ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
1. Introduction
2. LLPS in leukemia and myeloma
3. LLPS in solid cancers
4. LLPS and potentially...
5. Approaches that target...
6. Discussion
Abbreviations
Acknowledgements
References

Int J Biol Sci 2023; 19(13):4139-4156. doi:10.7150/ijbs.81521 This issue Cite
Review
Liquid‒liquid phase separation: roles and implications in future cancer treatment
Zheran Liu1*, Zijian Qin1*, Yingtong Liu2*, Xi Xia3, Ling He1, Na Chen4, Xiaolin Hu5 ![]() , Xingchen Peng1
, Xingchen Peng1 ![]()
1. Department of Biotherapy and National Clinical Research Center for Geriatrics, Cancer Center, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China.
2. Chengdu University of Traditional Chinese Medicine, Chengdu 610041, Sichuan, China.
3. Shanghai ETERN Biopharma Co., Ltd., Shanghai, China.
4. School of Pharmacy, Chengdu Medical College, Xindu Avenue No 783, Chengdu, 610500, Sichuan Province, China.
5. West China School of Nursing, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China.
* Zheran Liu, Zijian Qin and Yingtong Liu contributed equally to this work.
Abstract

Liquid‒liquid phase separation (LLPS) is a phenomenon driven by weak interactions between biomolecules, such as proteins and nucleic acids, that leads to the formation of distinct liquid-like condensates. Through LLPS, membraneless condensates are formed, selectively concentrating specific proteins while excluding other molecules to maintain normal cellular functions. Emerging evidence shows that cancer-related mutations cause aberrant condensate assembly, resulting in disrupted signal transduction, impaired DNA repair, and abnormal chromatin organization and eventually contributing to tumorigenesis. The objective of this review is to summarize recent advancements in understanding the potential implications of LLPS in the contexts of cancer progression and therapeutic interventions. By interfering with LLPS, it may be possible to restore normal cellular processes and inhibit tumor progression. The underlying mechanisms and potential drug targets associated with LLPS in cancer are discussed, shedding light on promising opportunities for novel therapeutic interventions.
Keywords: liquid‒liquid phase separation, biomolecular condensates, hematologic neoplasms, solid tumors
1. Introduction
Cancer development is an intricate process driven by randomly occurring mutations and genetic alterations. The complexities of cancer biology involve various factors, such as dysregulated signaling pathways leading to excessive cell proliferation, mechanisms that confer resistance to cell death, evasion of growth suppression mechanisms, sustained replicative potential, promotion of angiogenesis, and cellular invasive properties responsible for tissue invasion and metastasis (1,2). Despite the substantial progress in identifying the primary drivers of genetic mutations in cancer development, the precise pathological mechanisms underlying these processes are incompletely elucidated (3).
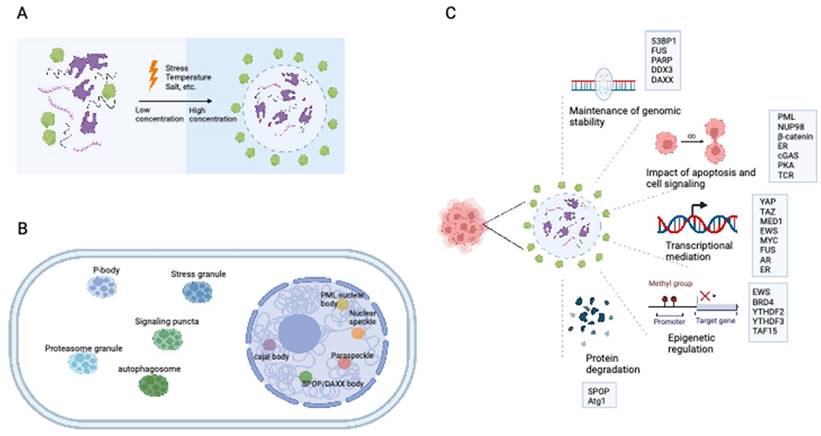
Eukaryotic cells are equipped with organelles that generate distinct chemical microenvironments to facilitate various biological processes. Through spatial segregation, various proteins and macromolecules are compartmentalized within subcellular structures, allowing the precise spatial and temporal control of complex biochemical reactions (4). In addition to membrane-bound organelles, membraneless structures, such as stress granules (SGs), Cajal bodies, and promyelocytic leukemia (PML) nuclear bodies (NBs),are essential for maintaining the normal functionality of eukaryotic cells. Liquid‒liquid phase separation (LLPS) is a cellular phenomenon characterized by the spontaneous segregation of macromolecules into dense and dilute phases, resulting in the formation of biomolecular condensates (Figure 1A) (5,6). These condensates generate a heterogeneous cellular environment, selectively enriching proteins and nucleic acids. They possess unique properties that facilitate biomolecule concentration and organization (Figure 1B) (7,8).
The establishment of interaction networks involving multivalent proteins or nucleic acids is crucial for LLPS and is facilitated primarily by peptides with folded modular domains, intrinsically disordered regions (IDRs), or polymerizing domains (9). Cancer-associated proteins and mutations can modulate the abundance and formation of condensates by impacting LLPS, thereby driving aberrant cellular activities and promoting tumorigenesis (Figure 1C) (10-13). This review presents an overview of the impact of LLPS on the development and progression of diverse cancer types, including hematological malignancies and solid tumors (Table 1). Furthermore, we discuss potential therapeutic approaches targeting LLPS that could be employed in cancer treatment strategies.
The formation of biomolecular condensates through LLPS. (A) The process of LLPS. Weakly multivalent interaction exists among scaffold proteins or between proteins and nucleic acids. External or internal stress can lead to LLPS. Biomolecules and their intercalating substrates maintain high concentrations in condensates while other proteins or substrates are excluded. (B) Nuclear and cytoplasmic condensates in eukaryotic cells. (C) Cellular functions and proteins that related to LLPS in cancer cells. IDR: intrinsically disordered regions; LLPS: liquid-liquid phase separation. AR: androgen receptor; ER: Estrogen receptor.
2. LLPS in leukemia and myeloma
2.1 NUcleoPorin 98 (NUP98) fusion protein and acute myeloid leukemia (AML)
LLPS is a pivotal molecular alteration that promotes tumorigenicity. Recurrent gene fusions involving IDRs and chromatin-binding proteins are commonly detected in various cancer types, notably, leukemia and sarcoma (14-16).
NUP98 is a constituent of the nuclear pore complex that facilitates macromolecule transport between the nucleus and cytoplasm (17). The discovery of the NUP98-HOXA9 fusion in AML patients with the t(7;11)(p15;p15) translocation marked the first identification of NUP98 rearrangement. Since that discovery, more than 31 distinct fusion partner genes of NUP98 have been identified in various hematological malignancies, including T-cell acute lymphoblastic leukemia (18), chronic myeloid leukemia (CML) (19), and AML (20).
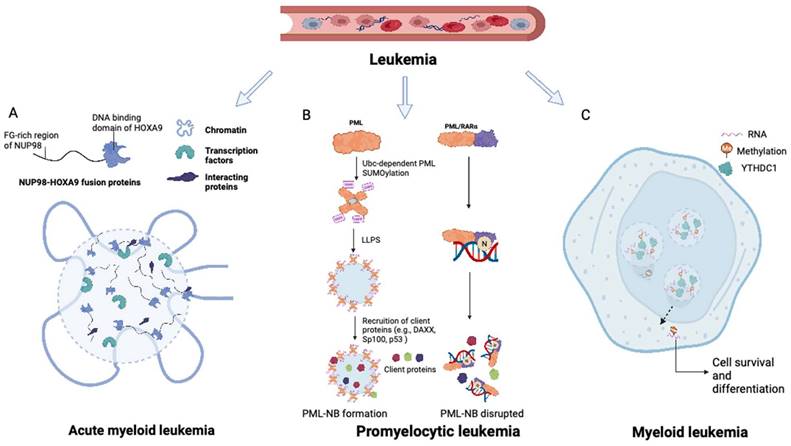
The intrinsically disordered FG domain within the NUP98 IDR-containing N-terminus has been shown to undergo spontaneous phase separation, forming FG particles (Figure 2A). These dense molecules repel inert macromolecules while allowing the entry of nuclear transport receptors carrying necessary cargo (21). NUP98 fusion proteins induce substantial modifications in the composition of biomolecular condensates, exhibiting characteristics distinct from those of native NUP98. Through associations with various cofactors, NUP98 fusion proteins modulate transcriptional changes. The FG domain of the NUP98 fusion protein is important for its specific subcellular localization and the induction of downstream leukemia-associated gene expression through LLPS (22). This process is essential for establishing a temporospatial subcellular environment, promoting chromatin remodeling, and altering gene expression patterns. Nuclear puncta containing multiple NUP98 fusion proteins (e.g., NUP98-NSD1, NUP98-HOXA9, and NUP98-HD) have been observed (23-25). It can thus be inferred that NUP98 fusion proteins associate with transcriptional cofactors in biomolecular condensates, which function as transcription centers to promote aberrant transcriptional programs (26).
Ahn et al. conducted a study demonstrating the essential LLPS-related role of IDRs in NUP98-HOXA9 fusion proteins (27). These authors elucidated the role of NUP98-HOXA9 phase separation in enhancing the ability of chimeric transcription factors to interact with and bind to specific chromatin domains, thus enabling long-range interactions between oncogene promoters and enhancer elements (27). Chandra et al. further showed that LLPS could drive the formation of NUP98-HOX9 nuclear puncta. The intermolecular interactions in the FG-repeat domain of NUP98-HOX9 and the heterotypic DNA binding regulated by the homeodomain of HOX9 are the driving forces of LLPS in vitro (28). The LLPS dynamics of NUP98-HOXA9 exhibited a direct correlation with the upregulation of genes associated with cell transformation and leukemogenesis (29). Similarly, fusion proteins such as NUP98-PRRX1, NUP98-KDM5A, and NUP98-LNP1 were observed to form nuclear condensates and induce the transformation of hematopoietic stem cells (28).
In summary, NUP98 fusion proteins play a leukemogenic role in hematopoietic cells, partially through LLPS. NUP98 fusion proteins are recognized as significant risk factors for leukemia. Thus, targeting NUP98 fusion proteins and NUP98-mediated LLPS constitutes a promising therapeutic approach for AML. Heikamp et al. demonstrated that inhibiting the menin-MLL1 protein complex using VTP50469 could inhibit leukemogenesis in NUP98-rearranged leukemia models by disrupting interactions between NUP98 fusion proteins and chromatin. Additionally, administration of VTP50469 resulted in improved survival outcomes in mice with NUP98-rearranged leukemias (29), highlighting the potential of VTP50469 as a targeted agent for the treatment of patients with NUP98-rearranged leukemias.
Main LLPS-related biomolecules and their roles in different cancer types.
| Cancer type | Biomolecule involved in LLPS | Related condensates | Biological role of biomolecules | Reference |
|---|---|---|---|---|
| Leukemia and myeloma | ||||
| Myeloid leukemia | NUP98 fusion proteins | Signaling puncta | Promotion of aberrant transcriptional programs | (21,22) |
| YTHDC1 | SEs | Suppression of myeloid leukemic differentiation | (63,64) | |
| PML | PML-RARα fusion proteins | PML NBs | Suppression of oncogenic pathways | (32,44) |
| T-cell acute lymphoblastic leukemia | TAL1 | SEs | Activation of oncogenic transcriptional programs | (153) |
| CML | BCR-ABL fusions | SGs | Inhibition of BCRA1 mRNA translation and recruitment of downstream signaling proteins | (57,58) |
| Myeloma | KRAS | SGs | Increased formation of SGs | (59) |
| Solid cancers | ||||
| Ewing sarcoma | EWS-FLI1 fusions | Transcription condensates | Activation of oncogenic transcriptional programs | (67-69) |
| Liposarcoma | FET family members | Transcription condensates | Activation of oncogenic transcriptional programs | (71) |
| Synovial sarcoma | SS18 | Transcription condensates | Activation of oncogenic transcriptional programs | (72) |
| Prostate cancer | SPOP and DAXX | Nuclear speckles | Degradation of oncoproteins | (76) |
| AR and MED1 | SEs | Androgen resistance | (79,81,82) | |
| Pancreatic cancer | KRAS | SGs | Enhancement of tumor cell adaptability through increased SG formation | (88,89) |
| Breast cancer | MED1 and BRD4 | SEs | Activation of oncogenic transcriptional programs; tamoxifen resistance | (94,95) |
| Lung cancer | EML4-ALK fusion | Signaling puncta | Initiation of lung tumorigenesis | (99,101) |
| Liver cancer | G6PC | Glycogen compartments | Activation of YAP signaling pathways | (103) |
| Multiple cancers | YAP and TAZ | Transcription condensates | Activation of YAP signaling pathways; anti-PD-1 immunotherapy resistance | (109-111) |
| SHP2 | Signaling puncta | Stimulation of downstream MAPK and ERK1 signaling pathways | (121) | |
| 53BP1 | DNA repair foci | DNA damage repair | (125,128) | |
| PARP | DNA repair foci | DNA damage repair | (131) | |
| mTORC1 | SGs | Cancer cell survival | (154) | |
| CDK7, CDK12, CDK13 | Transcription condensates | Activation of oncogenic transcriptional programs | (155,156) | |
| NEAT1 | Nuclear paraspeckles | Chemoresistance | (157) |
Roles of LLPS in leukemia and myeloma. (A) NUP98-HOXA9 fusion protein forms transcription condensates and leads to the activation of leukemogenesis-related genes. (B) The formation of PML NB was interrupted in promyelocytic leukemia with PML-RARα fusions. The disruption of PML NB in APL is mainly caused by the neddylation of the RARα part at the PML-RARα fusions, promoting its DNA-binding competency and hampering the further LLPS process. (C) M6A-mRNA needs YTHDC1 to undergo LLPS and form YTHDC1-m6A condensates in the nucleus to keep cell survival and maintain the undifferentiated states. YTHDC1-m6A condensates accumulate in acute myeloid leukemia, while if YTHDC1 is depleted in the AML cells, cell death and differentiation are promoted. LLPS: liquid-liquid phase separation; PML: promyelocytic leukemia. NB: nuclear body.
2.2 PML NBs and PML
PML NBs are phase-separated, stress-sensitive nuclear condensates implicated in multiple cellular functions, such as transcriptional regulation, cell proliferation control, stem cell self-renewal maintenance, and protein modification (30,31). The PML protein is an important component of and scaffold for PML NBs (32). It contains a conserved N-terminal RING finger/B-box/coiled-coil (RBCC) domain that governs the assembly of PML NBs (33-35), while its C-terminus contains a SUMO-interacting motif (SIM) for binding sumoylated proteins (36). These modular domains and interaction motifs are essential for driving LLPS and the formation of PML NBs (37). Extensive evidence indicates the tumor-suppressive functions of PML NBs mediated through multiple mechanisms, including sequestering proteins, serving as hubs for posttranslational modifications (PTMs), and regulating protein interactions (38,39). Furthermore, PML NBs influence critical cellular pathways such as the P53 and AKT signaling and DNA damage repair pathways (38,39).
In acute promyelocytic leukemia (APL), the PML gene undergoes a chromosomal translocation with retinoic acid receptor-α (RARα), resulting in the formation of the PML-RARα fusion protein, which is centrally involved in the initiation and progression of APL (40). PML-RARα fusions are found in over 95% of APL patients (40). Fusion of the PML gene with RARα in APL has been associated with two primary breakpoints, specifically, between exons 3-4 and exons 6-7, as evidenced by previous studies (41-43). Aberrant LLPS of PML-RARα resulting from neddylation is responsible for the impairment of PML NB assembly in APL. The neddylation-mediated enhancement of DNA binding of PML-RARα to the RARα domain interferes with LLPS of the PML domain, thereby hindering the assembly of PML NBs (Figure 2B) (44).
Potential therapeutic strategies to restore PML NBs have been explored. Retinoic acid (RA) and arsenic trioxide therapies have shown promise in inducing the re-formation of PML NBs and restoration of LLPS; these therapies promote apoptosis through activation of p53, localization of HIRA, and degradation of unwanted proteins (45). Various synthetic molecules such as emodin, MLN4924, and TSA, as well as natural compounds such as H2O2, EGF, and SFN, have also been found to increase the PML protein abundance (46-48). Notably, the neddylation inhibitor MLN4924 has demonstrated efficacy in suppressing the development of APL cells.
Current research efforts are focused on identifying the most effective combination regimens that maximize anticancer activity by enhancing PML protein accumulation. These studies offer valuable insights into potential cancer treatments based on targeting LLPS and PML NBs.
2.3 SGs, CML and myeloma
SGs are cytoplasmic ribonucleoprotein granules that form during cellular stress. SGs are formed via LLPS through a complex network of protein‒RNA interactions with the G3BP1 protein as the central node (49). Various stressors, such as extreme temperature, oxidation, UV radiation, and endoplasmic reticulum or mitochondrial stress, induce the formation of SGs (50). However, the precise mechanisms by which SGs contribute to tumor development and assist cancer cells in coping with tumor-related stress remain incompletely understood (51,52).
CML is associated with chromosomal translocation between the ABL gene on chromosome 9 and the BCR gene on chromosome 22. This genetic abnormality results in the production of the BCR-ABL fusion protein, which leads to persistent activation of the tyrosine kinase activity of BCR-ABL and is the pathophysiological cause of CML. Previous studies have demonstrated the cytoplasmic localization of BCR-ABL; however, its specific subcellular localization remains unclear and disputed (53). Evidence suggests that BCR-ABL proteins are present in granule-like structures in myeloid and lymphoid cells (53,54). These BCR-ABL proteins are predicted to possess long IDRs, which endow weak multivalency and potentially facilitate phase separation and granule formation (55,56). Kashiwagi et al. revealed that BCR-ABL-positive granules were SGs (57). The kinase activity of ABL and the N-terminal region of BCR are crucial for the phase separation of BCR-ABL (57). The granules formed as a result of BCR-ABL fusion play a pivotal role in promoting leukemogenic activity by inhibiting the mRNA translation of the tumor suppressor gene BCRA1 and recruiting downstream signaling proteins (57,58). Consequently, targeting the process of ABL-BCR phase separation could constitute a novel therapeutic strategy for CML.
The receptor tyrosine kinase/RAS/MAP kinase (MAPK) pathway is an oncogenic pathway with widespread aberrancies in human cancers; indeed, 20%-30% of cancer patients harbor RAS (KRAS, HRAS, or NRAS) mutations. Approximately 20% of multiple myeloma patients harbor KRAS mutations, leading to activation of the MEK/ERK pathway. Qiang et al. discovered that mutant KRAS significantly increases SG formation in multiple myeloma cells through 15-deoxy-delta(12,14)-prostaglandin J(2) (15d-PGJ2) (59). Additionally, inhibition of cyclooxygenase-2 (COX2) enhances the sensitivity of KRAS-mutant multiple myeloma cells to bortezomib (59). Therefore, targeting the formation of SGs could constitute an effective approach for treating KRAS-mutant myeloma (59).
2.4 N6-methyladenosine (m6A) and myeloid leukemia
m6A is the predominant chemical modification detected on mRNAs and critically influences myeloid leukemogenesis (60,61). However, the precise impact of m6A on different oncogenic processes across cellular contexts remains unknown. YTH domain-containing proteins (YTHDF 1-3), which serve as m6A readers, are involved in mediating the effects of m6A (62). These proteins also possess an N-terminal prion-like low complexity domain (LCD), which is believed to be able to undergo LLPS (63). Ries et al. demonstrated that m6A-binding proteins such as YTHDF1, YTHDF2, and YTHDF3 undergo LLPS facilitated by mRNAs containing multiple m6A-modified bases, both in cells and in vitro. During LLPS, mRNAs with multiple m6A modifications act as multivalent scaffolds for m6A-binding proteins and simultaneously bring their LCDs into proximity, facilitating LLPS (64).
Cheng et al. further revealed that the m6A reader protein YTHDC1 undergoes LLPS by binding to m6A, forming nuclear YTHDC-m6A condensates in myeloid leukemia cells. The IDR domain and m6A-binding capability of YTHDC1 are essential for this LLPS, and the abundance of YTHDC-m6A condensates serves as the basis for acute myeloid leukemogenesis (Figure 2C) (65). Given the high prevalence of abnormal m6A modifications in leukemia, targeting m6A and the associated LLPS process may constitute a promising therapeutic approach for myeloid leukemia.
3. LLPS in solid cancers
3.1 LLPS of fusion proteins in sarcomas
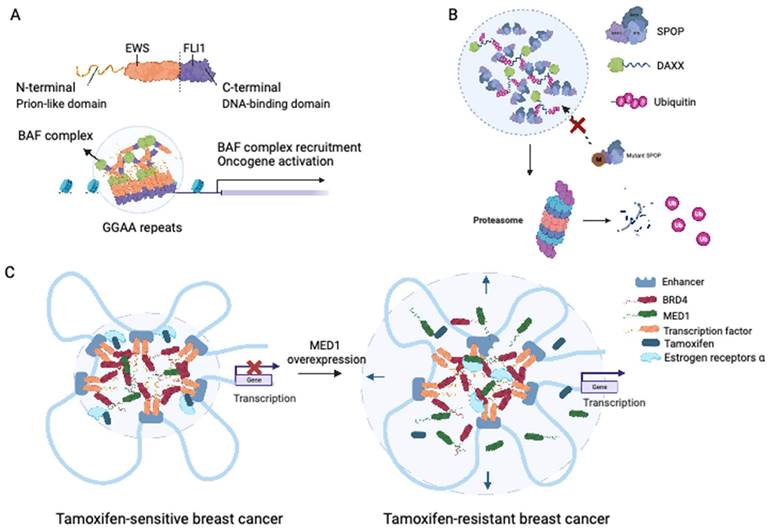
Ewing sarcoma is an aggressive malignancy that predominantly affects children and young adults and primarily involves bones and soft tissue. The presence of the EWS-FLI1 fusion protein, detected in approximately 85% of Ewing sarcoma cases, is considered a distinctive characteristic of this malignancy (66). The fusion protein responsible for Ewing sarcoma is generated by the fusion of the LCD of Ewing sarcoma RNA-binding protein 1 (EWSR1) with the transcription factor Friend leukemia virus integration 1 (FLI1) (67). Notably, fusions of the N-terminal prion-like domains of EWS to FLI1 have been implicated in facilitating protein aggregation through LLPS (68). Boulay et al. demonstrated that the interaction between EWS-FLI1 and BAF chromatin remodeling complexes is dependent on specific tyrosine residues within the prion-like domain of EWSR1 (69). LLPS-mediated recruitment of GGAA microsatellites to BAF complexes has been proposed as a mechanism that activates the aberrant transcriptional program driving Ewing sarcoma progression, encompassing processes such as DNA binding, BAF complex recruitment, enhancer activation, and target oncogene activation (69) (see Figure 3A).
In liposarcoma, chromosomal translocations often result in fusion events involving the prion-like domains of other proteins of the FET family and transcription factors (70). It is reasonable to speculate that fusion proteins associated with FUS and TAF15 may promote the formation of abnormal condensates at enhancer or promoter sites, leading to the activation of downstream tumorigenic transcriptional processes. Davis et al. demonstrated that the oncogenic fusion protein FUS-DDIT3 forms condensates through LLPS facilitated by its prion-like domain (71). These nuclear FUS-DDIT3 condensates recruit mammalian SWI/SNF complexes, thereby driving transcription through ectopic chromatin remodeling (71). The same study further indicated that other transcriptional regulators containing prion-like domains can interact with mSWI/SNF chromatin remodelers to orchestrate similar transcriptional programming (71).
Roles of LLPS in solid tumor. (A) EWS-FLI fusions acquire the ability of LLPS in Ewing's sarcoma. EWS-FLI1 fusion protein contains the prion-like domain on the N terminal and the DNA binding domain on the C terminal. The EWS-FLI1 fusion proteins undergo LLPS and form EWS-FLI1 condensates. These condensates then bind to the GGAA microsatellites of DNA, recruiting the BAF complex and acting as super-enhancers to activate oncogenic expression. BAF: BRG1-BRM-associated factor. (B) Mutant SPOP could not form biomolecular condensates in prostate cancer. Wild-type SPOP could self-associate to dimers, interact with the IDR domain of DAXX, and drive the LLPS. The formation of SPOP-DAXX condensates could accelerate the polyubiquitination of DAXX. For mutant SPOP in prostate cancer. For prostate cancers with mutations in the MATH region of SPOP, LLPS cannot proceed, and thus the ubiquitinated degradation of DAXX is terminated. (C) Sensitivity to tamoxifen in breast cancer patients is related to the magnitude of LLPS. In tamoxifen-sensitive breast cancer, MED1, BRD4, together with TFs and RNA polymerase II undergo LLPS and form transcriptional condensates at the site of super-enhancers. With the upregulation of MED1 expression, the volume of the condensate containing MED1 increases and relatively dilutes the tamoxifen concentration in the condensate. MED1: mediator complex subunit 1; BRD4: bromodomain-containing protein 4.
Given the notable involvement of FET family members in sarcomas, targeting the aberrant LLPS process mediated by FET fusion proteins could constitute a therapeutic approach. Disrupting the formation of abnormal condensates at enhancer or promoter sites holds promise for inhibiting downstream tumorigenic transcriptional processes facilitated by fusion proteins. This strategy may involve the development of small molecules or therapeutic agents designed to interfere with the LLPS process or disrupt interactions between fusion proteins and their associated complexes. Remarkably, recent studies by Ryan et al. demonstrated that engineered protein disaggregases, such as modified Hsp104 variants, can antagonize the accumulation of FUS-CHOP and EWS-FLI fusion protein condensates formed by LLPS, thus reducing the toxicity of these fusion proteins (68). These findings highlight the immense therapeutic potential of targeting the LLPS process in sarcoma.
Along with FET family members, LLPS has been shown to be involved in the oncogenicity of the SS18-SSX fusion protein. Phase separation of SS18 or SS18-SSX is initiated through self-association of their IDRs, which subsequently facilitates the recruitment of BRG1. Notably, the tyrosine residues within the QPGY domain are crucial for triggering this LLPS process. Disruptions in the LLPS of SS18-SSX or its interaction with BRG1 have been observed to impair the transformation of NIH3T3 cells, highlighting the potential of targeting this LLPS process as a promising therapeutic strategy (72).
3.2 Cancer-associated speckle-type pox virus and zinc finger (POZ) protein (SPOP) mutation disrupts protein degradation in prostate cancer
SPOP acts as a substrate adaptor for Cullin 3 RING E3 ubiquitin ligases, functioning as a tumor suppressor by facilitating the degradation of various oncoproteins. These oncoproteins include the androgen receptor (AR), BRD4, death-domain-associated protein (DAXX), PD-L1, and Myc (73). Through its involvement in the ubiquitination process, SPOP regulates various cellular activities, such as cellular metabolism, senescence, and hormone signaling (74). SPOP mutations are observed predominantly in prostate cancer (75). These cancer-associated mutations are frequently located in the MATH domain of SPOP, which is responsible for substrate binding. Consequently, these mutations disrupt the normal interaction with and degradation of substrates, leading to accumulation of the substrates (76,77).
Experimental evidence has demonstrated that SPOP exhibits self-association properties and can form oligomeric structures (78). Bouchard et al. observed the colocalization of SPOP and DAXX in liquid nuclear organelles, and in vitro, their binding triggered phase separation mediated by multiple weak SPOP-binding motifs in DAXX (79). Prostate cancer-associated mutations, such as W131F and F133V, within the MATH domain of SPOP have been found to disrupt the interaction between SPOP and DAXX. This disruption interferes with the phase separation process and inhibits substrate ubiquitination (Figure 3B) (79). These findings elucidate that substrate-directed LLPS may be a universal mechanism contributing to proteostasis and that mutations in SPOP can lead to dysregulated ubiquitination activity.
Bouchard et al. also observed that SPOP induces LLPS of AR (79), a critical transcription factor and the main oncogenic driver in prostate cancer. AR activation requires its interaction with the coactivator MED1, which contains IDRs and can undergo LLPS with itself or other transcription factors (80). Zhang et al. demonstrated that ARs are enriched in MED1-dependent condensates, promoting subsequent transcriptional regulation in prostate cancer cell models (81). The DNA-binding domain of full-length AR can bind RNA and drive phase separation in an RNA-dependent manner. However, in AR-v7 variants, the abundance of AR-rich condensates is diminished due to loss of the interaction between the ligand-binding domain and activation function 1 (AF1) of AR, resulting in impaired regulation of AR's transcriptional role in prostate cancer (82). Basu et al. developed a small molecule compound that covalently binds aromatic moieties to cysteines in the activation domain of AR and accumulates with LLPS of AR (83). This compound enhances the degradation of AR and suppresses AR-dependent transcriptional functions, exhibiting significant antitumor activity in prostate cancer models. Recently, through phase separation-based phenotypic screening, Jingjing Xie et al. identified a potential AR inhibitor, ET516, which selectively disrupts AR condensates and inhibits tumor growth in cells harboring resistance mutations in AR (84). These findings highlight the therapeutic potential of designing drugs that target the IDRs in oncogenic transcription factors to regulate their phase separation ability.
3.3 KRAS mutation upregulates SG formation in pancreatic cancer
KRAS mutation is a prevalent genetic alteration observed in pancreatic adenocarcinoma, lung adenocarcinoma, and colorectal cancer (85). Approximately 90% of pancreatic cancers harbor KRAS mutations, most commonly the G12D mutation (86,87). The study by Grabocka et al. provided the first evidence that KRAS mutation alone is sufficient to upregulate SG formation. SGs confer advantages on cancer cells by promoting tumorigenesis and progression, offering protection against tumor-related stress (88). The upregulation of SG formation induced by mutant KRAS is mediated by an increase in the level of 15d-PGJ2, a secreted molecule that acts in both cell-autonomous and non-cell-autonomous manners to modulate the expression of its targets (88). The increased abundance of SGs associated with KRAS mutation is linked to stress resistance properties in cancer cells; thus, SGs are potential biomarkers for tumor fitness and drug responses (88).
In pancreatic cancer cells carrying KRAS mutations, the activation of nuclear factor erythroid 2-related factor 2 (NRF2) leads to upregulation of glycolysis, increased glutamine uptake, and reprogrammed glutaminolysis (89). Mukhopadhyay et al. demonstrated that 15d-PGJ2 activates NRF2 and facilitates the formation of SGs. Activation of NRF2 by 15d-PGJ2 upregulates pathways involved in glutamine metabolism, thereby promoting chemoresistance in KRAS-mutant pancreatic cancers (90). Inhibitors of glutaminase, an enzyme involved in glutamine metabolism, can resensitize gemcitabine-resistant pancreatic cancer cells. Therefore, combination treatment with glutaminase inhibitors and chemotherapy holds promise as an effective therapeutic approach for patients with KRAS mutations.
3.4 LLPS of MED1 condensates in breast cancer
Estrogen receptor α (ERα)-positive breast cancer is the predominant subtype among hormone receptor-positive breast cancers (91). Treatment with tamoxifen, a selective modulator of ERs, is an effective therapeutic approach for managing ER-positive breast cancer in both pre- and postmenopausal individuals (92). However, the emergence of tamoxifen resistance presents a significant hurdle and is a primary contributor to mortality in breast cancer patients (93).
The IDRs of MED1 and BRD4 play a crucial role in the formation of transcriptional condensates, which concentrate the transcription machinery, at superenhancers (SEs) (94). Following estrogen stimulation, ERα becomes integrated into MED1 condensates (94). Klein et al. demonstrated that in breast cancer cells, ERα associates with MED1 and undergoes condensation in a tamoxifen-sensitive manner (95). Overexpression of MED1 has been linked to tamoxifen resistance and worse survival outcomes in breast cancer patients (96). Tamoxifen treatment drives the dissociation of ERα from MED1 condensates, as the drug selectively enters the condensates and competes with estrogen for binding (96). Upregulation of MED1 increases the volume of MED1-containing condensates, resulting in dilution of tamoxifen within the condensates and reducing the efficiency of ERα dissociation (Figure 3C) (96). These findings suggest that selective partitioning and concentration within condensates formed via LLPS may play a role in the pharmacodynamics of drugs and that alterations in these condensates could contribute to treatment resistance in breast cancer and other cancers.
3.5 LLPS of anaplastic lymphoma kinase (ALK) fusions in lung cancer
ALK fusions, particularly the EML4-ALK fusion, are recognized as oncogenic drivers in a subset of non-small cell lung cancers (97). The EML4-ALK fusion protein lacks a transmembrane domain and is localized primarily in the cytoplasm or microtubules (98). This fusion protein, constitutively active as a tyrosine kinase, plays a pivotal role in initiating lung tumorigenesis (99). While extensive studies have explored the oncogenic properties of EML4-ALK, the involvement of LLPS in regulating its function and contributing to lung cancer development has recently gained attention (100,101).
Recent studies by Qin et al. have demonstrated that EML4-ALK variant 1 can form liquid-like condensates in the cytoplasm, with the EML4 region of the fusion protein playing a crucial role in condensate formation (101). Truncation experiments have revealed that the EML4-N fragment alone is sufficient to drive phase separation, while the ALK-C fragment remains dispersed. Moreover, mutations in aromatic residues within the EML4 region significantly impair phase separation, underscoring the importance of these residues in this process. Functional characterization of EML4-ALK condensates has elucidated their role in activating downstream signaling pathways. Disruption of the LLPS process hampers the hyperactivation of key signaling molecules, such as AKT, ERK1/2, and STAT3, which are crucial for promoting cell survival, proliferation, and angiogenesis (101).
In summary, the emerging evidence of LLPS in ALK fusions, particularly EML4-ALK variant 1, emphasizes the significance of this cellular process in the pathogenesis of lung cancer. Understanding the role of LLPS in regulating the function of EML4-ALK and its downstream signaling pathways is expected to contribute to the development of novel therapeutic approaches for lung cancers with ALK fusions.
3.6 Glycogen accumulation and LLPS in liver carcinogenesis
Glycogen is the principal storage polysaccharide in hepatocytes, playing a vital role in liver metabolism and being linked to hepatocellular carcinoma development (102). Dysregulation of glycogen metabolism in premalignant liver lesions leads to impaired glycogen degradation and subsequent accumulation (103). The accumulated glycogen undergoes phase separation, leading to the formation of the Laforin-Mst1/2 complex. This complex sequesters the Hippo kinases Mst1/2 within liquid droplets composed of glycogen, thereby removing their inhibitory effect on Yes-associated protein (Yap) (103). Dysregulation of glycogen accumulation and phase separation has significant implications for liver tumorigenesis.
Research by Liu et al. has demonstrated that increased glycogen storage accelerates the development of liver tumors, while reducing the glycogen content decreases tumor incidence (103). Additionally, the interplay among glycogen accumulation, the Hippo signaling pathway, and downstream effectors such as Yap has been associated with liver enlargement and the formation of tumors (103).
These findings highlight the importance of glycogen accumulation and phase separation in liver tumorigenesis. Understanding the mechanisms underlying the dysregulation of glycogen metabolism and its impact on signaling pathways such as the Hippo and Yap pathways yields valuable insights into the development of hepatocellular carcinoma. Targeting these processes could offer new therapeutic strategies for liver cancer.
4. LLPS and potentially targetable proteins
4.1 LLPS, YAP, and transcriptional coactivator with PDZ-binding motif (TAZ)
LLPS plays an important role in the transcriptional programs regulated by YAP and TAZ, which are crucial regulators of the Hippo pathway (104). Activation of YAP and TAZ is involved in cancer cell survival and metastasis, tumor microenvironment remodeling, and immune evasion (105). However, in certain cases, these proteins have been found to exhibit tumor-suppressive activities in hematological malignancies and colorectal cancer (106-108).
Recent research has provided insights into the formation of YAP and TAZ condensates upon exposure to hyperosmotic conditions. The formation of YAP condensates, driven by the IDR transactivation domain, can occur in the nucleus and cytoplasm. These condensates contain TAZ and the transcription factor TEAD1, organized as SEs, and recruit RNA polymerase II to mediate the transcription of YAP target genes (109). Additionally, the interaction between YAP/TEAD and steroid receptor coactivator 1 (SRC-1) leads to the formation of SRC-1/YAP/TEAD condensates, which promote YAP transcription (110). Disruption of SRC-1 condensates by the anti-HIV drug elvitegravir could suppress the proliferation of YAP-dependent tumor cells, showing the potential of targeting phase-separated condensates as a therapeutic strategy (110) (Figure 4A).
In tumor cells, YAP condensates with formation driven by the coiled-coil domain and IDR are associated with anti-PD-1 immunotherapy resistance. These condensates act as transcriptional hubs in the tumor immune microenvironment, concentrating enzymes, transcription factors, and histone acetylases. Disruption of YAP condensates has been correlated with poor treatment outcomes and could be a potential target for combination treatment with immunotherapy (111).
Similar to YAP, TAZ also undergoes LLPS and forms nuclear condensates in vitro and in vivo (112). TAZ condensates concentrate TEAD4, BRD4, MED1, and CDK9, enhancing the efficiency of transcription and enabling the activation of TAZ-specific downstream pathways. Moreover, unlike YAP, TAZ can form condensates at various protein and salt concentrations and various temperatures, a phenomenon facilitated by its ability to homodimerize through its coiled-coil domain. The Hippo signaling pathway negatively regulates LLPS of TAZ by promoting its phosphorylation via LATS/NDR kinases (112). Furthermore, participation of the paraspeckle protein NONO is required for TAZ phase separation, as it interacts with TEAD and Rpb1. Notably, NONO plays an essential role in TAZ-associated tumorigenesis in glioma, and elevated expression of NONO is correlated with unfavorable survival outcomes in patients with glioblastoma (113).
4.2 LLPS and Src homology containing protein tyrosine phosphatase 2 (SHP2)
During cellular processes, protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) perform indispensable functions in controlling signaling cascades. SHP2 is a nonreceptor PTP encoded by PTPN11 that dephosphorylates targeted proteins (114). Functionally, SHP2 acts as a central hub connecting various subcellular signaling pathways involved in cancer, such as the RAS/MAPK and PD-1/PD-L1 pathways (115-118). SHP2 dysregulation is associated with several cancers, including breast cancer (115) and melanoma (119). Moreover, gain-of-function mutations in SHP2 are frequently observed in juvenile myelomonocytic leukemia and AML (120).
Disruption of LLPS by targeting related proteins. (A) Anti-HIV drug elvitegravir could bind to SRC-1 and prevent it from accessing the YAP/TEAD condensates. (B) SHP2 allosteric inhibitors moderate SHP2 mutant undergoing LLPS. Wild-type SHP2 has a well-folded PTP domain which is normally in a closed state. SHP2 with active or inactive mutants could transit the closed states of SHP2 conformation to open states. Mutant SHP2 could boost the activity of PTPase, drive the LLPS, and hyperactivate the Ras-ERK pathway. For SHP2 with active mutation, the high PTPase activity is acquired directly by the upregulated intrinsically PTPase activating, while for SHP2 with inactive mutation, it is mainly acquired from recruiting the wild-type SHP2. SHP2 allosteric inhibitors could stop the formation of SHP2 condensates by keeping SHP2 in closed states. EVG: elvitegravir; PTP: protein tyrosine phosphatase.
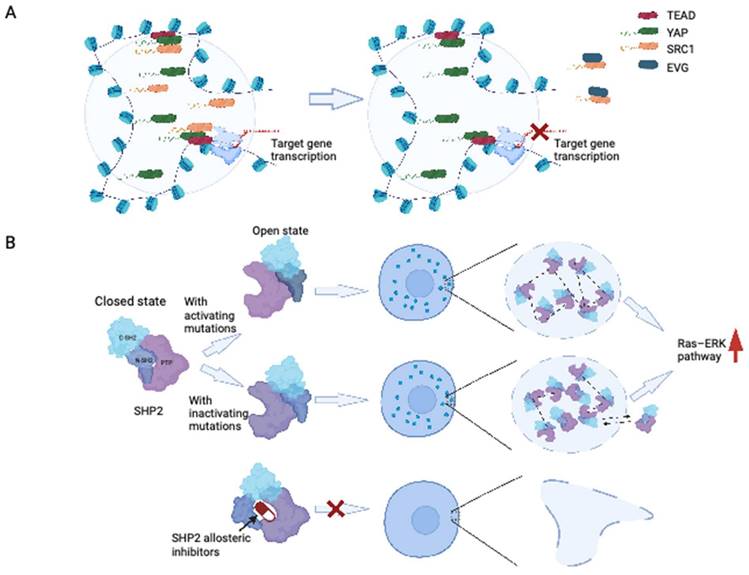
Recent investigations conducted by Zhu et al. provided important insights into the modulatory role of LLPS in PTP activity, with a specific emphasis on SHP2 (121). Researchers have demonstrated that wild-type SHP2 can form biomolecular condensates in response to specific factors such as EGF and FGF, indicating that LLPS may contribute to the regulation of SHP2 activity (120). Notably, cancer-associated SHP2 mutants have a higher propensity for LLPS. LLPS induced by SHP2 mutation represents a gain-of-function mechanism that promotes downstream MAPK signaling, leading to ERK1/2 activation (120). Importantly, Zhu et al. showed that SHP2 mutation-induced LLPS can be suppressed by allosteric inhibitors of SHP2 (120) (Figure 4B). These findings are particularly important considering that allosteric inhibitors of SHP2, including AB-3068, TNO155, RMC-4630, and RLY-1971, are currently under evaluation in clinical trials for solid cancers (114). Furthermore, Lin et al. have shown that LLPS could regulate the receptor tyrosine kinase (RTK) signaling through facilitating the formation of a signaling condensate comprising FGFR2, SHP2 and 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma 1 (PLCγ1) (122). Together, these studies highlight the therapeutic potential of targeting LLPS, particularly by inhibiting SHP2-mediated condensate formation.
4.3 LLPS and 53BP1
Genomic instability is a defining characteristic of cancer cells, and preservation of genomic integrity relies heavily on the functionality of DNA damage response pathways (123). A key regulator in this pathway is 53BP1, which acts as a scaffold and facilitates the DNA damage response (124). 53BP1 plays a role in binding to disrupted chromatin, recruiting other DNA double-strand break-responsive proteins, and promoting the synapsis of distal DNA ends during the repair process called nonhomologous end-joining (125). Furthermore, 53BP1 exerts direct regulatory control over the p53 tumor suppressor pathway, which plays a pivotal role in initiating cell cycle arrest, apoptosis, and the activation of other antitumorigenic signaling cascades (125).
Emerging research has substantiated the formation of DNA repair condensates facilitated by 53BP1 via LLPS upon encountering DNA damage, specifically DNA double-strand breaks (126,127). Formation of these condensates is enhanced by long noncoding RNAs that assemble at sites of DNA damage (126,127). The 53BP1 DNA repair condensates serve as platforms for recruiting p53 and its coactivators, leading to stabilization of p53 and facilitating its function (126). Disruption of 53BP1 condensates impairs downstream transcriptional induction and translation of p53 targets, indicating the necessity of these condensates for efficient DNA double-strand break repair (126). Furthermore, Ghodke et al. demonstrated that AHNAK, a component of the USP28-53BP1-p53-p21 network, can modulate G1/S checkpoint functions mediated by 53BP1 and p53 activation on chromatin. Depletion of AHNAK induces the accumulation of 53BP1 and intensifies LLPS, consequently enhancing the activation of the p53 response (128). These findings suggest that disrupting the assembly of 53BP1 condensates could dysregulate p53 and potentially impact the expression of downstream tumor suppressor genes.
Overall, the formation of 53BP1 condensates through LLPS is an important mechanism in DNA repair and p53-mediated cellular responses to DNA damage, and perturbations in these condensates can have significant implications for cancer development. Further exploration into the assembly and modulation of 53BP1 condensates may yield crucial insights for the discovery of novel therapeutic strategies aimed at selectively targeting DNA repair pathways in cancer.
4.4 LLPS and PARP family members
PARP1, a nuclear protein, is the first discovered member of the PARP protein family and is involved in poly(ADP-ribose) (PAR) polymerization (129). PARP1 functions as a writer of poly(ADP-ribosyl)ation (PARylation) by catalyzing the addition of ADP-ribose units to form negatively charged PAR chains on itself and other proteins, thereby regulating various pathways (130). PARP1 is indispensable for DNA repair mechanisms, stabilization of DNA replication forks, and alterations in chromatin structure (131). At loci undergoing DNA damage, PARP1 catalyzes the formation of extended PAR chains, thus initiating the assembly of DNA damage condensates through phase separation. This process is facilitated by the specific recruitment of FET proteins containing LCDs (132). Electrostatic interactions between PAR chains and LCDs promote phase separation, while aggregation-prone prion-like domains contribute to the amplification of this response, facilitating DNA repair processes (132).
The advent of PARP inhibitors constituted a substantial breakthrough in anticancer therapeutics, particularly for tumors characterized by impaired homologous recombination repair, including breast, ovarian, pancreatic, and prostate cancers carrying BRCA1/2 mutations (133). However, considering the functional complexity of PARP1, additional drugs that target the phase separation process and DNA damage repair mechanisms mediated by PARP1 could be developed. Further research in this area may provide insights into novel therapeutic strategies for cancer. By understanding and manipulating LLPS of PARP1, it might be possible to increase the effectiveness of PARP inhibitors or develop alternative approaches to target the DNA repair machinery in cancer cells.
5. Approaches that target proteins via LLPS
Traditional structure-based drug design relies on well-defined protein structures and pockets, which many oncoproteins, including transcription factors, may not have. Indeed, these proteins often lack folded structures and do not possess clearly defined pockets for drug binding. This limitation poses a substantial challenge in the development of therapies directed against these "undruggable" targets. However, the discovery of LLPS has provided new opportunities for targeting these proteins (Table 2).
5.1 Targeting IDRs
Targeting IDRs has emerged as a promising approach for disrupting LLPS and its associated pathogenic condensates. As previously described, IDRs are prevalent in proteins, especially oncoproteins and transcription factors that lack well-defined structures. These IDRs play critical roles in LLPS by mediating interactions between proteins and nucleic acids, contributing to the formation of biomolecular condensates (134).
Small molecule inhibitors that specifically bind to IDRs have shown potential in disrupting LLPS and modulating condensate formation. One approach to target IDRs is the use of small molecule inhibitors. These inhibitors are designed to bind specifically to the IDRs, disrupting their interactions and interfering with the assembly of condensates. For example, to target c-MYC, a well-known oncoprotein, highly specific small molecule inhibitors such as IIA4B20, IIA6B17, and mycmycin-1/2 have been developed. These inhibitors effectively target the IDRs of c-MYC, inhibiting its oncogenic activity and suppressing malignant cell transformation (134,135).
Strategies targeting LLPS and representative drugs.
| Targeting strategy | Representative drugs | Drug targets | Goal of drug targeting | Reference |
|---|---|---|---|---|
| Modification of condensates | Olaparib | PARP-1; members of the ADP-ribosyltransferase family | Suppression of DNA repair by inhibiting FUS condensate enrichment in regions of DNA damage | (140) |
| Chemical inhibitors of BET bromodomains (e.g., JQ1 and IBET) | BET protein BRD4 | Inhibition of gene-specific transcriptional activation by releasing the Mediator complex from SEs. | (158) | |
| Alteration of the drug partitioning process | Cisplatin; mitoxantrone; tamoxifen | SEs | Modulation of the characteristics of condensates and influencing drug concentration and efficacy through targeted localization within SE condensates to enhance therapeutic effectiveness | (95) |
| Targeting of IDRs | YK-4-279, a derivative of the lead compound | Interaction between EWS-FLI1 and RNA helicase A | Inhibiting the proliferation of Ewing sarcoma by induction of apoptosis in tumor cells | (159) |
| Tin(IV) oxochloride-derived cluster | IDR within the TAF2 subunit of TFIID | Specifically, disrupts transcription initiation by selectively impairing the function of TFIID | (136) | |
| PRIMA-1; ReACp53 | p53 mutants | Induction of cell cycle arrest in cancer cells with mutant p53 by restoring the native conformation of aggregated mutant p53 proteins | (160,161) | |
| IIA4B20; IIA6B17 | Transcription factor Myc | Neutralization of the oncogenic effects of Myc by disrupting Myc/Max dimerization | (135,162) | |
| Elvitegravir, an anti-HIV drug | SRC-1, a transcriptional coactivator for nuclear hormone receptors | Suppression of YAP transcriptional activity in cancer cells through inhibition of phase separation of SRC-1 condensates | (163) | |
| PCG | IDR of BRD4 | Suppression of BRD4-dependent gene transcription | (137) | |
| Dissolution of condensate | Sodium arsenate; vinblastine | Microtubules | Disruption of SG formation via inhibition of SG protein transport along microtubules | (164) |
| Allosteric inhibitors of SHP2 | SHP2 mutants | Suppression of the RAS-MAPK pathway via inhibition of LLPS of SHP2 mutants | (119,121) | |
| 4,4'-Dianilino-1,1'-binaphthyl-5,5'-disulfonic acid (bis-ANS) | LLPS of the TDP-43 LCD | Modulation of dysregulated LLPS: high concentrations inhibit TDP-43 condensate assembly, whereas low concentrations facilitate the formation of liquid droplets. | (165) | |
| C108 | GTPase-activating protein (SH3 domain)-binding protein 2, G3BP2 | Interference with breast tumor progression via modulation of SART3 mRNA regulation. | (166) |
The TFIID transcription complex is a critical component of the transcription initiation machinery in eukaryotes. Within this complex, the TAF2 subunit contains an IDR that plays a regulatory role in transcriptional activation. In an important discovery, a tin-based metal cluster was identified as a selective inhibitor that binds specifically to this IDR within the TAF2 subunit of TFIID. By targeting the IDR, the metal cluster acts as a selective inhibitor, modulating the activity of TFIID and subsequently impacting transcription initiation processes (136).
Recently, a noteworthy compound called PCG, derived from a natural source, has emerged as a potential regulator of LLPS. PCG has been shown to effectively convert phase-separated BRD4 into stable aggregates both in vivo and in vitro by specifically targeting the IDR of BRD4 (137). This targeted action of PCG results in the suppression of BRD4-dependent gene transcription and thus opens a promising avenue for therapeutic intervention.
Another approach to target IDRs is through peptide-based inhibitors. Peptides such as ReACp53 and polyarginine analogs have shown promise in blocking amyloid formation by p53 mutants. These peptides directly interact with the IDRs of p53 mutants, preventing their aggregation and restoring their tumor-suppressive function in cancer cells (138).
5.2 Targeting the modification of condensates
The regulation of LLPS dynamics is notably influenced by PTMs, modifications occurring after protein translation. Exploiting these PTMs as intervention targets holds considerable promise. The effectiveness of olaparib, a small molecule inhibitor of PARP1/2, has been demonstrated in terms of its ability to inhibit the formation of condensates involved in PARylation-related DNA repair and disrupt the DNA damage response. By inhibiting PARP activity, these inhibitors modulate the PTMs involved in condensate assembly, thereby impacting their functions and cellular outcomes (139,140). Furthermore, by inhibiting PARP1/2 activity, PARP inhibitors disrupt the PARylation process and alter the dynamics of LLPS. PARylation functions to generate a scaffold to facilitate the recruitment of proteins and nucleic acids to condensates, thereby promoting LLPS. Inhibition of PARP1/2 causes a reduction in the PAR level, affecting the assembly and stability of condensates formed through LLPS (141).
Kinases involved in LLPS regulation are another intervention target. For example, DYRK3, a kinase involved in LLPS, can drive the dissolution of SGs to release mTORC1. By inhibiting DYRK3 activity with GSK-626616, the recondensation process can be promoted in cells. Thus, the disrupted SGs can re-form, leading to sequestration of RNA and proteins within the recondensed granules. Additionally, DYRK3 inhibitors can effectively suppress mTORC1 signaling by preventing the release of mTORC1 from dissolved SGs (142).
Furthermore, the small molecule inhibitor JQ1 has gained considerable attention for its ability to target LLPS. JQ1 acts primarily on BET proteins, particularly BRD4. By binding to the bromodomain of BRD4, JQ1 disrupts the interaction between BRD4 and acetylated lysine residues, thereby interfering with its recruitment to hyperacetylated promoter and enhancer regions (143). Moreover, BRD4 can physically associate with the Mediator complex, and the application of JQ1 can lead to rapid release of Mediator. This dissociation of Mediator from chromatin is strongly correlated with repressed transcription of nearby genes. Notably, these genes exhibit significant enrichment as targets of the MYB transcription factor, a key regulator of leukemogenesis, as well as for functions associated with the development and progression of leukemia (144).
Targeting modifications of condensates is a valuable strategy to modulate their assembly, stability, and function. By specifically interfering with the PTMs involved in LLPS, these approaches can regulate condensate behavior and cellular outcomes. However, it is crucial to consider the specificity and selectivity of these inhibitors to minimize off-target effects and potential disruption of normal cellular processes.
5.3 Targeting the drug partitioning process
In traditional approaches, small molecule inhibitors disrupt LLPS by directly interacting with the components of condensates. However, an alternative strategy focuses on the process of drug partitioning into phase-separated condensates, which changes their physicochemical properties and functional outcomes. As Klein et al. reported, several antineoplastic compounds, such as cisplatin, mitoxantrone, tamoxifen, THZ1, and JQ1, can become highly concentrated into biomolecular condensates (e.g., MED1 condensates), which influences their therapeutic activity. This selective partitioning into SGs may occur even in the absence of a compound's targets (95). This group further found that molecules with aromatic rings are more likely to accumulate within MED1 condensates, suggesting that the physicochemical properties of small molecules, such as pi-pi or pi-cation interactions, contribute to their selective partitioning into MED1 condensates (95). In addition, cisplatin treatment was found to induce gradual and specific disruption of MED1 condensates, thus clarifying the mechanisms through which platinum drugs effectively target tumor cells strongly dependent on SE-driven oncogene expression.
5.4 Advantages of and challenges in targeting LLPS
Targeting phase separation as a therapeutic strategy offers important advantages in the development of novel interventions (Table 2). By modulating the assembly and dynamics of biomolecular condensates, precise control over cellular processes can be achieved. This approach provides a unique opportunity to restore normal cellular functions and attenuate disease progression. Moreover, targeting phase separation offers a new approach to overcome challenges in traditional drug discovery. Conventional drug development often focuses on inhibition of specific protein targets with well-defined structures. However, many proteins lack well-defined structures and are challenging to target using traditional approaches. By targeting the process of phase separation itself, rather than specific protein structures, a broader range of potential therapeutic targets becomes accessible (145).
However, targeting phase separation also presents challenges. Biomolecular condensates are complex and heterogeneous; thus, understanding their precise mechanisms and identifying specific targets requires comprehensive characterization techniques and advanced computational methods. Achieving selectivity while preserving normal condensates is another challenge, as modulating phase separation may impact essential cellular structures and functions. Robust experimental and computational approaches are crucial for target identification, validation, and optimization of drug candidates.
6. Discussion
LLPS has recently emerged as a novel biophysical paradigm, offering valuable insights into the spontaneous formation of membraneless organelles (146,147). The causal relationship between aberrant LLPS and cancer is a crucial issue that needs to be addressed to establish the importance of LLPS in oncogenesis.
As mentioned earlier in this review, evidence has shown that the LLPS-competent IDR in NUP98-HOXA9 plays a pivotal role in leukemogenesis. The IDR facilitates the formation of puncta, which leads to enhanced chromatin occupancy of the chimeric transcription factor and promotes transcriptional activation (27). Other evidence for a causal role of LLPS is that LLPS of SHP2 can be targeted therapeutically using allosteric inhibitors, which attenuate LLPS of SHP2 mutants and enhance the enzymatic activity of SHP2. This observation provides evidence that LLPS can regulate the activity of SHP2, suggesting its potential as a therapeutic target (121). A very recent study conducted by Song et al. further demonstrated that hotspot mutations in the structured Yaf9, ENL, AF9, Taf14, Sas5 (YEATS) domain of the chromatin reader eleven-nineteen-leukemia (ENL) played a pivotal role in the formation of aberrant transcriptional condensates associated with cancer (148). The condensates formed by ENL mutants at physiological levels are functionally associated with upregulation of oncogenes. Moreover, excessive expression of ENL mutants can result in the formation of dysfunctional condensates (148). Collectively, the results of these studies, among others, support the idea that aberrant LLPS plays a causal role in cancer. Through the utilization of mutants with impaired LLPS capacity, investigators have established a direct connection between dysregulated LLPS and the acquisition of oncogenic phenotypes (149).
Understanding LLPS presents an opportunity to design drugs with new targeting strategies. One of the greatest advantages of LLPS is that it presents a novel approach to address the limitations encountered in traditional drug discovery. Conventional drug development revolves primarily around inhibiting specific protein targets with well-defined structures. However, proteins involved in LLPS, such as IDR proteins, often lack distinct structures, posing challenges for conventional targeting methods (145). By directing efforts toward the modulation of phase separation itself, rather than individual protein structures, a wider array of potential therapeutic targets can be explored, circumventing the limitations of traditional approaches. For example, the SRC1 inhibitor elvitegravir disrupts the formation of SRC-1 condensates in cancer cells, presenting a promising LLPS-based strategy for targeting the traditionally challenging SRC-1/YAP/TEAD complex and suppressing YAP-dependent cancer proliferation (110). This approach provides new possibilities for overcoming challenges in drug discovery and expanding the scope of therapeutic interventions. the future development of effective therapeutics requires a comprehensive understanding of the biophysical principles and regulatory mechanisms underlying LLPS.
A key challenge in studying LLPS lies in the development of novel conceptual frameworks, cutting-edge tools, and sophisticated probes that can effectively modulate the physicochemical properties of specific condensates. Methods for studying LLPS have yielded important insights regarding the properties and dynamics of phase-separated condensates. Ensemble experiments, such as imaging and fluorescence techniques, allow the direct measurement of droplet properties over time and the systematic testing of various factors. Single-molecule techniques such as single-molecule Förster resonance energy transfer (smFRET) offer the ability to observe conformational changes and dynamic processes at the molecular level. Nuclear magnetic resonance (NMR) spectroscopy provides atomic-level structural and dynamic information. The development of novel techniques that can provide both high-resolution structural information and real-time dynamic properties of condensates is needed (150). These techniques could involve the integration of complementary methods, such as cryo-electron microscopy (151) and superresolution microscopy (152), to visualize condensate structures at high resolution. Additionally, the combination of single-molecule techniques with NMR or other spectroscopic approaches could impart a more comprehensive understanding of condensate behavior (5). Furthermore, given the intricate complexity and regulatory mechanisms in the intracellular environment, the effectiveness and safety of anticancer agents that modulate LLPS must be evaluated in animal models.
In conclusion, targeting phase separation offers promising opportunities for therapeutic interventions in various diseases, including cancer. This strategy provides a means to disrupt pathological condensates and restore normal cellular function. However, the complexity of LLPS, the dynamic nature of condensates and the need for specificity pose challenges that must be carefully addressed. Future research and innovative approaches are necessary to overcome these challenges and fully exploit the therapeutic opportunities associated with manipulating LLPS for disease treatment.
Abbreviations
LLPS: liquid‒liquid phase separation; IDRs: intrinsically disordered regions; AML: acute myeloid leukemia; NUP98: NUcleoPorin 98; CML: chronic myeloid leukemia; Lin-HSPC: lineage-negative hematopoietic stem and progenitor cell; PML: Promyelocytic leukemia; NBs: nuclear bodies; APL: acute promyelocytic leukemia; RBCC: RING finger/B-box/coiled-coil; SIM: SUMO-interacting motif; RARα: retinoic acid receptor-α; RA: retinoic acid; SGs: Stress granules; MAPK: MAP kinase; 15d-PGJ2: 15-deoxy-delta (12,14)-prostaglandin J (2); RTK: receptor tyrosine kinase; COX2: Cyclooxygenase-2; m6A: N6-methyladenosine; YTH: YT521-B homology; LCD: low complexity domain; EWSR1: Ewing sarcoma RNA-binding protein 1; ER: Estrogen; FLI1: Friend leukemia virus integration 1; POZ: pox virus and zinc finger; SPOP: Speckle-type pox virus and zinc finger (POZ) protein; DAXX: death-domain-associated protein; AR: androgen receptor; AF1: activator function 1; NRF2: nuclear factor erythroid 2-related factor 2; Erα: estrogen receptor α; YAP: Yes-associated protein; TAZ: transcriptional coactivator with PDZ-binding motif; SRC-1: steroid receptor coactivator 1; SHP2: Src homology containing protein tyrosine phosphatase 2; PTP: protein tyrosine phosphatases; 53BP1: p53-binding protein 1; PARP1: Poly(ADP-ribose) polymerase 1; PARylation: poly(ADP-ribosyl)ation; YEATS: Yaf9, ENL, AF9, Taf14, Sas5; ENL: eleven-nineteen-leukemia.
Acknowledgements
All figures were created with BioRender.com.
Funding Sources
The work was supported by the National Key Research and Development Program of China (2021YFE0206600), National Natural Science Foundation of China (82172842 and 81672386), the Sichuan Province Science and Technology Support grant and TianFu Laboratory (2022SYSX0064, 2021YFSY0008, 2020YFS0276), West China Nursing Discipline Development Special Fund Project (HXHL21008), the Post-Doctor Research Project, West China Hospital, Sichuan University (2020HXBH119), Translational medicine fund of West China Hospital (2020HXBH119 and CGZH19002). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011Mar4;144(5):646-74
2. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer Genome Landscapes. Science. 2013Mar29;339(6127):1546-58
3. Taniue K, Akimitsu N. Aberrant phase separation and cancer. FEBS J. 2022Jan;289(1):17-39
4. Ismail H, Liu X, Yang F, Li J, Zahid A, Dou Z. et al. Mechanisms and regulation underlying membraneless organelle plasticity control. J Mol Cell Biol. 2021Aug4;13(4):239-58
5. Alberti S, Gladfelter A, Mittag T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell. 2019Jan24;176(3):419-34
6. Che X, Wu J, Liu H, Su J, Chen X. Cellular liquid-liquid phase separation: Concept, functions, regulations, and detections. J Cell Physiol. 2023May;238(5):847-65
7. Zhang JZ, Mehta S, Zhang J. Liquid-liquid phase separation: a principal organizer of the cell's biochemical activity architecture. Trends Pharmacol Sci. 2021Oct;42(10):845-56
8. Peng Q, Tan S, Xia L, Wu N, Oyang L, Tang Y. et al. Phase separation in Cancer: From the Impacts and Mechanisms to Treatment potentials. Int J Biol Sci. 2022;18(13):5103-22
9. Li P. et al. Phase transitions in the assembly of multivalent signalling proteins. Nature. 2012;483:336-340
10. Peng PH, Hsu KW, Wu KJ. Liquid-liquid phase separation (LLPS) in cellular physiology and tumor biology. Am J Cancer Res. 2021;11(8):3766-76
11. Spegg V, Altmeyer M. Biomolecular condensates at sites of DNA damage: More than just a phase. DNA Repair (Amst). 2021Oct;106:103179
12. Dao TP, Castañeda CA. Ubiquitin-Modulated Phase Separation of Shuttle Proteins: Does Condensate Formation Promote Protein Degradation?. Bioessays. 2020Nov;42(11):e2000036
13. Hnisz D, Shrinivas K, Young RA, Chakraborty AK, Sharp PA. A Phase Separation Model for Transcriptional Control. Cell. 2017Mar23;169(1):13-23
14. Kovar H. Dr. Jekyll and Mr. Hyde: The Two Faces of the FUS/EWS/TAF15 Protein Family. Sarcoma. 2011;2011:837474
15. Mendes A, Fahrenkrog B. NUP214 in Leukemia: It's More than Transport. Cells. 2019Jan21;8(1):76
16. Gough SM, Slape CI, Aplan PD. NUP98 gene fusions and hematopoietic malignancies: common themes and new biologic insights. Blood. 2011Dec8;118(24):6247-57
17. Chatel G, Desai SH, Mattheyses AL, Powers MA, Fahrenkrog B. Domain topology of nucleoporin Nup98 within the nuclear pore complex. J Struct Biol. 2012Jan;177(1):81-9
18. Hussey DJ, Nicola M, Moore S, Peters GB, Dobrovic A. The (4;11)(q21;p15) translocation fuses the NUP98 and RAP1GDS1 genes and is recurrent in T-cell acute lymphocytic leukemia. Blood. 1999Sep15;94(6):2072-9
19. Yamamoto K, Nakamura Y, Saito K, Furusawa S. Expression of the NUP98/HOXA9 fusion transcript in the blast crisis of Philadelphia chromosome-positive chronic myelogenous leukaemia with t(7;11)(p15;p15). Br J Haematol. 2000May;109(2):423-6
20. Struski S, Lagarde S, Bories P, Puiseux C, Prade N, Cuccuini W. et al. NUP98 is rearranged in 3.8% of pediatric AML forming a clinical and molecular homogenous group with a poor prognosis. Leukemia. 2017Mar;31(3):565-72
21. Schmidt HB, Görlich D. Nup98 FG domains from diverse species spontaneously phase-separate into particles with nuclear pore-like permselectivity. Elife. 2015Jan6;4:e04251
22. Terlecki-Zaniewicz S, Humer T, Eder T, Schmoellerl J, Heyes E, Manhart G. et al. Biomolecular condensation of NUP98 fusion proteins drives leukemogenic gene expression. Nat Struct Mol Biol. 2021Feb;28(2):190-201
23. Hollink IHIM, van den Heuvel-Eibrink MM, Arentsen-Peters STCJM, Pratcorona M, Abbas S, Kuipers JE. et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood. 2011Sep29;118(13):3645-56
24. Kasper LH, Brindle PK, Schnabel CA, Pritchard CE, Cleary ML, van Deursen JM. CREB binding protein interacts with nucleoporin-specific FG repeats that activate transcription and mediate NUP98-HOXA9 oncogenicity. Mol Cell Biol. 1999Jan;19(1):764-76
25. Fahrenkrog B, Martinelli V, Nilles N, Fruhmann G, Chatel G, Juge S. et al. Expression of Leukemia-Associated Nup98 Fusion Proteins Generates an Aberrant Nuclear Envelope Phenotype. PLoS One. 2016;11(3):e0152321
26. Boija A, Klein IA, Sabari BR, Dall'Agnese A, Coffey EL, Zamudio AV. et al. Transcription Factors Activate Genes through the Phase-Separation Capacity of Their Activation Domains. Cell. 2018Dec13;175(7):1842-1855.e16
27. Ahn JH, Davis ES, Daugird TA, Zhao S, Quiroga IY, Uryu H. et al. Phase separation drives aberrant chromatin looping and cancer development. Nature. 2021Jul;595(7868):591-5
28. Chandra B. et al. Phase Separation Mediates NUP98 Fusion Oncoprotein Leukemic Transformation. Cancer discovery. 2022Jan4;12(4):1152-1169
29. Heikamp EB, Henrich JA, Perner F, Wong EM, Hatton C, Wen Y. et al. The menin-MLL1 interaction is a molecular dependency in NUP98-rearranged AML. Blood. 2022Feb10;139(6):894-906
30. Li Y, Ma X, Wu W, Chen Z, Meng G. PML Nuclear Body Biogenesis, Carcinogenesis, and Targeted Therapy. Trends Cancer. 2020Oct;6(10):889-906
31. Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017May;18(5):285-98
32. Corpet A, Kleijwegt C, Roubille S, Juillard F, Jacquet K, Texier P. et al. PML nuclear bodies and chromatin dynamics: catch me if you can!. Nucleic Acids Res. 2020Dec2;48(21):11890-912
33. Jensen K, Shiels C, Freemont PS. PML protein isoforms and the RBCC/TRIM motif. Oncogene. 2001Oct29;20(49):7223-33
34. Hsu KS, Kao HY. PML: Regulation and multifaceted function beyond tumor suppression. Cell Biosci. 2018;8:5
35. Lallemand-Breitenbach V, de Thé H. PML nuclear bodies. Cold Spring Harb Perspect Biol. 2010May;2(5):a000661
36. Shen TH, Lin HK, Scaglioni PP, Yung TM, Pandolfi PP. The mechanisms of PML-nuclear body formation. Mol Cell. 2006Nov3;24(3):331-9
37. Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T. et al. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J Cell Biol. 1999Oct18;147(2):221-34
38. Guan D, Kao HY. The function, regulation and therapeutic implications of the tumor suppressor protein, PML. Cell Biosci. 2015;5:60
39. Bernardi R, Pandolfi PP. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol. 2007Dec;8(12):1006-16
40. de Thé H, Le Bras M, Lallemand-Breitenbach V. The cell biology of disease: Acute promyelocytic leukemia, arsenic, and PML bodies. J Cell Biol. 2012Jul9;198(1):11-21
41. Kakizuka A, Miller WH, Umesono K, Warrell RP, Frankel SR, Murty VV. et al. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell. 1991Aug23;66(4):663-74
42. de Thé H, Lavau C, Marchio A, Chomienne C, Degos L, Dejean A. The PML-RAR alpha fusion mRNA generated by the t(15;17) translocation in acute promyelocytic leukemia encodes a functionally altered RAR. Cell. 1991Aug23;66(4):675-84
43. Nisole S. Differential Roles of PML Isoforms. Frontiers in oncology. 2013May22;3:125
44. Shao X, Chen Y, Xu A, Xiang D, Wang W, Du W. et al. Deneddylation of PML/RARα reconstructs functional PML nuclear bodies via orchestrating phase separation to eradicate APL. Cell Death Differ. 2022Aug;29(8):1654-68
45. de Thé H. et al. Acute Promyelocytic Leukemia: A Paradigm for Oncoprotein-Targeted Cure. Cancer cell. 2017Nov13;32(5):552-560
46. Guo S, Cheng X, Lim JH, Liu Y, Kao HY. Control of antioxidative response by the tumor suppressor protein PML through regulating Nrf2 activity. Mol Biol Cell. 2014Aug15;25(16):2485-98
47. Sahin U, Ferhi O, Jeanne M, Benhenda S, Berthier C, Jollivet F. et al. Oxidative stress-induced assembly of PML nuclear bodies controls sumoylation of partner proteins. J Cell Biol. 2014Mar17;204(6):931-45
48. Cheng X, Kao HY. Post-translational modifications of PML: consequences and implications. Front Oncol. 2012;2:210
49. Yang P, Mathieu C, Kolaitis RM, Zhang P, Messing J, Yurtsever U. et al. G3BP1 Is a Tunable Switch that Triggers Phase Separation to Assemble Stress Granules. Cell. 2020Apr16;181(2):325-345.e28
50. Aulas A, Finetti P, Lyons SM, Bertucci F, Birnbaum D, Acquaviva C. et al. Revisiting the Concept of Stress in the Prognosis of Solid Tumors: A Role for Stress Granules Proteins?. Cancers (Basel). 2020Sep1;12(9):2470
51. Asadi MR, Rahmanpour D, Moslehian MS, Sabaie H, Hassani M, Ghafouri-Fard S. et al. Stress Granules Involved in Formation, Progression and Metastasis of Cancer: A Scoping Review. Front Cell Dev Biol. 2021;9:745394
52. Miroshnychenko D, Dubrovska A, Maliuta S, Telegeev G, Aspenström P. Novel role of pleckstrin homology domain of the Bcr-Abl protein: analysis of protein-protein and protein-lipid interactions. Exp Cell Res. 2010Feb15;316(4):530-42
53. Patel H, Marley SB, Greener L, Gordon MY. Subcellular distribution of p210(BCR-ABL) in CML cell lines and primary CD34+ CML cells. Leukemia. 2008Mar;22(3):559-71
54. Skourides PA, Perera SA, Ren R. Polarized distribution of Bcr-Abl in migrating myeloid cells and co-localization of Bcr-Abl and its target proteins. Oncogene. 1999Feb4;18(5):1165-76
55. Gregor T, Bosakova MK, Nita A, Abraham SP, Fafilek B, Cernohorsky NH. et al. Elucidation of protein interactions necessary for the maintenance of the BCR-ABL signaling complex. Cell Mol Life Sci. 2020Oct;77(19):3885-903
56. Kong Y, Jiang C, Wei G, Sun K, Wang R, Qiu T. Small Molecule Inhibitors as Therapeutic Agents Targeting Oncogenic Fusion Proteins: Current Status and Clinical. Molecules. 2023Jan;28(12):4672
57. Kashiwagi S, Fujioka Y, Kondo T, Satoh AO, Yoshida A, Fujioka M. et al. Localization of BCR-ABL to Stress Granules Contributes to Its Oncogenic Function. Cell Struct Funct. 2019Dec26;44(2):195-204
58. Podszywalow-Bartnicka P, Wolczyk M, Kusio-Kobialka M, Wolanin K, Skowronek K, Nieborowska-Skorska M. et al. Downregulation of BRCA1 protein in BCR-ABL1 leukemia cells depends on stress-triggered TIAR-mediated suppression of translation. Cell Cycle. 2014;13(23):3727-41
59. Qiang YW, Ye S, Chen Y, Epstein J, Walker BA, van Rhee F. et al. Mutant KRAS and Brafs Upregulate Stress Granules and Mediate Drug Resistance, Which Can be Modulated By Cox2 Inhibition in Multiple Myeloma. Blood. 2018Nov29;132(Supplement 1):3166-3166
60. Wang J, Li Y, Wang P, Han G, Zhang T, Chang J. et al. Leukemogenic Chromatin Alterations Promote AML Leukemia Stem Cells via a KDM4C-ALKBH5-AXL Signaling Axis. Cell Stem Cell. 2020Jul2;27(1):81-97.e8
61. Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L. et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell. 2020Jul2;27(1):64-80.e9
62. Patil DP, Pickering BF, Jaffrey SR. Reading m6A in the Transcriptome: m6A-Binding Proteins. Trends Cell Biol. 2018Feb;28(2):113-27
63. Gao Y. et al. Multivalent m6A motifs promote phase separation of YTHDF proteins. Cell research. 2019Sep;29(9):767-769
64. Ries RJ, Zaccara S, Klein P, Olarerin-George A, Namkoong S, Pickering BF. et al. m6A enhances the phase separation potential of mRNA. Nature. 2019Jul;571(7765):424-8
65. Cheng Y, Xie W, Pickering BF, Chu KL, Savino AM, Yang X. et al. N6-Methyladenosine on mRNA facilitates a phase-separated nuclear body that suppresses myeloid leukemic differentiation. Cancer Cell. 2021Jul12;39(7):958-972.e8
66. Ludwig JA, Meyers PA, Dirksen U. Ewing's Sarcoma. N Engl J Med. 2021Apr15;384(15):1476
67. Delattre O. et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature. 1992Oct9;359(6391):162-165
68. Ryan JJ, Sprunger ML, Holthaus K, Shorter J, Jackrel ME. Engineered protein disaggregases mitigate toxicity of aberrant prion-like fusion proteins underlying sarcoma. J Biol Chem. 2019Jul19;294(29):11286-96
69. Boulay G, Sandoval GJ, Riggi N, Iyer S, Buisson R, Naigles B. et al. Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell. 2017Sep21;171(1):163-178.e19
70. Tan AY, Manley JL. The TET family of proteins: functions and roles in disease. J Mol Cell Biol. 2009Dec;1(2):82-92
71. Davis RB, Kaur T, Moosa MM, Banerjee PR. FUS oncofusion protein condensates recruit mSWI/SNF chromatin remodeler via heterotypic interactions between prion-like domains. Protein Sci. 2021Jul;30(7):1454-66
72. Cheng Y, Shen Z, Gao Y, Chen F, Xu H, Mo Q. et al. Phase transition and remodeling complex assembly are important for SS18-SSX oncogenic activity in synovial sarcomas. Nat Commun. 2022May18;13(1):2724
73. Wang Z, Song Y, Ye M, Dai X, Zhu X, Wei W. The diverse roles of SPOP in prostate cancer and kidney cancer. Nat Rev Urol. 2020Jun;17(6):339-50
74. Clark A, Burleson M. SPOP and cancer: a systematic review. Am J Cancer Res. 2020;10(3):704-26
75. Kim MS, Je EM, Oh JE, Yoo NJ, Lee SH. Mutational and expressional analyses of SPOP, a candidate tumor suppressor gene, in prostate, gastric and colorectal cancers. APMIS. 2013Jul;121(7):626-33
76. Geng C, Kaochar S, Li M, Rajapakshe K, Fiskus W, Dong J. et al. SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene. 2017Aug17;36(33):4767-77
77. Gan W, Dai X, Lunardi A, Li Z, Inuzuka H, Liu P. et al. SPOP Promotes Ubiquitination and Degradation of the ERG Oncoprotein to Suppress Prostate Cancer Progression. Mol Cell. 2015Sep17;59(6):917-30
78. Marzahn MR, Marada S, Lee J, Nourse A, Kenrick S, Zhao H. et al. Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles. EMBO J. 2016Jun15;35(12):1254-75
79. Bouchard JJ, Otero JH, Scott DC, Szulc E, Martin EW, Sabri N. et al. Cancer Mutations of the Tumor Suppressor SPOP Disrupt the Formation of Active, Phase-Separated Compartments. Mol Cell. 2018Oct4;72(1):19-36.e8
80. Vijayvargia R, May MS, Fondell JD. A coregulatory role for the mediator complex in prostate cancer cell proliferation and gene expression. Cancer Res. 2007May1;67(9):4034-41
81. Zhang F, Biswas M, Massah S, Lee J, Lingadahalli S, Wong S. et al. Dynamic phase separation of the androgen receptor and its coactivators key to regulate gene expression. Nucleic Acids Res. 2023Jan11;51(1):99-116
82. Ahmed J, Meszaros A, Lazar T, Tompa P. DNA-binding domain as the minimal region driving RNA-dependent liquid-liquid phase separation of androgen receptor. Protein Sci. 2021Jul;30(7):1380-92
83. Basu S, Martínez-Cristóbal P, Pesarrodona M, Frigolé-Vivas M, Szulc E, Lewis M. et al. Androgen receptor condensates as drug targets. bioRxiv; 2022. p. 2022 08.18.504385
84. Xie J, He H, Kong W, Li Z, Gao Z, Xie D. et al. Targeting androgen receptor phase separation to overcome antiandrogen resistance. Nat Chem Biol. 2022Dec;18(12):1341-50
85. Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible?. Nat Rev Drug Discov. 2014Nov;13(11):828-51
86. Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016Jan12;23(1):27-47
87. Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014Feb;39(2):91-100
88. Grabocka E, Bar-Sagi D. Mutant KRAS Enhances Tumor Cell Fitness by Upregulating Stress Granules. Cell. 2016Dec15;167(7):1803-1813.e12
89. Hamada S, Matsumoto R, Tanaka Y, Taguchi K, Yamamoto M, Masamune A. Nrf2 Activation Sensitizes K-Ras Mutant Pancreatic Cancer Cells to Glutaminase Inhibition. Int J Mol Sci. 2021Feb14;22(4):1870
90. Mukhopadhyay S, Goswami D, Adiseshaiah PP, Burgan W, Yi M, Guerin TM. et al. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020Apr15;80(8):1630-43
91. Dai X, Xiang L, Li T, Bai Z. Cancer Hallmarks, Biomarkers and Breast Cancer Molecular Subtypes. J Cancer. 2016;7(10):1281-94
92. Fan W, Chang J, Fu P. Endocrine therapy resistance in breast cancer: current status, possible mechanisms and overcoming strategies. Future Med Chem. 2015Aug;7(12):1511-9
93. Mills JN, Rutkovsky AC, Giordano A. Mechanisms of resistance in estrogen receptor positive breast cancer: overcoming resistance to tamoxifen/aromatase inhibitors. Curr Opin Pharmacol. 2018Aug;41:59-65
94. R Sabari B. et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science (New York, NY). 2018Jul27;361(6400):eaar3958
95. Klein IA, Boija A, Afeyan LK, Hawken SW, Fan M, Dall'Agnese A. et al. Partitioning of cancer therapeutics in nuclear condensates. Science. 2020Jun19;368(6497):1386-92
96. Nagalingam A, Tighiouart M, Ryden L, Joseph L, Landberg G, Saxena NK. et al. Med1 plays a critical role in the development of tamoxifen resistance. Carcinogenesis. 2012Apr;33(4):918-30
97. Horn L, Pao W. EML4-ALK: honing in on a new target in non-small-cell lung cancer. J Clin Oncol. 2009Sep10;27(26):4232-5
98. Sabir SR, Yeoh S, Jackson G, Bayliss R. EML4-ALK Variants: Biological and Molecular Properties, and the Implications for Patients. Cancers. 2017Sep5;9(9):118
99. Mossé YP, Wood A, Maris JM. Inhibition of ALK signaling for cancer therapy. Clin Cancer Res. 2009Sep15;15(18):5609-14
100. Tulpule A, Guan J, Neel DS, Allegakoen HR, Lin YP, Brown D. et al. Kinase-mediated RAS signaling via membraneless cytoplasmic protein granules. Cell. 2021May13;184(10):2649-2664.e18
101. Qin Z, Sun H, Yue M, Pan X, Chen L, Feng X. et al. Phase separation of EML4-ALK in firing downstream signaling and promoting lung tumorigenesis. Cell Discov. 2021May11;7(1):33
102. Soon GST, Torbenson M. The Liver and Glycogen: In Sickness and in Health. International Journal of Molecular Sciences. 2023Jan;24(7):6133
103. Liu Q. et al. Glycogen accumulation and phase separation drives liver tumor initiation. Cell. 2021Oct28;184(22):5559-5576.e19
104. Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005Aug12;122(3):421-34
105. Lopez-Hernandez A. Emerging Principles in the Transcriptional Control by YAP and TAZ. Cancers. 2021Aug23;13:4242
106. Cottini F, Hideshima T, Xu C, Sattler M, Dori M, Agnelli L. et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat Med. 2014Jun;20(6):599-606
107. Grieve S, Wajnberg G, Lees M, Chacko S, Weir J, Crapoulet N. et al. TAZ functions as a tumor suppressor in multiple myeloma by downregulating MYC. Blood Adv. 2019Nov26;3(22):3613-25
108. Cheung P, Xiol J, Dill MT, Yuan WC, Panero R, Roper J. et al. Regenerative Reprogramming of the Intestinal Stem Cell State via Hippo Signaling Suppresses Metastatic Colorectal Cancer. Cell Stem Cell. 2020Oct1;27(4):590-604.e9
109. Cai D, Feliciano D, Dong P, Flores E, Gruebele M, Porat-Shliom N. et al. Phase separation of YAP reorganizes genome topology for long-term YAP target gene expression. Nat Cell Biol. 2019Dec;21(12):1578-89
110. Zhu G, Xie J, Fu Z, Wang M, Zhang Q, He H. et al. Pharmacological inhibition of SRC-1 phase separation suppresses YAP oncogenic transcription activity. Cell Res. 2021Sep;31(9):1028-31
111. Yu M, Peng Z, Qin M, Liu Y, Wang J, Zhang C. et al. Interferon-γ induces tumor resistance to anti-PD-1 immunotherapy by promoting YAP phase separation. Mol Cell. 2021Mar18;81(6):1216-1230.e9
112. Lu Y, Wu T, Gutman O, Lu H, Zhou Q, Henis YI. et al. Phase separation of TAZ compartmentalizes the transcription machinery to promote gene expression. Nat Cell Biol. 2020Apr;22(4):453-64
113. Wei Y, Luo H, Yee PP, Zhang L, Liu Z, Zheng H. et al. Paraspeckle Protein NONO Promotes TAZ Phase Separation in the Nucleus to Drive the Oncogenic Transcriptional Program. Adv Sci (Weinh). 2021Dec;8(24):e2102653
114. Yuan X, Bu H, Zhou J, Yang CY, Zhang H. Recent Advances of SHP2 Inhibitors in Cancer Therapy: Current Development and Clinical Application. J Med Chem. 2020Oct22;63(20):11368-96
115. Easton JB, Royer AR, Middlemas DS. The protein tyrosine phosphatase, Shp2, is required for the complete activation of the RAS/MAPK pathway by brain-derived neurotrophic factor. J Neurochem. 2006May;97(3):834-45
116. Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell. 2017Jun29;170(1):17-33
117. Li J, Jie HB, Lei Y, Gildener-Leapman N, Trivedi S, Green T. et al. PD-1/SHP-2 inhibits Tc1/Th1 phenotypic responses and the activation of T cells in the tumor microenvironment. Cancer Res. 2015Feb1;75(3):508-18
118. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA. et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017Mar31;355(6332):1428-33
119. Zhang RY, Yu ZH, Zeng L, Zhang S, Bai Y, Miao J. et al. SHP2 phosphatase as a novel therapeutic target for melanoma treatment. Oncotarget. 2016Nov8;7(45):73817-29
120. Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A. et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003Jun;34(2):148-50
121. Zhu G, Xie J, Kong W, Xie J, Li Y, Du L. et al. Phase Separation of Disease-Associated SHP2 Mutants Underlies MAPK Hyperactivation. Cell. 2020Oct15;183(2):490-502.e18
122. Lin CC, Suen KM, Jeffrey PA, Wieteska L, Lidster JA, Bao P. et al. Receptor tyrosine kinases regulate signal transduction through a liquid-liquid phase separated state. Mol Cell. 2022Mar17;82(6):1089-1106.e12
123. Nakad R, Schumacher B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front Genet. 2016;7:147
124. Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014Jan;15(1):7-18
125. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181-211
126. Kilic S, Lezaja A, Gatti M, Bianco E, Michelena J, Imhof R. et al. Phase separation of 53BP1 determines liquid-like behavior of DNA repair compartments. EMBO J. 2019Aug15;38(16):e101379
127. Pessina F, Giavazzi F, Yin Y, Gioia U, Vitelli V, Galbiati A. et al. Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat Cell Biol. 2019Oct;21(10):1286-99
128. Ghodke I, Remisova M, Furst A, Kilic S, Reina-San-Martin B, Poetsch AR. et al. AHNAK controls 53BP1-mediated p53 response by restraining 53BP1 oligomerization and phase separation. Mol Cell. 2021Jun17;81(12):2596-2610.e7
129. Langelier MF, Planck JL, Roy S, Pascal JM. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science. 2012May11;336(6082):728-32
130. Kamaletdinova T, Fanaei-Kahrani Z, Wang ZQ. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells. 2019Dec12;8(12):1625
131. Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010Apr;10(4):293-301
132. Altmeyer M, Neelsen KJ, Teloni F, Pozdnyakova I, Pellegrino S, Grøfte M. et al. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat Commun. 2015Aug19;6:8088
133. Rose M, Burgess JT, O'Byrne K, Richard DJ, Bolderson E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front Cell Dev Biol. 2020;8:564601
134. Martin EW, Holehouse AS. Intrinsically disordered protein regions and phase separation: sequence determinants of assembly or lack thereof. Emerg Top Life Sci. 2020Dec11;4(3):307-29
135. Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J. et al. Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc Natl Acad Sci U S A. 2002Mar19;99(6):3830-5
136. Zhang Z, Boskovic Z, Hussain MM, Hu W, Inouye C, Kim HJ. et al. Chemical perturbation of an intrinsically disordered region of TFIID distinguishes two modes of transcription initiation. Elife. 2015Aug28;4:e07777
137. Wang C, Lu H, Liu X, Gao X, Tian W, Chen H. et al. A natural product targets BRD4 to inhibit phase separation and gene transcription. iScience. 2022Jan21;25(1):103719
138. Soragni A, Janzen DM, Johnson LM, Lindgren AG, Thai-Quynh Nguyen A, Tiourin E. et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell. 2016Jan11;29(1):90-103
139. Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM. et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017Sep;18(9):1274-84
140. S Singatulina A. et al. PARP-1 Activation Directs FUS to DNA Damage Sites to Form PARG-Reversible Compartments Enriched in Damaged DNA. Cell reports. 2019Jul5;27(6):1809-1821.e5
141. Leung AKL. Poly(ADP-ribose): A Dynamic Trigger for Biomolecular Condensate Formation. Trends Cell Biol. 2020May;30(5):370-83
142. Wippich F. et al. Dual specificity kinase DYRK3 couples stress granule condensation/dissolution to mTORC1 signaling. Cell. 2013Feb14;152(4):791-805
143. Roe JS, Mercan F, Rivera K, Pappin DJ, Vakoc CR. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol Cell. 2015Jun18;58(6):1028-39
144. Bhagwat AS, Roe JS, Mok BYL, Hohmann AF, Shi J, Vakoc CR. BET Bromodomain Inhibition Releases the Mediator Complex from Select cis-Regulatory Elements. Cell Rep. 2016Apr19;15(3):519-30
145. Conti BA, Oppikofer M. Biomolecular condensates: new opportunities for drug discovery and RNA therapeutics. Trends in Pharmacological Sciences. 2022Oct1;43(10):820-37
146. Mir M, Bickmore W, Furlong EEM, Narlikar G. Chromatin topology, condensates and gene regulation: shifting paradigms or just a phase?. Development. 2019Sep25;146(19):dev182766
147. A P, Weber SC. Evidence for and against Liquid-Liquid Phase Separation in the Nucleus. Noncoding RNA. 2019Nov1;5(4):50
148. Song L, Yao X, Li H, Peng B, Boka AP, Liu Y. et al. Hotspot mutations in the structured ENL YEATS domain link aberrant transcriptional condensates and cancer. Mol Cell. 2022Nov3;82(21):4080-4098.e12
149. Hahn S. Phase Separation, Protein Disorder, and Enhancer Function. Cell. 2018Dec13;175(7):1723-5
150. Ganser LR, Myong S. Methods to Study Phase-Separated Condensates and the Underlying Molecular Interactions. Trends Biochem Sci. 2020Nov;45(11):1004-5
151. Zhang M, Díaz-Celis C, Onoa B, Cañari-Chumpitaz C, Requejo KI, Liu J. et al. Molecular organization of the early stages of nucleosome phase separation visualized by cryo-electron tomography. Mol Cell. 2022Aug18;82(16):3000-3014.e9
152. Zuo L, Lai L, Qi Z. Single-Molecule Imaging of the Phase Separation-Modulated DNA Compaction to Study Transcriptional Repression. Methods Mol Biol. 2023;2563:215-23
153. Mansour MR, Abraham BJ, Anders L, Berezovskaya A, Gutierrez A, Durbin AD. et al. Oncogene regulation. An oncogenic super-enhancer formed through somatic mutation of a noncoding intergenic element. Science. 2014Dec12;346(6215):1373-7
154. Rai AK, Chen JX, Selbach M, Pelkmans L. Kinase-controlled phase transition of membraneless organelles in mitosis. Nature. 2018Jul;559(7713):211-6
155. Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK. et al. Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat Chem Biol. 2016Oct;12(10):876-84
156. Nilson KA, Guo J, Turek ME, Brogie JE, Delaney E, Luse DS. et al. THZ1 Reveals Roles for Cdk7 in Co-transcriptional Capping and Pausing. Molecular Cell. 2015Aug20;59(4):576-87
157. Barra J, Gaidosh GS, Blumenthal E, Beckedorff F, Tayari MM, Kirstein N. et al. Integrator restrains paraspeckles assembly by promoting isoform switching of the lncRNA NEAT1. Science Advances. 6(27):eaaz9072.
158. Bhagwat AS, Roe JS, Mok BYL, Hohmann AF, Shi J, Vakoc CR. BET Bromodomain Inhibition Releases the Mediator Complex from Select cis-Regulatory Elements. Cell Rep. 2016Apr19;15(3):519-30
159. Erkizan HV, Kong Y, Merchant M, Schlottmann S, Barber-Rotenberg JS, Yuan L. et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing's sarcoma. Nat Med. 2009Jul;15(7):750-6
160. Rangel LP, Ferretti GDS, Costa CL, Andrade SMMV, Carvalho RS, Costa DCF. et al. p53 reactivation with induction of massive apoptosis-1 (PRIMA-1) inhibits amyloid aggregation of mutant p53 in cancer cells. J Biol Chem. 2019Mar8;294(10):3670-82
161. Soragni A, Janzen DM, Johnson LM, Lindgren AG, Thai-Quynh Nguyen A, Tiourin E. et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell. 2016Jan11;29(1):90-103
162. Shi J, Stover JS, Whitby LR, Vogt PK, Boger DL. Small molecule inhibitors of Myc/Max dimerization and Myc-induced cell transformation. Bioorg Med Chem Lett. 2009Nov1;19(21):6038-41
163. Zhu G, Xie J, Fu Z, Wang M, Zhang Q, He H. et al. Pharmacological inhibition of SRC-1 phase separation suppresses YAP oncogenic transcription activity. Cell Res. 2021Sep;31(9):1028-31
164. Ivanov PA, Chudinova EM, Nadezhdina ES. Disruption of microtubules inhibits cytoplasmic ribonucleoprotein stress granule formation. Exp Cell Res. 2003Nov1;290(2):227-33
165. Babinchak WM, Dumm BK, Venus S, Boyko S, Putnam AA, Jankowsky E. et al. Small molecules as potent biphasic modulators of protein liquid-liquid phase separation. Nat Commun. 2020Nov4;11(1):5574
166. Gupta N, Badeaux M, Liu Y, Naxerova K, Sgroi D, Munn LL. et al. Stress granule-associated protein G3BP2 regulates breast tumor initiation. Proc Natl Acad Sci U S A. 2017Jan31;114(5):1033-8
Author contact
![]() Corresponding authors: Prof. Xingchen Peng is to be contacted at Department of Biotherapy, Cancer Center, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China. E-mail address: pxx2014com or pxx2014edu.cn. Prof. Xiaolin Hu is to be contacted at West China School of Nursing, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China. E-mail address: huxiaolincn.
Corresponding authors: Prof. Xingchen Peng is to be contacted at Department of Biotherapy, Cancer Center, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China. E-mail address: pxx2014com or pxx2014edu.cn. Prof. Xiaolin Hu is to be contacted at West China School of Nursing, West China Hospital, Sichuan University, Chengdu 610041, Sichuan, China. E-mail address: huxiaolincn.
Received 2022-12-4
Accepted 2023-7-23
Published 2023-8-6