pathway, including inhibitor of DNA binding 1 (ID1), cyclooxygenase-2 (Cox-2), Slug and EGFR, was consistently decreased (Fig. 5d and 5e). Therefore, we hypothesized that TRIP13 was involved in the regulation of EGFR signaling in BCa.
Inhibition of TRIP13 induces the differential expression of genes involved in mesenchyme development and suppresses EGFR signaling. (a) The canonical pathway analysis of all genes with altered expression. A positive z-score indicates a predicted activation of the enriched pathway, and a negative z-score indicates a predicted inactivation of the enriched pathway. (b) and (c) Gene Ontology enrichment analysis of the differentially expressed genes upon TRIP13 knockdown. (d) qPCR and western blot (e) analyses validating the microarray findings.
To identify the interacting partner of TRIP13, we performed LC-tandem mass spectrometry. Flag-TRIP13-immunoprecipitated complexes were purified from T24 cells stably overexpressing Flag-TRIP13, stained and immunoblotted (Fig. 6a). A total of 2476 proteins were identified in association with TRIP13, including tubulins and several key components in the EGFR signaling pathway, such as EGFR, JAK2, and AKT1 (Table S2 and Table S3). Furthermore, coimmunoprecipitation revealed that TRIP13 directly interacted with EGFRbut not JAK2 or AKT1 (Fig. 6b).
EGFR is the direct target of TRIP13, and its expression is positively correlated with TRIP13 expression in human BCa specimens. (a) Immunoprecipitates eluted with the Flag peptide were verified by SDS-PAGE. Candidate gel bands were excised and used for mass spectrometry. Control: T24 cells transduced with lentiviruses expressing the control vector. OE: T24 cells transduced with lentiviruses expressing Flag-TRIP13. (b) Immunoprecipitates were immunoblotted with anti-EGFR, -AKT1, -JAK2 and -Flag antibodies. (c) Correlation analysis of TRIP13 and EGFR mRNA expression in 165 BCa specimens. (d) The association between TRIP13 and EGFR expression and the overall survival rate in BCa patients. The patients (n=165) were divided into three groups according to their gene expression. Patients with high expression of both TRIP13 and EGFR had a significantly worse overall survival rate than the other two groups (P=0.016). (e) The association between TRIP13 and EGFR expression and the disease-specific survival rate in BCa patients (P=0.013).
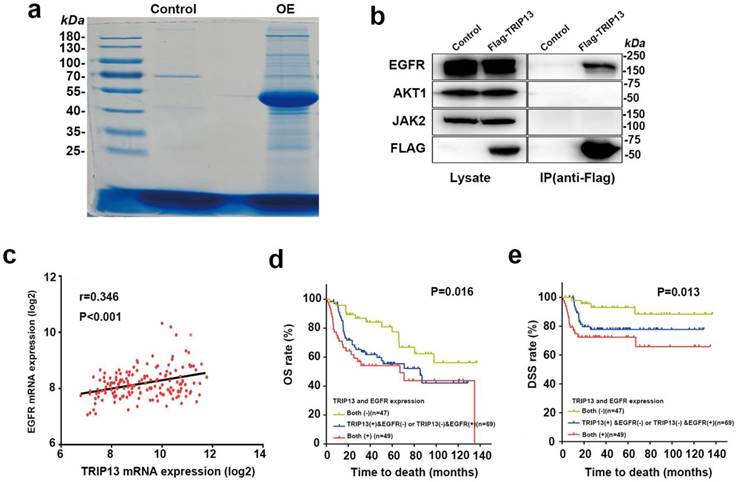
High expression of TRIP13 and EGFR is associated with poor survival in BCa
To further determine whether this finding is clinically relevant, we investigated the expression of TRIP13 and EGFR in the BCa dataset GSE13507 [12]. Pearson correlation analysis revealed that the correlation between TRIP13 and EGFR was significant (Fig. 6c, r=0.346, P<0.001). Importantly, Kaplan-Meier analysis demonstrated that patients with high expression of both TRIP13 and EGFR had a significantly shorter survival time (OS: 71.7 months; DSS: 95.1 months), whereas patients with low expression of both TRIP13 and EGFR had a significantly longer survival time (OS: 100.3 months; DSS: 125.4 months). Patients with high expression of either TRIP13 or EGFR had a survival time (OS: 75.0 months; DSS: 103.6 months) that was between that of the other two groups (Fig. 6d and 6e, P<0.05).
Discussion
Our results revealed for the first time that the expression of TRIP13 was higher in BCa tissues than in normal bladder tissues (Fig. 1a and 1b). Moreover, cohorts from the Oncomine (https://www.oncomine.org/) [12, 13] and TCGA databases (https://cancergenome.nih.gov/) verified our results (Fig. 1c and 1d). More importantly, elevated TRIP13 expression was significantly correlated with the clinicopathological characteristics of BCa, including advanced AJCC stage, lymph node metastasis and distant metastasis (Table 1), indicating its potential role as a biomarker for metastasis in BCa.
It is estimated that nearly 80% of BCa patients are non-muscle-invasive at the time of initial presentation and that patients with NMIBC have a relatively high five-year survival rate. However, at least one-third of patients with NMIBC may progress to MIBC with a high rate of metastasis, resulting in a decreased survival rate despite aggressive therapy [3, 14, 15]. When considering the prognosis of NMIBC, it is necessary to consider not only the clinicopathological parameters but also the molecular alterations in the tumors. Several studies have attempted to use molecular biomarkers, such as Ki-67, FGFR3, and p53, to predict outcomes in patients with NMIBC [16-19]. Our results indicated that TRIP13 expression was correlated with reduced OS and DSS (Fig. 1e and 1f). Moreover, NMIBC patients, rather than MIBC patients, with high expression of TRIP13 tended to have a shorter overall survival time (Fig. 1g and 1h). Therefore, TRIP13 may be a potential risk factor in patients with NMIBC, and its predictive accuracy requires further characterization. Although we cannot rule out that the relatively small sample size may have reduced the statistical significance of TRIP13, multivariate Cox regression analysis suggested that, at least for the samples we used as models, TRIP13 was not an independent prognostic factor.
TRIP13 has been identified as a key regulator of the spindle assembly checkpoint, which ensures the proper segregation of chromosomes until all sister chromatids are attached to opposite spindle poles by microtubules [20]. Mechanistically, TRIP13 can catalyze MAD2 conformation switchingwith the help of p31comet, thereby allowing anaphase progression and mitotic exit [21]. In our study, the mass spectrometry results showed that the major proteins that interacted with TRIP13 were tubulins (Table S2). In addition, GO enrichment analysis revealed that the cellular components of differentially expressed genes upon TRIP13 knockdown included cytoplasmic microtubules (Fig. 5c). Taken together, these data indicated the likely interaction between TRIP13 and microtubules. In the current study, knockdown of TRIP13 in BCa cells inhibited cell proliferation. This observation can be explained by the possible imbalance between MAD2 and TRIP13. MAD2 is a common spindle assembly checkpoint protein that is overexpressed in multiple cancers, including BCa [22]. Marks et al. [23] demonstrated that TRIP13 and MAD2 were cooverexpressed in multiple tumor types and that TRIP13 buffered the effects of MAD2 overexpression. Most importantly, the loss of TRIP13 in MAD2-overexpressing cells led to severe mitotic arrest and an almost complete block of cell proliferation. Thus, the inhibition of bladder cancer cell growth we observed upon TRIP13 knockdown may be attributed to the regulation of microtubules and the imbalance between TRIP13 and MAD2. However, severe mitotic arrest was not observed upon TRIP13 knockdown in our study. One possibility is that TRIP13 is not an integral part of mitotic regulation in BCa. Another possibility is that the overexpression of other mitotic checkpoint genes potentially buffers the effects of TRIP13 knockdown.
Proposed mechanisms of TRIP13 functions in BCa. TRIP13 directly binds to EGFR and promotes the activation of EGFR signaling and its downstream signaling pathways, thus contributing to the aggressive characteristics of BCa.
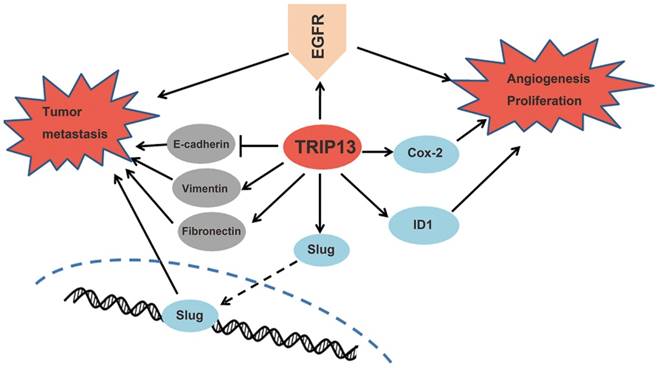
Over the past decade, the role of the EGFR signaling pathway in tumorigenesis and cancer progression has been a hot research area. It was reported that the hyperactivation of EGFR pathway signaling was correlated with grade/stage progression and shorter survival in bladder cancer [24-26]. Our results demonstrated that among the key components in the EGFR signaling pathway, EGFR, rather than JAK2 or AKT1, was able to bind to TRIP13 (Fig. 6b). EGFR tyrosine kinase inhibitors are in clinical use for several malignancies [27, 28], and those inhibitors alone or along with other conventional chemotherapeutic agents resultin suppressed tumor growth. Although EGFR inhibitors have had limited effects in BCa clinical trials, it has been predicted that carefully stratifying patients based on their molecular profiles will help improve the efficacy of anti-EGFR therapy [29]. Recently, a consensus about the molecular subclassification of BCa was reached regarding the existence of the basal-squamous-like subtype [30]. Basal-squamous-like tumors are associated with a worse outcome, presumably because this subtype tends to be more aggressive [31]. This cluster has a higher expression of stem cell markers and immune response genes, as well as signaling molecules, including EGFR [32]. Furthermore, erlotinib, an EGFR inhibitor, was shown to have better antitumor efficacy for this phenotype in vivo [33]. Accordingly, it is possible that TRIP13 may contribute to the malignant evolution of basal-squamous-like tumors through its interaction with EGFR. Considering its role as an AAA-ATPase, TRIP13 is a more readily druggable target, which may have a better toxicity profile than traditional chemotherapeutic agents, and targeting TRIP13 along with EGFR antagonists may be a new therapeutic strategy for this subtype.
In summary, our study showed for the first time that TRIP13 expression was increased in human BCa, particularly in patients in late stage or with metastasis, indicating that TRIP13 may serve as a biomarker to predict progression and metastasis. Our data also demonstrated that the increased expression of TRIP13 exerted a crucial role in the tumorigenesis of BCa both in vitro and in vivo by modulating the EGFR signaling pathway via a direct interaction with EGFR. Therefore, TRIP13 is a promising biomarker, expanding the potential therapeutic options for BCa treatment.
Materials and Methods
Clinical samples and immunohistochemical staining
A total of 342 BCa tissues were obtained from patients with BCa who underwent surgical resection in the Department of Urology, Lanzhou University Second Hospital. A tissue microarray (TMA) containing a total of 46 BCa samples was obtained from the National Engineering Center for Biochip in Shanghai, China. All experimental procedures in this study were approved by the Institutional Review Board of Lanzhou University Second Hospital. Informed written consent was obtained from each patient. The clinicopathological characteristics of the patients are shown in Table 1. The paraffin-embedded tissue sections and the TMA were baked at 65 °C for 30 minutes and then dewaxed in xylene and rehydrated using graded percentages of ethanol. After incubation in 3% H2O2 for 10 minutes, sections were boiled in citrate buffer (pH=6.0) for 20 minutes, followed by incubation with 10% bovine serum albumin. Rabbit anti-TRIP13 (Sigma, Carlsbad, CA, USA) was used for TRIP13 staining. After washing, the sections were incubated with a goat anti-rabbit secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) for one hour at room temperature. The tissue sections were then stained with 3, 3'-diaminobenzidine, counterstained with hematoxylin, dehydrated, cleared and mounted. The sections were reviewed and scored independently by two pathologists based on both the proportion of positive cells and the intensity of staining. The proportion of positively stained cells was scored according to the following criteria: 0 (<10% positive cells), 1 (10-25% positive cells), 2 (26-50% positive cells), 3 (51-75% positive cells) and 4 (>75% positive cells). The intensity of staining was scored as follows: 0 (no staining), 1 (weak staining), 2 (moderate staining) and 3 (strong staining). The final staining score was calculated as the staining intensity score × the proportion score and ranged from 0 to 12. A score ≤ 3 was considered low TRIP13 expression (-) and a score ≥ 4 was considered high TRIP13 expression (+).
Cell culture
Bladder cancer cell lines (5637 and T24) were obtained from the Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. These cells were authenticated by short tandem repeat (STR) DNA profiling analysis at Shanghai Genechem Co., Ltd. and cultured in RPMI 1640 medium (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Beyotime, Shanghai, China). All cells examined in this study were used within six months of thawing of the original cultures and maintained at 37 °C with a 5% CO2 atmosphere. Mycoplasma testing was routinely performed.
Cell transduction
The sequences of the shRNA targeting TRIP13 (shTRIP13) and the negative control shRNA (shCtrl), with no definite target, are as follows: shTRIP13-1: 5′- TGGGTTCTACCTGCAGCTGAA -3′, shTRIP13-2: 5′- CGATTATGTGATGACAACTTT -3′, and shCtrl: 5′- TTCTCCGAACGTGTCACGT-3′. For the overexpresssion of TRIP13, T24 cells were transduced with lentiviruses expressing Flag-TRIP13 or a control vector. Both the shRNA sequences and the TRIP13 construct were subcloned into the lentiviral expression vector with GFP by Shanghai Genechem Co., Ltd. (Shanghai, China). Lentiviral transduction was performed according to the user's manual.
Quantitative real-time PCR
Total RNA was isolated with TRIzol Reagent (Thermo Fisher Scientific, Beijing, China) according to the manufacturer's protocol. cDNA was synthesized from total RNA using the PrimeScript RT Reagent Kit with gDNA Eraser (Takara Biomedical Technology, Dalian, China). Quantitative real-time PCR analysis was performed using the SYBR Premix Ex Taq kit (Takara Biomedical Technology). Each sample was run in triplicate, and the data were analyzed using the relative quantification method with normalization to GAPDH. The primers used are listed in Table S1.
Western blot analysis
Western blot analysis was performed as previously described [34]. Anti-GAPDH (SC-32233), goat anti-rabbit IgG (sc-2004) and goat anti-mouse IgG (sc-2005) were purchased from Santa Cruz Biotechnology. Anti-JAK2 (#3230), anti-AKT1 (#9272) and anti-vimentin (#5741) were purchased from CST (Cell Signaling Technology, Beverly, MA, USA). Anti-TRIP13 (HPA005727) was purchased from Sigma. Anti-E-cadherin (ab1416), anti-fibronectin (ab6328), anti-caveolin-1 (ab2910) and anti-EGFR (ab52894) were purchased from Abcam (Abcam, Cambridge, MA, USA). Anti-Flag (PSI-TA-001) was purchased from Shanghai PSI Biotechnology Co., Ltd. (Shanghai, China).
MTT and colony formation assays
Cell viability was assessed using the MTT Cell Proliferation and Cytotoxicity Detection Kit (Beyotime). For the colony formation assay, cells were seeded into 6-well plates at a concentration of 1×103 cells per well and incubated at 37 °C with a 5% CO2 atmosphere. After two weeks, cells were fixed and stained with Giemsa stain, modified solution (Sigma). Colonies containing more than 50 individual cells were counted.
Apoptosis analysis
Apoptosis was measured by flow cytometry using the Annexin V Apoptosis Detection Kit (eBioscience, San Diego, CA, USA) according to the manufacturer's protocol. All experiments were performed in triplicate.
Cell migration and wound-healing assays
The cell migration assay was performed in 24-well transwell chambers (Corning, Shanghai, China) according to the manufacturer's instructions. For the wound-healing assay, cells were seeded into 96-well plates at a density of 5 ×104 per well and cultured overnight. Wounds were created with a 96 Wounding Replicator (VP Scientific, San Diego, CA, USA). The cells were washed gently with culture medium and further cultured with medium containing 1% FBS. Images were acquired at different time points with a microscope at 100x magnification. The gap distance was evaluated using Adobe Photoshop CS6 (v13.0).
Mouse subcutaneous xenograft model
All experimental procedures were approved by the Animal Ethics Committee of Lanzhou University Second Hospital. T24 cells transduced with shTRIP13 lentiviruses or shCtrl lentiviruses were incubated for 72 hours and then treated with 2 µg/ml puromycin (Clontech) for 96 hours. The puromycin-resistant cells were harvested and injected subcutaneously into the right dorsal flank of 4-week-old female BALB/c nude mice (n=10 per group). Two weeks after injection, the tumor diameters were measured twice each week for four weeks. The tumor volume (mm3) was calculated using the formula volume= (π/6) ×length× (width) 2, where the length and width were the longest and shortest diameters, respectively. Forty days after inoculation, fluorescent images of the tumor were obtained with the IVIS Lumina LT in vivo imaging system (Perkin Elmer, Chicago, IL, USA). Then, the mice were sacrificed, and the tumors were isolated for further analysis.
Mass spectrometry and coimmunoprecipitation
The total protein of T24 cells stably transduced with lentiviruses expressing Flag-TRIP13 or control viruses was extracted with RIPA buffer supplemented with a proteinase inhibitor and a phosphatase inhibitor. Lysates were incubated with anti-Flag magnetic beads (Sigma) at 4 °C overnight, and the immune complexes were eluted with Flag peptide (Sigma), precipitated and separated by SDS-PAGE. Candidate gel bands were excised and cut into 1-2 mm3 cubes. The cubes were destained with 50% acetonitrile /50 mM ammonium bicarbonate solution and 100% acetonitrile. A 25 mM ammonium bicarbonate solution modified with 12.5 ng/μl sequencing grade trypsin (Promega, Madison, WI, USA) was used for digestion of the cubes. The tryptic peptides were harvested with 50% acetonitrile/0.1% trifluoroacetic acid and subjected to protein identification using the EASY-nLC 1000 Liquid Chromatograph system (Thermo Fisher Scientific, Waltham, MA, USA) and an Orbitrap Fusion Mass Spectrometer (Thermo Fisher Scientific, San Jose, CA, USA). Proteins were identified by searching the data against the UniProtKB/Swiss-Prot database appended with decoy sequences using Proteome Discoverer software (version 1.4, Table S2 and Table S3). Whole-cell lysates and immunoprecipitates were immunoblotted with the anti-EGFR, anti-JAK2 anti-AKT1 and anti-Flag antibodies.
Microarray assay and bioinformatics analysis
The microarray experiments were performed at Shanghai Genechem Co., Ltd. Total RNA was isolated from three replicate samples of T24/shCtrl and T24/shTRIP13 cells using TRIzol Reagent (Thermo Fisher Scientific) and subjected to hybridization on the Affymetrix Human Gene Expression Array (Affymetrix, San Jose, CA, USA) according to the manufacturer's instructions. Differentially expressed genes and additional details are available at NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE109029. Data analyses, including the canonical pathways and putative network analysis, were performed using the Ingenuity Pathway Analysis tool (www.ingenuity.com). Gene Ontology enrichment analysis was performed using iPathwayGuide (https://www.advaitabio.com/).
The BCa datasets from the Oncomine database (https://www.oncomine.org/) used for the analysis were Lee Bladder [12] and Sanchez-Carbayo Bladder [13].
To identify the prognostic role of TRIP13 and EGFR in BCa, the limma R package (v 3.2.3) was used to extract the clinical and microarray gene expression data from the BCa dataset GSE13507, available in the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). The cohort was divided by the median value of gene expression.
Statistical analysis
Data analyses were performed using SPSS 19.0 (SPSS Inc., Chicago, IL, USA). A two-tailed Student's t-test was used to compare mean values. Categorical data were analyzed using either the Fisher exact test or the χ2 test. Survival analysis was performed using the Kaplan-Meier method and the log-rank test. Cox proportional hazard regression analysis was used to calculate the relative risk ratios. P < 0.05 was considered statistically significant.
Supplementary Material
Supplementary figure, tables 1-2 and table legends.
Supplementary table 3.
Abbreviations
TRIP13: thyroid hormone receptor interactor 13.
BCa: bladder cancer.
NMIBC: non-muscle-invasive bladder cancer.
MIBC: muscle-invasive bladder cancer.
EGFR: epidermal growth factor receptor.
AAA+: ATPases associated with diverse cellular activities.
TMA: tissue microarray.
STR: short tandem repeat.
GEO: Gene Expression Omnibus.
TCGA: The Cancer Genome Atlas.
IHC: immunohistochemistry.
AJCC: American Joint Committee on Cancer.
OS: overall survival.
DSS: disease-specific survival.
EMT: epithelial-mesenchymal transition.
IPA: ingenuity pathway analysis.
ID1: inhibitor of DNA binding 1.
Cox-2: cyclooxygenase-2.
MAD2: mitotic arrest deficient 2.
qPCR: quantitative real-time polymerase chain reaction.
Acknowledgements
We are grateful to Jianzhong Lu, Yan Tao, Shengjun Fu, and Baoliang Ma for their excellent technical assistance. We would also like to acknowledge the TCGA (https://cancergenome.nih.gov/), Oncomine (https://www.oncomine.org/) and Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) databases for the related data available for free use.
This work was supported by the National Natural Science Foundation of China (Grants: 81672519, 8137273 and 81172437), the Natural Science Foundation Project of Gansu Province (Grant: 1606RJZA044) and the Cuiying Scientific and Technological Innovation Program of Lanzhou University Second Hospital (Grant: CY2017-QN15).
Authorship
Z.W., Y.G. (Yanjun Gao) and Q.G. conceived of and designed the experiments. Y.G. (Yanjun Gao), S.L., Q.G., S.Z., Y.Z., T.L., Y.G. (Yuwen Gong), Y.W. and T.Z. performed the experiments. Y.G. (Yanjun Gao), S.L., H.W., L.X. and T.L. analyzed the data. Y.G. (Yanjun Gao), L.X., and Q.G. wrote the manuscript. D.B., W.H.C., R.R., Z.D. and Z.W. critically revised the manuscript.
Proteomics data are available in the iProX database (http://www.iprox.org) under accession number IPX0001274000.
The microarray datasets are available at the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) database under accession numbers GSE109029and GSE13507.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Svatek RS, Hollenbeck BK, Holmang S. et al. The economics of bladder cancer: costs and considerations of caring for this disease. European urology. 2014;66:253-62
2. Netto GJ. Molecular biomarkers in urothelial carcinoma of the bladder: are we there yet? Nature reviews Urology. 2011;9:41-51
3. Kaplan AL, Litwin MS & Chamie K. The future of bladder cancer care in the USA. Nature reviews Urology. 2014;11:59-62
4. Li XC & Schimenti JC. Mouse pachytene checkpoint 2 (trip13) is required for completing meiotic recombination but not synapsis. PLoS genetics. 2007;3:e130
5. Vader G. Pch2(TRIP13): controlling cell division through regulation of HORMA domains. Chromosoma. 2015;124:333-9
6. Ma HT & Poon RY. TRIP13 Regulates Both the Activation and Inactivation of the Spindle-Assembly Checkpoint. Cell reports. 2016;14:1086-99
7. Banerjee R, Russo N, Liu M. et al. TRIP13 promotes error-prone nonhomologous end joining and induces chemoresistance in head and neck cancer. Nature communications. 2014;5:4527
8. Zhang Y, Xue Q, Pan G. et al. Integrated Analysis of Genome-Wide Copy Number Alterations and Gene Expression Profiling of Lung Cancer in Xuanwei, China. PloS one. 2017;12:e0169098
9. Dazhi W, Mengxi Z, Fufeng C. et al. Elevated expression of thyroid hormone receptor-interacting protein 13 drives tumorigenesis and affects clinical outcome. Biomark Med. 2017;11:19-31
10. Maurizio E, Wisniewski JR, Ciani Y. et al. Translating Proteomic Into Functional Data: An High Mobility Group A1 (HMGA1) Proteomic Signature Has Prognostic Value in Breast Cancer. Mol Cell Proteomics. 2016;15:109-23
11. Yost S, de Wolf B, Hanks S. et al. Biallelic TRIP13 mutations predispose to Wilms tumor and chromosome missegregation. Nat Genet. 2017;49:1148-1151
12. Kim WJ, Kim EJ, Kim SK. et al. Predictive value of progression-related gene classifier in primary non-muscle invasive bladder cancer. Mol Cancer. 2010;9:3
13. Sanchez-Carbayo M, Socci ND, Lozano J. et al. Defining molecular profiles of poor outcome in patients with invasive bladder cancer using oligonucleotide microarrays. J Clin Oncol. 2006;24:778-89
14. Stein JP, Lieskovsky G, Cote R. et al. Radical cystectomy in the treatment of invasive bladder cancer: long-term results in 1,054 patients. J Clin Oncol. 2001;19:666-75
15. Kaufman DS, Shipley WU & Feldman AS. Bladder cancer. Lancet. 2009;374:239-49
16. Qureshi KN, Naguib RN, Hamdy FC. et al. Neural network analysis of clinicopathological and molecular markers in bladder cancer. The Journal of urology. 2000;163:630-3
17. Shariat SF, Zippe C, Ludecke G. et al. Nomograms including nuclear matrix protein 22 for prediction of disease recurrence and progression in patients with Ta, T1 or CIS transitional cell carcinoma of the bladder. The Journal of urology. 2005;173:1518-25
18. van Rhijn BW, Vis AN, van der Kwast TH. et al. Molecular grading of urothelial cell carcinoma with fibroblast growth factor receptor 3 and MIB-1 is superior to pathologic grade for the prediction of clinical outcome. J Clin Oncol. 2003;21:1912-21
19. Sarkis AS, Dalbagni G, Cordon-Cardo C. et al. Nuclear overexpression of p53 protein in transitional cell bladder carcinoma: a marker for disease progression. J Natl Cancer Inst. 1993;85:53-9
20. Ma HT & Poon RYC. TRIP13 Functions in the Establishment of the Spindle Assembly Checkpoint by Replenishing O-MAD2. Cell reports. 2018;22:1439-1450
21. Ye Q, Rosenberg SC, Moeller A. et al. TRIP13 is a protein-remodeling AAA+ ATPase that catalyzes MAD2 conformation switching. eLife. 2015:4
22. Choi JW, Kim Y, Lee JH. et al. High expression of spindle assembly checkpoint proteins CDC20 and MAD2 is associated with poor prognosis in urothelial bladder cancer. Virchows Arch. 2013;463:681-7
23. Marks DH, Thomas R, Chin Y. et al. Mad2 Overexpression Uncovers a Critical Role for TRIP13 in Mitotic Exit. Cell reports. 2017;19:1832-1845
24. Miyamoto H, Kubota Y, Noguchi S. et al. C-ERBB-2 gene amplification as a prognostic marker in human bladder cancer. Urology. 2000;55:679-83
25. Latif Z, Watters AD, Dunn I. et al. HER2/neu gene amplification and protein overexpression in G3 pT2 transitional cell carcinoma of the bladder: a role for anti-HER2 therapy? Eur J Cancer. 2004;40:56-63
26. Bryan RT, Regan HL, Pirrie SJ. et al. Protein shedding in urothelial bladder cancer: prognostic implications of soluble urinary EGFR and EpCAM. Br J Cancer. 2015;112:1052-8
27. Bonner JA, Harari PM, Giralt J. et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567-78
28. Ji H, Sharpless NE & Wong KK. EGFR targeted therapy: view from biological standpoint. Cell cycle. 2006;5:2072-6
29. van Kessel KE, Zuiverloon TC, Alberts AR. et al. Targeted therapies in bladder cancer: an overview of in vivo research. Nature reviews Urology. 2015;12:681-94
30. Lerner SP, McConkey DJ, Hoadley KA. et al. Bladder Cancer Molecular Taxonomy: Summary from a Consensus Meeting. Bladder cancer (Amsterdam, Netherlands). 2016;2:37-47
31. Choi W, Porten S, Kim S. et al. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25:152-65
32. Volkmer JP, Sahoo D, Chin RK. et al. Three differentiation states risk-stratify bladder cancer into distinct subtypes. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:2078-83
33. Rebouissou S, Bernard-Pierrot I, de Reynies A. et al. EGFR as a potential therapeutic target for a subset of muscle-invasive bladder cancers presenting a basal-like phenotype. Sci Transl Med. 2014;6:244ra91
34. Walter KR, Goodman ML, Singhal H. et al. Interferon-Stimulated Genes Are Transcriptionally Repressed by PR in Breast Cancer. Mol Cancer Res. 2017;15:1331-1340
Author contact
![]() Corresponding authors: Zhiping Wang; Phone: +86-0931-8942498; E-mail: gaoyj13edu.cn and Zhilong Dong; Phone: +86-13919780635; Email: 562691313com
Corresponding authors: Zhiping Wang; Phone: +86-0931-8942498; E-mail: gaoyj13edu.cn and Zhilong Dong; Phone: +86-13919780635; Email: 562691313com