ISSN: 1449-2288International Journal of Biological Sciences
- Current issue
- Volume 20; 2024
- Volume 19; 2023
- Volume 18; 2022
- Volume 17; 2021
- Volume 16; 2020
- Archive
- Advance articles
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
The components and functions of...
The γ-secretase substrates...
The correlation between...
The potential and challenges of...
Summary and outlook
Acknowledgements
References

Int J Biol Sci 2023; 19(16):5089-5103. doi:10.7150/ijbs.87334 This issue Cite
Review
The critical role of γ-secretase and its inhibitors in cancer and cancer therapeutics
Congkuan Song1,#, Jinjin Zhang2,#, Chenzhen Xu1,#, Minglang Gao1, Ning Li1, ![]() , Qing Geng1,
, Qing Geng1, ![]()
1. Department of Thoracic Surgery, Renmin Hospital of Wuhan University, Wuhan, China.
2. Department of Emergency, Taihe Hospital, Shiyan, China.
# These authors contributed equally to this work.
Abstract

As a multi-substrate transmembrane protease, γ-secretase exists widely in various cells. It controls multiple important cellular activities through substrate cleavage. γ-secretase inhibitors (GSIs) play a role in cancer inhibition by blocking Notch cleavage, and are considered as potential therapeutic strategies for cancer. Currently, GSIs have encouraging performance in preclinical models, yet this success does not translate well in clinical trials. In recent years, a number of breakthrough discoveries have shown us the promise of targeting γ-secretase for the treatment of cancer. Here, we integrate a large amount of data from γ-secretase and its inhibitors and cancer in nearly 30 years, comb and discuss the close connection between γ-secretase and cancer, as well as the potential and problems of current GSIs in cancer treatment. We analyze the possible reasons for the failure performance of current GSIs in clinical trials, and make recommendations for future research areas.
Keywords: γ-secretase, γ-secretase inhibitors (GSIs), cancer
Introduction
γ-secretase is a multi-substrate transmembrane protease associated with Alzheimer's disease (AD), and is widespread in a variety of cells. It consists of four different integral membrane proteins: presenilin (PS1 or PS2), anterior pharynx-defective 1 (APH1A or APH1B), presenilin enhancer protein 2 (Pen-2), and nicastrin, containing 20 transmembrane domains (TMDs) and a large extracellular domain (ECD). γ-secretase engages in various biological pathways through substrate cleavage. Inhibition of γ-secretase activity has been considered as a potential therapeutic strategy for cancer. γ-secretase inhibitors (GSIs) have shown encouraging performance in preclinical models; however, their performance in clinical trials has been unsatisfactory. This may be partly attributed to a poor understanding of the γ-secretase and its inhibitors.
In recent years, Yigong Shi et al. analyzed the structure of γ-secretase (including the sequence of each subunit) and the cryo-electron microscopic structure of γ-secretase binding Notch and amyloid precursor protein (APP) under the condition of high-resolution by using ultra-low temperature electron microscopy [1-4], and also reported for the first time the four atomic resolution cryo-electron microscopic structure of γ-secretase binding three small molecule inhibitors and one regulator, elucidating the molecular mechanism of γ-secretase in recognizing different kinds of inhibitors and modulators [5]. These breakthrough findings are exciting and will greatly advance the design and optimization of the next generation of γ-secretase inhibitors and modulators. We seem to see promising prospects for targeting γ-secretase against various human diseases, including cancer. Therefore, it is necessary to further expand our understanding on the critical role of γ-secretase and its inhibitors in cancer and cancer therapeutics.
The components and functions of γ-secretase complex
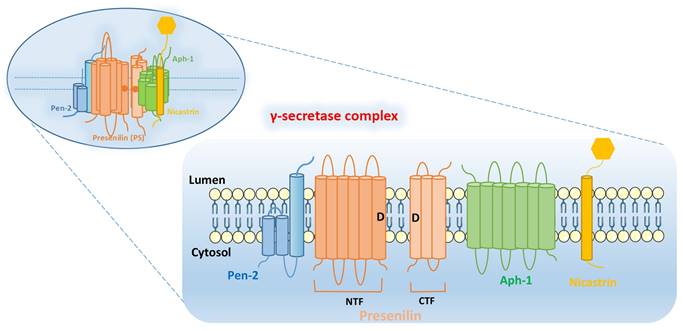
γ-secretase complex is composed of four subunits: PS, Pen-2, Aph-1, and Nicastrin. The subunits of γ-secretase are closely arranged and each subunit contains at least one TMD. The intact γ-secretase also contains the glycation part of the ECD in Nicastrin subunit (Figure 1).
PS: PS is found in mammals in two subtypes PS1 and PS2, encoded by PSEN1 and PSEN2, respectively [6, 7]. PS contains 9 TMDs, which are decomposed by endogenous proteins between the 6th and 7th TMDs into two parts: N-terminal fragment (NTF) and C-terminal fragment (CTF). The aspartate residues (Asp257 and Asp385) in the 6th and 7th TMDs are essential for their enzymatic activity. The precursor of PS1 is an inactive holoprotein, which is subsequently hydrolyzed to a heterodimer composed of PS1-CTF and PS1-NTF with the synergistic action of other subunits [8, 9]. The whole-protein form of PS1 is barely detectable in the organism, while the catalytically active PS1 is often present as a heterodimer. Notably, not all of the catalytically active PS1 exists as a heterodimer, such as ΔE9 PSEN1[10-12].
Pen-2: Pen-2, encoded by PSENEN, was previously thought to be a “U-shaped” hairpin protein with N- and C-terminus exposed to the lumen, containing two TMDs [13, 14]. However, further researches have yielded different findings. These studies [15, 16] found that Pen-2 harbors three TMDs, two of which traverse the membrane only half-way from the intracellular side, with N-terminus of Pen-2 facing the cytoplasm and the C-terminus exposed to the lumen (Figure 1). Pen-2 is closely linked to PS, facilitating the automatic catalytic cleavage of PS (located between TMD6 and TMD7) to produce two fragments, NTF and CTF [10, 17, 18]. The C-terminal hydrophilic region of Pen-2 was reported to be critical for stabilizing PS1-NTF and -CTF, but it is not necessary [16]. And HP1 (the first of the two hydrophobic regions contained by Pen-2) is essential for determining the topology of Pen-2, which is required for promoting Pen-2-mediated endoproteolysis of PS1 and γ-secretase activity [19].
Aph-1: Aph-1 can be encoded by APH1A or APH1B, and contains seven TMDs, with the N-terminal domain facing the lumen and the C-terminal domain facing the cytoplasm. Together with Nicastrin, it plays a supporting role in facilitating the assembly and transport of γ-secretase complex. At the same time, it is also responsible for supporting the proteolytic activity of γ-secretase [20, 21].
Nicastrin: Nicastrin, a glycoylated protein encoded by NCSTN, contains a large ECD and is thought to serve as a complement of enzyme substrate, which can provide docking sites for γ-secretase substrates [22, 23]. In addition, Nicastrin binds well to both NTF and CTF of PS, and is the component that maintains the stability of PS, and also have to rely on the PS to leave the endoplasmic reticulum (ER) to reach the cell surface [9, 24].
Structural integrity of γ-secretase complex. It is composed of four subunits: PS, Pen-2, Aph-1, and Nicastrin. PS can be decomposed by endogenous proteins between the 6th and 7th TMDs into two parts: N-terminal fragment (NTF) and C-terminal fragment (CTF). The aspartate residues (Asp257 and Asp385: as shown by the “D” in Figure) in the 6th and 7th TMDs are essential for their enzymatic activity.
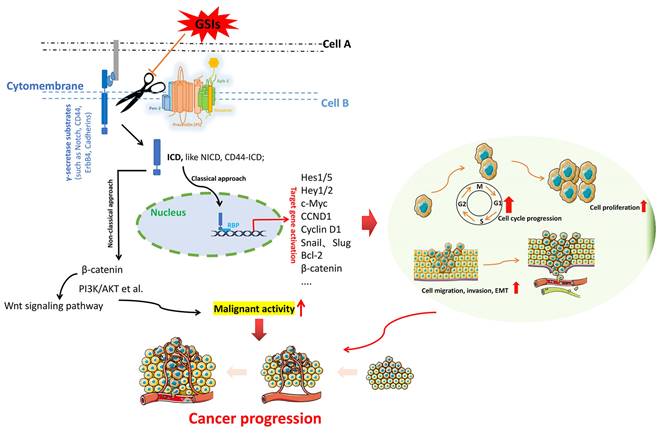
The fundamental process by which γ-secretase acts on substrates to exert essential functions. γ-secretases cleave the key substrate (e.g. Notch, CD44, ErbB 4, Cadherins) to release an active ICD, which allows ICD to migrate to the nucleus, where it binds to the transcription factors (e.g. CSL and MAML) to activate downstream effector factors. Activation of downstream target genes triggers multiple oncogenic pathways, which triggering cancer progression.
Overall, γ-secretase complex contains twenty TMDs and a large ECD from Nicastrin [4]. PS is the active center of γ-secretase, and the other three subunits (Nicastrin, Aph-1 and Pen-2) are essential components for the maturation and stability of γ-secretase [3]. These four subunits have different functions, and they need to be properly assembled, modified, matured and transported to the corresponding sites to play their normal physiological functions. They interact with each other to play hydrolytic activity and shear function together.
The γ-secretase substrates associated with cancer
γ-secretase is a kind of multi-substrate protease complex widely existing in various cells. It is mainly involved in the cleavage and hydrolysis of a variety of transmembrane proteins, such as APP, Notch, ErbB4, CD44, Cadherins, etc [25]. The intracellular domains (ICDs) of these substrates are released from the membrane into the cytoplasm under the action of γ-secretase, and these ICDs have different physiological functions associated with regulating the transcription of downstream genes. Figure 2 illustrates the general process by which γ-secretase acts on the substrates to exert important functions. The γ-secretase substrates associated with cancer mainly include Notch, ErbB4, CD44, Cadherins, VEGFR1, IGF1R, MUC1, etc. They are involved in various cellular pathways, including the regulation of cell fate, transcriptional regulation, cell adhesion, and neurotrophin signal transduction [26].
Notch: As a transmembrane receptor protein, Notch is one of the main targets of γ-secretase activity. It is a cell fate sensor that serves as a receptor for a variety of classical and non-classical ligands (such as Delta, Jagged), and upon binding, Notch is cleaved by γ-secretase and releases an active ICD (here called NICD). This allows NICD to transfer to the nucleus, where it binds to the transcription factor CSL and MAML, which in turn activates downstream effectors (such as Hes1), preventing irreversible cell differentiation and cell cycle exit [27-29]. There are four members of the human Notch receptor family, namely Notch 1, Notch 2, Notch 3, and Notch 4, whose ligands are also transmembrane proteins including Delta-like-1, Delta-like-2, Delta-like-3, Delta-like-4, Jagged-1, and Jagged-2 [30]. The Notch signaling pathway is an evolutionarily conserved pathway whose dysregulation has been implicated in various human cancers [31]. Current data [32, 33] also confirm that the oncogenic range of Notch signaling is partly due to its crosstalk with other signaling pathways, such as NF-kB, Hedgehog, JAK/STAT, MAPK, HIF-1α, Wnt, TGF-p, VEGF, PI3K/Akt, Ras, etc. In fact, based on the overwhelming evidence for the role of Notch signaling in cancer, this pathway has clearly become an important target for cancer therapy.
CD44: CD44, a non-kinase transmembrane glycoprotein involved in cell-cell interactions, is a receptor for hyaluronic acid and is overexpressed in a variety of cell types, including cancer stem cells [34, 35]. It is a major adhesion molecule and can also activate cell signal pathways, and mediate various biological processes, including lymphocyte homing, cell proliferation, migration and metastasis [36, 37]. CD44 can release the active intracellular domain (CD44-ICD) upon cleavage of γ-secretase [38, 39], which can translocate to the nucleus and trigger downstream signaling pathway. Overall, as an important cell surface adhesion molecule, the association of CD44 with human malignancies has been extensively reported.
ErbB4: ErbB4 is a type I transmembrane receptor tyrosine kinase that binds to its ligand (heregulin) and is cleaved by metalloproteinases to produce ECD containing transmembrane and cytoplasmic domains. The ECD is then cleaved by γ-secretase and releases the cytoplasmic domain into the cytoplasm, thereby regulating cell proliferation and differentiation [40, 41]. Previous studies [42, 43] have also confirmed the important role of ErbB4 in cancer.
E-cadherins and N-cadherins: Cadherins, a class of cell adhesion molecules, are essential for maintaining cell-cell contact and for regulating cytoskeletal complexes [44, 45]. Cadherins rely on calcium ions (Ca2+) to function. And they regulate the development and function of most tissues and have important roles in cell signaling, proliferation, and differentiation. Epithelial cadherins (E-cadherins) bind to PS1 and are treated by γ-secretase [46]. Neurocadherins (N-cadherins) experience PS1-mediated cleavage of γ-secretase to produce ICDs, which are potent repressor of the cAMP response element binding protein (CREB) and CREB binding protein (CBP)-mediated transcription [46, 47]. E-cadherins and N-cadherins play a key role in maintaining the structural integrity and polarity of epithelial tissues, and their close relationship with tumor progression as important markers of epithelial mesenchymal transformation (EMT) has been demonstrated by numerous studies.
Overall, many γ-secretase substrates are closely related to the occurrence and development of cancer. Here we list only a few substrates that are widely reported and relatively well studied. Other substrates, such as VEGFR, IGF1R, and MUC1, have also been shown to be associated with neoangiogenesis and cell adhesion, and γ-secretase may also regulate tumorigenesis by affecting these effects of the above substrates.
The correlation between components of γ-secretase and cancer
At present, related studies on γ-secretase have mainly focused on the function of substrates or the role of inhibitors/modulators as well as spatial structure of γ-secretase. However, the potential role of individual γ-secretase components, including potential links to cancer, has not been fully understood. Here, we pool the forefront data from γ-secretase studies to attempt to answer this question.
PS: PS is the major component of the γ-secretase complex. It is so named because its mutation is closely related to early-onset familial inherited AD and belongs to the member of the evolutionary conserved gene family. PS (PS-1 or PS-2) can be encoded by PSEN1 or PSEN2. PSEN1 is the most commonly mutated gene in patients with familial inherited AD, accounting for 70% to 80%. While mutations in PSEN2 gene are rare [48]. PSEN2 has been reported to play an important role in promoting the progression of lung tumors [49] and gliomas [50]. However, studies on the relationship between PSEN2 and cancer are still lacking, and whether PSEN2 plays similar roles in other tumors is still unknown.
As the highly homologous gene of PSEN2, PSEN1 has been shown to be associated with various tumorigenic processes, such as cell proliferation, apoptosis, and cell adhesion [51, 52]. As the core catalytic subunit of the γ-secretase complex, PSEN1 can generate activated Notch intracellular domain (NICD) by cleaving Notch family proteins, and then translocate NICD to the nucleus to regulate the transcriptional expression of a series of target genes. Besides, it interacts with Wnt/β-catenin, PI3K/AKT/mTOR and RAS/RAF/MEK pathways [53, 54], and is involved in cell proliferation, invasion, metastasis and neovasculangiogenesis of malignant tumors. Similarly, CD44, cadherins, as important transmembrane proteins, can also participate in their intracellular signal transduction through a similar mechanism to regulate the biological behaviors of tumor cells, thus affecting the progression of tumor [38, 39].
Additionally, as the catalytic core of γ-secretase complex, the expression level and mutation of PSEN1 obviously directly affect the activity and mode of action of γ-secretase. Importantly, γ-secretase has many substrates, and there are many signaling pathways affected by it. Thus, this undoubtedly highlights the broad and complex roles of PSEN1. It should also be noted that PSEN1 plays an important role in various cancers, both dependent on and independent of γ-secretase activity. For example, PSEN1/γ-secretase can, on the one hand, generate activated Notch-ICD and CD44-ICD by cutting Notch and CD44, translocate them to the nucleus, and bind with its activation transcription factors to activate downstream target genes. On the other hand, it can act on β-catenin independently of γ-secretase activity to activate the WNT signaling pathway [53, 55].
Although most genes involved in tumorigenesis can be divided into tumor suppressor genes and oncogenes, PSEN1 cannot be clearly classified, because it exhibits two functions of promoting and suppressing cancer in different tumor-specific genetic damage. A previous study [56] found that PSEN1 expression was significantly up-regulated in both head and neck squamous cell carcinoma (HNSCC) cell lines and tissue samples, and was associated with poor prognosis and radiotherapy resistance in HNSCC. Similar findings were also seen in hepatocellular carcinoma [57] and oesophageal cancer [58]. In addition, the down-regulation of PSEN1 expression in cell line U937 led to slower proliferation and increased apoptosis of tumor cells, and the down-regulation of PSEN1 expression also reduced the tumor-causing ability in nude mice [59]. These studies suggest that PSEN1 plays a “driving” role in some cancer diseases, acting as an oncogene. However, other studies have found that PSEN1 plays an opposite role in some tumors. For example, in glioblastoma, PSEN1 can inhibit tumor cell invasiveness [60]. Additionally, Xia et al. [61] found that PSEN1 knockout mice spontaneously formed skin malignations due to the absence of PSEN1, which leads to the accumulation of β-catenin in the cytoplasm and nucleus, thus activating the β-catenin signaling pathway and resulting in increased cyclin D1 expression. It was also shown that increased expression of PSEN1 was associated with good disease-free survival in patients with breast cancer [62], suggesting that PSEN1 might play a role in inhibiting the formation and progression of some tumors. To sum up, it is not difficult to find that PSEN1 plays different or even opposite roles in different cancer diseases, which may be related to the tissue-specific microenvironment in which different cancers occur.
Pen-2/PSENEN: Pen-2/PSENEN is the minimal subunit of the γ-secretase complex. Current data indicate that PSENEN is involved in the occurrence and development of many human diseases, such as hidradenitis suppurative (HS) and Dowling's disease (DDD). PSENEN has also been found to play a vital role in adipocyte differentiation [63]. Moreover, PSENEN deletion can inhibit HES1 and activate STAT3 to trigger GFAP activation, thereby promoting the differentiation of oligodendrocyte progenitors into astrocytes [64]. Up to now, the study of PSENEN in cancer is still scarce, and the only studies are mostly stuck in bioinformatics analysis and lack of in-depth exploration of wet experiments. Gu et al. [65] found that the expression of PSENEN was increased in low-grade gliomas based on bioinformatics methods, which was corresponding to the poor prognosis of patients. Similarly, a similar phenomenon was observed in pancreatic cancer [66], suggesting that PSENEN may play a “bad” role as an oncogene in some tumors. However, based on the same bioinformatics analysis, Chen et al. [67] found that PSENEN was downregulated in gastric cancer tissues, and its low expression level was associated with worse prognosis. This seems to suggest that PSENEN as a tumor suppressor plays a “good” role in gastric cancer. Notably, these findings are still unsupported by adequate evidence. The specific role of PSENEN in different human cancers still needs further study.
Nicastrin/NCSTN: NCSTN is the largest subunit of the γ-secretase complex, with a single TMD and a large ECD. These domains were identified as functional sites for the recruitment of γ-secretase substrates [68]. NCSTN is mainly synthesized by fibroblasts and neurons, and is widely distributed in the body. A growing number of studies have reported a close relationship between NCSTN and tumorigenesis and progression. NCSTN was highly expressed in breast cancer and had carcinogenic effects [69, 70]. Overexpression of NCSTN can regulate the properties of breast cancer stem cells and induce the epithelial-mesenchymal transition (EMT) by cleaving the Notch1 protein [69-71]. Moreover, NCSTN can also regulate AKT activation in hepatocellular carcinoma, which in turn affects cellular malignant behaviors [72]. The process by which NCSTN controls cell death through the PI3K/Akt pathway is independent of γ-secretase, that is, NCSTN can independently perform some functions [73, 74]. Filipovic A et al. [74] found that specific monoclonal antibodies against NCSTN had anti-tumor effects on invasive triple-negative breast cancer cells. Furthermore, siRNA-NCSTN has been found to prevent the induction of the Notch1 intracellular domain (NICD) after oxaliplatin, thereby affecting the response to chemotherapy in colon cancer [75]. Another study also demonstrated that siRNA-NCSTN in basal-like breast cancer could enhance the anti-tumor effect of EGFR inhibitors by blocking the Notch and AKT signaling pathways [76]. Taken all together, NCSTN does participate in the occurrence and progression of some tumors. However, on the whole, the study of NCSTN in tumor is still insufficient, and a lot of research is needed to reveal the more specific roles of NCSTN in tumor.
Aph-1: Increasing evidence indicates that γ-secretase plays a critical role in cancer development and progression. Although Aph-1, as an important member of the γ-secretase complex, is important in performing biological processes such as cleaving transmembrane proteins, there is still a significant lack of research on Aph-1 in cancers. Human Aph-1 is encoded by two genes, APH1A and APH1B, of which APH1A appears to be more important [77]. APH1A was overexpressed in diffuse large B-cell lymphoma with poor prognosis [78], and its expression level was also positively correlated with the grade of hepatocellular carcinoma [79]. Peltonen HM et al. [80] investigated the expression levels of γ-secretase subunits in breast cancer, and found that the mRNA expression of APH1B, PSENEN, and NCSTN was significantly reduced in breast cancer cases with higher tumor grade. In addition, APH1B was also reported to be involved in the maintenance of spherical cell stemness in cervical cancer [81]. Importantly, although these studies have observed some association of Aph-1 with some tumors, the specific mechanism remains poorly investigated. The role of Aph-1 as an important component of γ-secretase in cancer occurrence and progression remains to be uncovered.
The potential and challenges of γ-secretase inhibitors as anticancer therapeutic strategies
In the previous section, we have discussed the association of substrates and each component of γ-secretase with cancer. The intriguing question is whether targeting γ-secretase can be an effective anticancer strategy, given its close association with cancer. In fact, γ-secretases have been proposed as therapeutic targets for human diseases, including cancer. Two candidates for targeting the γ-secretase complex have emerged: inhibitors and modulators. γ-secretase inhibitors (GSIs) were originally developed as a treatment for AD. However, to date, GSIs has not gained a good indication for the treatment of AD. This is well illustrated by the adverse reactions of Semagacestat (a broad-spectrum GSI), in the Phase III clinical trial of AD [82]. GSIs has been repurposing as an anticancer drug, due to its ability to block γ-secretase activity and inhibit Notch cleavage. The anti-tumor effects of GSIs in various cancers have been extensively studied. In Table 1, we summarize the GSIs and their associated information in the preclinical models. Globally, the anti-cancer efficacy of GSIs in preclinical models is promising, which is believed that GSIs can drive tumor cell differentiation and apoptosis through multiple mechanisms, reduce the burden of cancer stem cells, and also hinder EMT, and overcome resistance to conventional therapies. Disappointingly, however, these GSIs have not performed well in clinical trials. Because most solid tumors do not derive clinical benefit from them. J.Bart Rose and Tyler R.McCaw et al. [26] reviewed the relevant information of clinical research of GSIs in various cancer types in detail. Here, we update and sort out this data (Table 2). It can be rationally seen, that although GSIs do not confer significant clinical activity in the majority of patients in some cancer types, it is undeniable that GSIs do show anti-cancer effects in some patients. For example, in a phase II clinical trial of RO4929097 monotherapy for metastatic refractory pancreatic cancer [83], 3 of 12 (25%) evaluable patients had stable disease, while the other 9 patients did not benefit from RO4929097. In another phase I trial of MK-0752 in adult patients with advanced solid tumors, 5 of the 21 evaluable glioma patients showed clinical benefit, although none of the patients with other tumors (breast cancer, colorectal cancer, ovarian cancer, sarcoma cancer, etc.) gained clinical benefit [84]. Encouragingly, a recently published Phase 3 international, double-blind, randomized, placebo-controlled trial of nirogacestat (a potent, orally active, reversible, non-competitive and selective GSI) for progressive desmoid tumors has yielded exciting results [85]. Globally, the therapeutic efficacy of GSIs varies by cancer type and individual patient. The reasons responsible for the apparently different clinical activities of GSIs in patients of different cancer types as well as in different patients of the same cancer type are diverse and complex. In addition to individual heterogeneity among patients, the dose and duration of GSIs, as well as the treatment strategies of patients prior to this treatment may be important reasons for this difference.
According to the currently available data, the mechanism of anti-tumor action of GSIs is still elusive. Since many γ-secretase substrates are directly involved in carcinogenesis or tumor progression, GSIs should theoretically be an ideal anti-cancer strategy. However, this is not the case (as noted above). These disappointing clinical manifestations reflect important issues that still deserve our consideration. The first is the non-selective suppression of GSIs. γ-secretase as a protease complex can cleave a variety of transmembrane proteins with different biological functions, which adds diversity and complexity to the biological pathways in which γ-secretases participate. Non-selective inhibition of this complex is bound to cause off-target toxicity, which may be refractory.
Performance of GSIs in preclinical models.
| GSIs | Cancer Types | Intervention objects | Effect | Reference |
|---|---|---|---|---|
| GSI and LY-411,575 | Kaposi's sarcoma | Kaposi's sarcoma tumor cells | Induction of apoptosis | PMID: 15940249 [91] |
| GSI-XII (Z-IL-CHO) and GSI-IX (DAPT) | Multiple myeloma | NCI-H929, U266 and RPMI-8226 | Induction of apoptosis | PMID: 21965140 [92] |
| Z-LLNle-CHO | Breast cancer | MCF-7, BT474, T47D, SKBR3, MDA-MB-231, and MDA-MB-468 | Z-LLNle-CHO mediates the damage to breast cancer cells through proteasome inhibition (rather than γ -secretase inhibition) | PMID: 19660128 [93] |
| LLNle | Glioblastoma | human glioblastoma tumor-initiating cells (GBM TICs) | LLNle mediates the GBM TICs apoptotic cell death through γ-secretase and proteasome inhibition | PMID: 19861404 [94] |
| MRK-003 | Pancreatic cancer | Pa03C, Pa14C, Pa16C and Pa29C; patient-derived PDAC xenografts | MRK-003 can reduce tumor cell proliferation, induce apoptosis and intratumoral necrosis | PMID: 22752426 [95] |
| DAPT | Ovarian cancer | SKOV3 and HO8910 | DAPT prevents ovarian cancer stem cells (OCSCs) formation, and inhibits OCSC self-renewal and proliferation | PMID: 23482909 [96] |
| RO4929097 | Melanoma | WM35, WM98.1, WM115, WM983A, WM3248, A375, WM239A/131/4-5B1 (5B1); human primary melanoma xenograft in NOD/SCID/IL2gammaR-/- mice | RO4929097 can weaken cell proliferation and tumor growth of Melanoma. | PMID: 21980408 [97] |
| MRK-003, MRK-006 | T-cell acute lymphoblastic leukemias (T-ALL) | T-All cells | Combination of GSI with a CDK4 inhibitor results in potent cell cycle arrest and death. | PMID: 19318552 [98] |
| RO4929097 | NSCLC | A549, H460a cells, A549 NSCLC xenograft model | Significant tumor growth inhibition | PMID: 19773430 [99] |
| PF-03084014 | Prostate Cancer | Du145, PC3 and Du145R, PC3R; 7-8-week-old male NOD.CB17-Prkdcscid/NCrCrl (NOD/SCID) mice | PF-03084014 enhanced the docetaxel-mediated tumor response | PMID: 26202948 [100] |
| MK-0725 | Ovarian cancer | A2780, OVCAR3, SKOV3, HO8910PM; Mouse xenograft model of A2780 | Induction of apoptosis; Significant tumor growth inhibition | PMID: 26704638 [101] |
| GSI I and GSI XX | NSCLC | H460, A549 and H1395 | Treatment with GSIs after radiation can significantly enhance radiation-mediated tumour cytotoxicity and delay tumor progression. | PMID: 22596234 [102] |
| DAPT | Ovarian cancer | A2780, A2780/CP70 and OV2008, OV2008/C13 | DAPT pretreatment can improve the sensitivity of cisplatin-resistant human ovarian cancer cells to cisplatin. | PMID: 24535252 [103] |
| BMS-708163 | Lung Cancer | PC9, PC9/AB2, PC9/AB2 xenografts | BMS-708163 can sensitize PC9/AB2 cells to gefitinib-induced cytotoxicity. BMS-708163 combined with gefitinib can induce high level of apoptosis. And the combination of gefitinib and BMS-708163 can inhibit the growth of PC9/AB2 xenografts. | PMID: 25561332 [104] |
| GSI I (cbz-IL-CHO) | Gastric cancer | AGS, SNU601, SNU638, SNU-668, SNU-719, MKN28, and YCC-2; orthotopically transplanted gastric cancer mouse models | GSI I can significantly inhibit the proliferation of gastric cancer cells and reduce the tumor load of orthotopic transplantation mouse models, and the combination of GSI I and 5-FU can enhance the therapeutic effect | PMID: 26134677 [105] |
A typical example is that the GSIs currently used for anti-cancer therapy works primarily by blocking Notch signaling. However, the impact of Notch signaling on the body is comprehensive and profound, and in addition to its impact on cancer cells, it is also critical to the fate of various cells during embryonic development and adulthood. In addition to counteracting the pro-cancer effects of Notch signaling, GSIs may also affect other important signaling and cellular events, leading to unacceptable toxic effects. Of course, we should also note that some types of cancer cells do not rely excessively on Notch signaling for survival. Conversely, activation of Notch signaling even blocks the formation of some malignancies [86]. Aaron Proweller et al also reported that impaired Notch signaling promoted the formation of nascent squamous cell carcinoma [87]. Another question worth considering is why the success of GSIs in preclinical research is difficult to translate in clinical trials. Going back to the essential differences between preclinical research and clinical trials, this phenomenon is not difficult to explain. First, preclinical studies are largely conducted on various cell lines, which are in vitro experiments that lack the involvement of the body's immune metabolic processes. Moreover, animal models in preclinical studies are unable to fully mimic the pathophysiological features of the human body, and even some animal models are not immunocompetent. It is worth noting that, although GSIs may hinder tumor progression through Notch signaling, at the same time it may in turn impair the body's anti-tumor immune response. LI-CHO (a GSI) has been reported to inhibit the proliferation of mouse CD8 T cells (a major effector of anti-tumor immunity) in a dose-dependent manner [88]. While the expression of effector molecules in CD8 T cells also requires the triggering of Notch signaling [89]. As highlighted by J.Art Rose and Tyler R. McCaw et al. [26], the large number of immunosuppressive cells in the tumor microenvironment can also be a great challenge. Charbonnier LM et al. [90] found that Notch signaling can disrupt the stability of Treg, and GSIs as a blocker of Notch signaling may promote Treg-mediated immunosuppression and impair anti-tumor immunity. The mechanism by which GSIs weaken the body's anti-tumor immunity may be much more than that, and further investigations are pending.
The implementation of GSIs may require individualized and targeted management. Addressing the current unfavorable situation of GSIs requires a better understanding of the fine structure and specific mechanisms of γ-secretases, which may facilitate the development of more efficient and selective GSIs. At the same time, the optimization of GSIs anticancer strategy should also include the dose, frequency and duration of drug use, as well as the rational combination strategy with other drugs.
Performance of GSIs as anti-cancer strategies in clinical trials.
| Cancer Types | Name | Phase | Case selection | Usage | Outcome | Reference |
|---|---|---|---|---|---|---|
| Lung cancer | MK-0752 | I | Age ≥ 18 years; patients with histologically confirmed solid tumors that had failed to respond to standard therapies or for which no proven treatments existed. | Oral administration; Specific dose and time are not given (Despite providing the drug dose and duration of use in each schedule, the specific cancer type was not specified); | None of the patients received any clinical benefit (n=3). | PMID: 22547604 [84] |
| PF-03084014 | I | Advanced patients who were resistant to standard therapy or for which no therapy was available. | Oral administration for 21 days in a test dose range of 20 to 330 mg BID (Please refer to this article for specific usage); | None of the patients received any clinical benefit (n=5). | PMID: 25231399 [106] | |
| LY900009 | I | Age ≥ 18 years; patients with advanced cancer refractory to standard therapy (or no available standard therapy) and a 12-week expectancy. | LY900009 was administered orally thrice weekly (Monday, Wednesday, and Friday) on a 28-d cycle; Dose escalation was performed at a pre-specified dose level (2 - 60 mg); | Of the 2 patients evaluable for response, 1 patient was observed with SD (73d). | PMID: 26798966 [107] | |
| RO4929097 + Cediranib | I | Age ≥ 18 years; Patients had histologically or cytologically documented advanced solid malignancy, refractory to standard therapy or for which conventional therapy was not effective. | Patients received a progressively increased dose of RO4929097 (on a 3 days-on and 4 days-off schedule) in combination with cediranib (once daily). The first cycle, which lasted 42 days, was given RO4929097 alone for the first 3 weeks, followed by RO4929097 and cediranib in combination from day 22. The second and subsequent periods were 21 days. | None of the patients received any clinical benefit (n=1). | PMID: 23868004 [108] | |
| Pancreatic cancer | MK-0752 | I | Age ≥ 18 years; patients with histologically confirmed solid tumors that had failed to respond to standard therapies or for which no proven treatments existed. | Oral administration; Specific dose and time are not given (Despite providing the drug dose and duration of use in each schedule, the specific cancer type was not specified). | None of the patients received any clinical benefit (n=2). | PMID: 22547604 [84] |
| LY900009 | I | Age ≥ 18 years; patients with advanced cancer refractory to standard therapy (or no available standard therapy) and a 12-week expectancy. | LY900009 was administered orally thrice weekly (Monday, Wednesday, and Friday) on a 28-d cycle; Dose escalation was performed at a pre-specified dose level (2-60 mg). | None of the patients received any clinical benefit (n=3). | PMID: 26798966 [107] | |
| PF-03084014 | I | Advanced patients who were resistant to standard therapy or for which no therapy was available. | Oral administration for 21 days in a test dose range of 20 to 330 mg BID (Please refer to this article for specific usage); | None of the patients received any clinical benefit (n=2). | PMID: 25231399 [106] | |
| RO4929097 | II | Age ≥ 18 years; patients with previously treated metastatic pancreatic adenocarcinoma. | Oral administration; 20 mg daily on days 1-3, 8-10 and 15-17 of 21-day cycles; | Three (25%) of 12 evaluable patients achieved stable disease. Median PFS was 1.5 months. | PMID: 24668033 [83] | |
| R04929097 + Gemcitabine | II | Patient was 18 years or older, had histologically or cytologically proven advanced solid tumors with no further standard treatment options available. | RO4929097 was administered orally, once daily on days 1-3, 8-10, 15-17, 22-24. RO4929097 dose levels were 20 mg, 30 mg, 45 mg and 90 mg; Gemcitabine was administered at 1000 mg/m2 on d1, 8, and 15 in 28 d cycles. | One in three patients with pancreatic cancer achieved long-term stable disease (> 4 months). | PMID: 23645447 [109] | |
| MK-0752 + Gemcitabine | I | Patients with stage III (inoperable) and stage IV pancreatic ductal adenocarcinoma (Of the 44 patients included, 93% had stage IV pancreatic cancer, and 30% had received prior chemotherapy) | MK-0752 was administered orally weekly; Gemcitabine was administered intravenously at 800 or 1000 mg m-2 on days 1,8, and 15 (28-day cycles). | Of the 19 patients undergoing response assessment, one confirmed partial response and 13 had stable disease. | PMID: 29438372 [110] | |
| Melanoma | RO4929097 | I | Age ≥ 18 years; patients with histologically confirmed solid tumors refractory to standard therapy or for which no standard therapy exists. | Oral increasing doses of RO4929097 by two regimens: (A) 3 consecutive days per week for 2 weeks every 3 weeks; (B) 7 consecutive days every 3 weeks. | Of the 24 melanoma patients with evaluable efficacy, one nearly complete FDG-PET response. | PMID: 22529266 [111] |
| RO4929097 | II | Patient had stage IV melanoma of histologically confirmed skin or unknown origin (excluding ocular and mucosal sources), had not received chemotherapy (immunotherapy and adjuvant therapy were allowed) and had no history of central nervous system metastasis. | Taken orally on an empty stomach at a dose of 20 mg daily 3 consecutive days per week. | Of the 32 evaluable patients, 1 confirmed partial response persisted for 7 months and 8 patients had stable disease until at least week 12, with 1 continuing for 31 months. | PMID: 25250858 [112] | |
| MK-0752 | I | Age ≥ 18 years; patients with histologically confirmed solid tumors that had failed to respond to standard therapies or for which no proven treatments existed. | Oral administration; Specific dose and time are not given (Despite providing the drug dose and duration of use in each schedule, the specific cancer type was not specified) | None of the patients received any clinical benefit (n=3). | PMID: 22547604 [84] | |
| LY3039478 | I | Age ≥ 20 years; Japanese patients with advanced solid tumors for whom standard therapies failed or would not be appropriate. | 2 dose levels of crenigacestat (25 mg and 50 mg) were administered orally 3 times weekly (TIW) over a 28-day cycle. | None of the patients received any clinical benefit (n=2). | PMID: 32939607 [113] | |
| Ovarian cancer | RO4929097 | I | Age ≥ 18 years; patients with histologically confirmed solid tumors refractory to standard therapy or for which no standard therapy exists. | Oral increasing doses of RO4929097 by two regimens: (A) 3 consecutive days per week for 2 weeks every 3 weeks; (B) 7 consecutive days every 3 weeks. | Of the 9 ovarian cancer patients with evaluable efficacy, 0 showed clinical benefit. | PMID: 22529266 [111] |
| LY900009 | I | Age ≥ 18 years; patients with advanced cancer refractory to standard therapy (or no available standard therapy) and a 12-week expectancy. | LY900009 was administered orally thrice weekly (Monday, Wednesday, and Friday) on a 28-d cycle; Dose escalation was performed at a pre-specified dose level (2-60 mg). | None of the patients received any clinical benefit (n=11). | PMID: 26798966 [107] | |
| RO4929097 | II | Age ≥ 18 years; Women with progressive platinum-resistant epithelial ovarian cancer treated with ≤ 2 chemotherapy regimens for recurrent disease; | RO4929097 administered orally at 20 mg once daily, 3 days on/4 days off each week in a three-week cycle. | No objective responses were observed. 15 patients (33%) had SD as their best response, with a median duration of 3.1 months. | PMID: 25769658 [114] | |
| MK-0752 | I | Age ≥ 18 years; patients with histologically confirmed solid tumors that had failed to respond to standard therapies or for which no proven treatments existed. | Oral administration; Specific dose and time are not given (Despite providing the drug dose and duration of use in each schedule, the specific cancer type was not specified) | None of the patients received any clinical benefit (n=3). | PMID: 22547604 [84] | |
| R04929097 + Gemcitabine | II | Patient was 18 years or older, had histologically or cytologically proven advanced solid tumors with no further standard treatment options available. | RO4929097 was administered orally, once daily on days 1-3, 8-10, 15-17, 22-24. RO4929097 dose levels were 20 mg, 30 mg, 45 mg and 90 mg; Gemcitabine was administered at 1000 mg/m2 on d1, 8, and 15 in 28 d cycles. | None of the patients received any clinical benefit (n=2). | PMID: 23645447 [109] | |
| RO4929097 + Temsirolimus | Ib | Age ≥ 18 years; patients with histologically confirmed advanced, incurable solid malignancy refractory to conventional therapy or for which no standard therapy existed. | RO4929097 and Temsirolimus were given in three progressively incremented dose levels. RO4929097 was orally administered on an empty stomach on a 3 days on/4 days off schedule, weekly; Intravenous temsirolimus every week. | No objective responses were observed. | PMID: 23860641 [115] | |
| RO4929097 + Cediranib | I | Age ≥ 18 years; Patients had histologically or cytologically documented advanced solid malignancy, refractory to standard therapy or for which conventional therapy was not effective. | Patients received a progressively increased dose of RO4929097 (on a 3 days-on and 4 days-off schedule) in combination with cediranib (once daily). The first cycle, which lasted 42 days, was given RO4929097 alone for the first 3 weeks, followed by RO4929097 and cediranib in combination from day 22. The second and subsequent periods were 21 days. | None of the patients received any clinical benefit (n=1). | PMID: 23868004 [108] | |
| Sarcoma | RO4929097 | I | Age ≥ 18 years; patients with histologically confirmed solid tumors refractory to standard therapy or for which no standard therapy exists. | Oral increasing doses of RO4929097 by two regimens: (A) 3 consecutive days per week for 2 weeks every 3 weeks; (B) 7 consecutive days every 3 weeks. | Of the 12 sarcoma patients with evaluable efficacy, 1 showed mixed response (stable disease). | PMID: 22529266 [111] |
| MK-0752 | I | Age ≥ 18 years; patients with histologically confirmed solid tumors that had failed to respond to standard therapies or for which no proven treatments existed. | Oral administration; Specific dose and time are not given (Despite providing the drug dose and duration of use in each schedule, the specific cancer type was not specified) | None of the patients received any clinical benefit (n=3). | PMID: 22547604 [84] | |
| LY900009 | I | Age ≥ 18 years; patients with advanced cancer refractory to standard therapy (or no available standard therapy) and a 12-week expectancy. | LY900009 was administered orally thrice weekly (Monday, Wednesday, and Friday) on a 28-d cycle; Dose escalation was performed at a pre-specified dose level (2-60 mg). | Of the 2 patients evaluable for response, 1 patient was observed with SD (113d). | PMID: 26798966 [107] | |
| Glioma | MK-0752 | I | Patients, aged between 3 and 21 years, were histologically proven malignant central nervous system tumor (diffuse pontine glioma does not require histology) and refractory to conventional therapy with Lansky or Karnofsky score 60. | MK-0752 was taken orally in a starting dose of 200 mg/m2 once every 7 days for three consecutive days. | Most patients experienced disease progression after 1 or 2 courses. Prolonged SD was observed only in 2 patients (≥3 courses). | PMID: 21825264 [116] |
| RO4929097 +Bevacizumab | I | Patient was 18 years or older, had histologically proven malignant glioma, and progressed after radiotherapy and chemotherapy with temozolomide. | RO4929097 was taken orally for 3 days on/4 days off each week for 4 consecutive cycles (days 1-3, 8-10, 15-17, and 22-24), and intravenous infusion of bevacizumab (Day 1 and Day 15, 10mg /kg) every 2 weeks. | Two of the 12 patients had radiological responses (one patient gained CR and the other PR). | PMID: 27826680 [117] | |
| MK-0752 | I | Age ≥ 18 years; patients with histologically confirmed solid tumors that had failed to respond to standard therapies or for which no proven treatments existed. | Oral administration; Specific dose and time are not given (Despite providing the drug dose and duration of use in each schedule, the specific cancer type was not specified) | Of the 21 patients with evaluable efficacy, 5 showed SD. | PMID: 22547604 [84] | |
| Breast cancer | MK-0752 | I | Age ≥ 18 years; patients with histologically confirmed solid tumors that had failed to respond to standard therapies or for which no proven treatments existed. | Oral administration; Specific dose and time are not given (Despite providing the drug dose and duration of use in each schedule, the specific cancer type was not specified) | None of the patients received any clinical benefit (n=24). | PMID: 22547604 [84] |
| RO4929097 | I | Age ≥ 18 years; patients with histologically confirmed solid tumors refractory to standard therapy or for which no standard therapy exists. | Oral increasing doses of RO4929097 by two regimens: (A) 3 consecutive days per week for 2 weeks every 3 weeks; (B) 7 consecutive days every 3 weeks. | None of the patients received any clinical benefit (n=10). | PMID: 22529266 [111] | |
| PF-03084014 | I | Advanced patients who were resistant to standard therapy or for which no therapy was available. | Oral administration for 21 days in a test dose range of 20 to 330 mg BID (Please refer to this article for specific usage); | None of the patients received any clinical benefit (n=7). | PMID: 25231399 [106] | |
| RO4929097 + Exemestane | Ib | Patients with ER+/HER2- metastatic breast cancer | RO4929097 was taken orally every day for 3 consecutive days, followed by 4 days of discontinuation, and the cycle was 21 days. Exemestane was used at a dose of 25 mg daily. | Of the 14 evaluable patients, 8 patients showed clinical responses (1 PR and 7 SD). The overall clinical benefit rate (CR + PR + SD >= 6 months) was 20% and PFS was 3.2 months. | PMID: 34903452 [118] | |
| R04929097 + Gemcitabine | II | Patient was 18 years or older, had histologically or cytologically proven advanced solid tumors with no further standard treatment options available. | RO4929097 was administered orally, once daily on days 1-3, 8-10, 15-17, 22-24. RO4929097 dose levels were 20 mg, 30 mg, 45 mg and 90 mg; Gemcitabine was administered at 1000 mg/m2 on d1, 8, and 15 in 28 d cycles. | Of the 5 patients, 1 patient showed SD (> 4 months). | PMID: 23645447 [109] | |
| PF-03084014 + Docetaxel | I | Adult women with advanced or metastatic triple-negative breast cancer or hormone-refractory ER/PR-positive breast cancer. | PF-03084014 was taken orally twice daily continuously in combination with intravenous docetaxel given on day 1 of each 21-day cycle. | 4 of the 25 evaluable patients achieved a confirmed partial response. 9 (36%) patients had stable disease, 5 of whom had unconfirmed partial responses. 11 (44%) patients had the best overall response to progressive disease. The median PFS was 4.1 months, and the 6-month PFS rate was 17.1%. | PMID: 27906684 [119] | |
| MK-0752 + Docetaxel | Ib | Male or female patients with advanced breast cancer not responsive to first-line anthracycline chemotherapy. | MK-0752 was used on days 1 to 3, with the dose determined by a dose-escalation protocol, followed by docetaxel on day 8 of each 21-day cycle. | Of the 24 patients evaluable for response, 11 patients were observed with PR, 9 SD, and 3 PD. | PMID: 23340294 [120] | |
| RO4929097 + Cediranib | I | Age ≥ 18 years; Patients had histologically or cytologically documented advanced solid malignancy, refractory to standard therapy or for which conventional therapy was not effective. | Patients received a progressively increased dose of RO4929097 (on a 3 days-on and 4 days-off schedule) in combination with cediranib (once daily). The first cycle, which lasted 42 days, was given RO4929097 alone for the first 3 weeks, followed by RO4929097 and cediranib in combination from day 22. The second and subsequent periods were 21 days. | None of the patients received any clinical benefit (n=1). | PMID: 23868004 [108] | |
| Colorectal cancer | MK-0752 | I | Age ≥ 18 years; patients with histologically confirmed solid tumors that had failed to respond to standard therapies or for which no proven treatments existed. | Oral administration; Specific dose and time are not given (Despite providing the drug dose and duration of use in each schedule, the specific cancer type was not specified) | None of the patients received any clinical benefit (n=16). | PMID: 22547604 [84] |
| PF-03084014 | I | Advanced patients who were resistant to standard therapy or for which no therapy was available. | Oral administration for 21 days in a test dose range of 20 to 330 mg BID (Please refer to this article for specific usage); | None of the patients received any clinical benefit (n=11). | PMID: 25231399 [106] | |
| LY900009 | I | Age ≥ 18 years; patients with advanced cancer refractory to standard therapy (or no available standard therapy) and a 12-week expectancy. | LY900009 was administered orally thrice weekly (Monday, Wednesday, and Friday) on a 28-d cycle; Dose escalation was performed at a pre-specified dose level (2-60 mg). | Of the 5 patients evaluable for response, 1 patient (rectal carcinoma) was observed with SD (55d). | PMID: 26798966 [107] | |
| RO4929097 + Cediranib | I | Age ≥ 18 years; Patients had histologically or cytologically documented advanced solid malignancy, refractory to standard therapy or for which conventional therapy was not effective. | Patients received a progressively increased dose of RO4929097 (on a 3 days-on and 4 days-off schedule) in combination with cediranib (once daily). The first cycle, which lasted 42 days, was given RO4929097 alone for the first 3 weeks, followed by RO4929097 and cediranib in combination from day 22. The second and subsequent periods were 21 days. | Of the 6 patients evaluable for response, 2 patients were observed with SD (7 and 11 cycles). | PMID: 23868004 [108] | |
| RO4929097 | I | Age ≥ 18 years; patients with histologically confirmed solid tumors refractory to standard therapy or for which no standard therapy exists. | Oral increasing doses of RO4929097 by two regimens: (A) 3 consecutive days per week for 2 weeks every 3 weeks; (B) 7 consecutive days every 3 weeks. | None of the patients received any clinical benefit (n=12). | PMID: 22529266 [111] | |
| LY3039478 | I | Age ≥ 20 years; Japanese patients with advanced solid tumors for whom standard therapies failed or would not be appropriate. | 2 dose levels of crenigacestat (25 mg and 50 mg) were administered orally 3 times weekly (TIW) over a 28-day cycle. | None of the patients received any clinical benefit (n=5). | PMID: 32939607 [113] | |
| Desmoma | PF-03084014 | I | Advanced patients resistant to standard therapy or for which no therapy was available. | The oral dose of PF-03084014 ranges from 20 to 330 mg twice daily. | Of the 7 patients, 5 achieved a PR, with a mean time to achieve a response of 11.9 months. All patients who achieved PR continued responding over 47.9 to 73 months. | PMID: 28887726 [121] |
| PF-03084014 | II | Age ≥ 18 years; Patients with histologically confirmed desmoid tumors who were not candidates for surgical resection or definitive radiation therapy and whose disease progressed aggressively after at least one line of standard treatment. | PF-03084014 was taken orally at a dose of 150mg twice daily in 21-day cycles. | Of the 16 patients evaluable, 5 achieved a confirmed partial response and had been on study for more than 2 years, and another 5 patients with prolonged SD remained on study. | PMID: 28350521 [122] | |
| LY3039478 | I | Age ≥ 20 years; Japanese patients with advanced solid tumors for whom standard therapies failed or would not be appropriate. | 2 dose levels of crenigacestat (25 mg and 50 mg) was administered orally 3 times weekly (TIW) over a 28-day cycle. | None of the patients had a complete or partial response to the treatment. One patient with a desmoid tumor in the 50-mg treatment arm showed tumor size shrinkage of 22.4% and had stable disease for 22.5 months. | PMID: 32939607 [113] | |
| PF-03084014 | I | Advanced patients who were resistant to standard therapy or for which no therapy was available. | Oral administration for 21 days in a test dose range of 20 to 330 mg BID (Please refer to this article for specific usage); | Of the 7 patients evaluable, 5 achieved a partial response (71.4% objective response rate). | PMID: 25231399 [106] | |
| Nirogacestat | III | Adults with progressing desmoid tumors | Oral administration in a test dose of 150mg BID | Patients receiving Nirogacestat performed better on progression-free survival, objective response, pain, symptom burden, physical function, role function, and health-related quality of life. Although adverse events with Nirogacestat are frequent, they are mostly low grade. | PMID: 36884323[85] |
Summary and outlook
As a multisubstrate protease complex, γ-secretase is involved in several biological pathways of the organism. Among the substrates of γ-secretase action, many are closely associated with the occurrence and progression of cancer, of which Notch receptor is the most concerned. Given the promoting role of Notch in multiple cancers, GSIs have also been developed in anticancer therapy. However, it is frustrating that the success of GSIs in preclinical models is not replicated in clinical trials. Although there has been significant progress in the understanding of γ-secretase (especially the three-dimensional structure), there is no denying that we still do not fully understand it. Several key issues still remain to be elucidated. For example, what are the specific biological mechanisms by which γ-secretase acts on substrates? Is the effect of GSIs on anti-tumor immunity related to cancer context? Since GSIs can somehow affect multiple receptors and block several key signaling pathways, is it necessary to target the interference of these non-destination signals simultaneously? Further dissecting and revealing the complexity of γ-secretase structure and function, as well as selecting better diseased animal models to track the multidimensional dynamic effects of γ-secretase in vivo, should be the direction of future efforts. Here, we systematically review the important components of γ-secretases and their relevance to cancer, focusing on combing and discussing the potential and problems of current GSIs in cancer treatment. Taken together recent breakthrough findings in the field of γ-secretase research, we believe that despite the unsatisfactory performance of GSIs in clinical trials, after overcoming these challenges, GSIs will remain a promising strategy for anticancer therapy.
Acknowledgements
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81770095).
Author contributions
CKS, JJZ and CZX: made significant contributions to the study design, and article writing. CKS: literature search, and data interpretation. JJZ summarized the Tables and draw the Figures in the article. MLG, and NL: participated in drafting and revising the article critically. QG and NL: funding acquisition, investigation, project administration, resources. QG and other co-authors read and approved the final manuscript.
Data availability
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres S. et al. Three-dimensional structure of human gamma-secretase. NATURE. 2014;512(7513):166-170
2. Yang G, Zhou R, Zhou Q, Guo X, Yan C, Ke M, Lei J, Shi Y. Structural basis of Notch recognition by human gamma-secretase. NATURE. 2019;565(7738):192-197
3. Bai XC, Yan C, Yang G, Lu P, Ma D, Sun L, Zhou R, Scheres S, Shi Y. An atomic structure of human gamma-secretase. NATURE. 2015;525(7568):212-217
4. Sun L, Zhao L, Yang G, Yan C, Zhou R, Zhou X, Xie T, Zhao Y, Wu S, Li X. et al. Structural basis of human gamma-secretase assembly. Proc Natl Acad Sci U S A. 2015;112(19):6003-6008
5. Yang G, Zhou R, Guo X, Yan C, Lei J, Shi Y. Structural basis of gamma-secretase inhibition and modulation by small molecule drugs. CELL. 2021;184(2):521-533
6. Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. CELL. 2006;126(5):981-993
7. Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, Kelleher RR. Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer's disease. NEURON. 2015;85(5):967-981
8. Luo WJ, Wang H, Li H, Kim BS, Shah S, Lee HJ, Thinakaran G, Kim TW, Yu G, Xu H. PEN-2 and APH-1 coordinately regulate proteolytic processing of presenilin 1. J BIOL CHEM. 2003;278(10):7850-7854
9. LaVoie MJ, Fraering PC, Ostaszewski BL, Ye W, Kimberly WT, Wolfe MS, Selkoe DJ. Assembly of the gamma-secretase complex involves early formation of an intermediate subcomplex of Aph-1 and nicastrin. J BIOL CHEM. 2003;278(39):37213-37222
10. Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M. et al. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. NEURON. 1996;17(1):181-190
11. Capell A, Grunberg J, Pesold B, Diehlmann A, Citron M, Nixon R, Beyreuther K, Selkoe DJ, Haass C. The proteolytic fragments of the Alzheimer's disease-associated presenilin-1 form heterodimers and occur as a 100-150-kDa molecular mass complex. J BIOL CHEM. 1998;273(6):3205-3211
12. Wolfe MS. Unraveling the complexity of gamma-secretase. SEMIN CELL DEV BIOL. 2020;105:3-11
13. Crystal AS, Morais VA, Pierson TC, Pijak DS, Carlin D, Lee VM, Doms RW. Membrane topology of gamma-secretase component PEN-2. J BIOL CHEM. 2003;278(22):20117-20123
14. Bergman A, Hansson EM, Pursglove SE, Farmery MR, Lannfelt L, Lendahl U, Lundkvist J, Naslund J. Pen-2 is sequestered in the endoplasmic reticulum and subjected to ubiquitylation and proteasome-mediated degradation in the absence of presenilin. J BIOL CHEM. 2004;279(16):16744-16753
15. Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J. et al. aph-1 and pen-2 are required for Notch pathway signaling, gammasecretase cleavage of betaAPP, and presenilin protein accumulation. DEV CELL. 2002;3(1):85-97
16. Zhang X, Yu CJ, Sisodia SS. The topology of pen-2, a gamma-secretase subunit, revisited: evidence for a reentrant loop and a single pass transmembrane domain. MOL NEURODEGENER. 2015;10:39
17. Prokop S, Shirotani K, Edbauer D, Haass C, Steiner H. Requirement of PEN-2 for stabilization of the presenilin N-/C-terminal fragment heterodimer within the gamma-secretase complex. J BIOL CHEM. 2004;279(22):23255-23261
18. Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, Iwatsubo T. The role of presenilin cofactors in the gamma-secretase complex. NATURE. 2003;422(6930):438-441
19. Kim SH, Sisodia SS. A sequence within the first transmembrane domain of PEN-2 is critical for PEN-2-mediated endoproteolysis of presenilin 1. J BIOL CHEM. 2005;280(3):1992-2001
20. Pei J, Mitchell DA, Dixon JE, Grishin NV. Expansion of type II CAAX proteases reveals evolutionary origin of gamma-secretase subunit APH-1. J MOL BIOL. 2011;410(1):18-26
21. Steiner H, Winkler E, Haass C. Chemical cross-linking provides a model of the gamma-secretase complex subunit architecture and evidence for close proximity of the C-terminal fragment of presenilin with APH-1. J BIOL CHEM. 2008;283(50):34677-34686
22. Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CR, Sudhof T, Yu G. Nicastrin functions as a gamma-secretase-substrate receptor. CELL. 2005;122(3):435-447
23. Dries DR, Shah S, Han YH, Yu C, Yu S, Shearman MS, Yu G. Glu-333 of nicastrin directly participates in gamma-secretase activity. J BIOL CHEM. 2009;284(43):29714-29724
24. Dries DR, Yu G. Assembly, maturation, and trafficking of the gamma-secretase complex in Alzheimer's disease. CURR ALZHEIMER RES. 2008;5(2):132-146
25. Haapasalo A, Kovacs DM. The many substrates of presenilin/gamma-secretase. J ALZHEIMERS DIS. 2011;25(1):3-28
26. McCaw TR, Inga E, Chen H, Jaskula-Sztul R, Dudeja V, Bibb JA, Ren B, Rose JB. Gamma Secretase Inhibitors in Cancer: A Current Perspective on Clinical Performance. ONCOLOGIST. 2021;26(4):e608-e621
27. Liu H, Kiseleva AA, Golemis EA. Ciliary signalling in cancer. NAT REV CANCER. 2018;18(8):511-524
28. O'Rourke CJ, Matter MS, Nepal C, Caetano-Oliveira R, Ton PT, Factor VM, Andersen JB. Identification of a Pan-Gamma-Secretase Inhibitor Response Signature for Notch-Driven Cholangiocarcinoma. HEPATOLOGY. 2020;71(1):196-213
29. Man J, Yu X, Huang H, Zhou W, Xiang C, Huang H, Miele L, Liu Z, Bebek G, Bao S. et al. Hypoxic Induction of Vasorin Regulates Notch1 Turnover to Maintain Glioma Stem-like Cells. CELL STEM CELL. 2018;22(1):104-118
30. Luca VC, Kim BC, Ge C, Kakuda S, Wu D, Roein-Peikar M, Haltiwanger RS, Zhu C, Ha T, Garcia KC. Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. SCIENCE. 2017;355(6331):1320-1324
31. Pine SR. Rethinking Gamma-secretase Inhibitors for Treatment of Non-small-Cell Lung Cancer: Is Notch the Target?. CLIN CANCER RES. 2018;24(24):6136-6141
32. Chen X, Jung JG, Shajahan-Haq AN, Clarke R, Shih I, Wang Y, Magnani L, Wang TL, Xuan J. ChIP-BIT: Bayesian inference of target genes using a novel joint probabilistic model of ChIP-seq profiles. NUCLEIC ACIDS RES. 2016;44(7):e65
33. Cheng YL, Choi Y, Sobey CG, Arumugam TV, Jo DG. Emerging roles of the gamma-secretase-notch axis in inflammation. Pharmacol Ther. 2015;147:80-90
34. Zoller M. CD44: can a cancer-initiating cell profit from an abundantly expressed molecule?. NAT REV CANCER. 2011;11(4):254-267
35. Dhar D, Antonucci L, Nakagawa H, Kim JY, Glitzner E, Caruso S, Shalapour S, Yang L, Valasek MA, Lee S. et al. Liver Cancer Initiation Requires p53 Inhibition by CD44-Enhanced Growth Factor Signaling. CANCER CELL. 2018;33(6):1061-1077
36. Chen RY, Yen CJ, Liu YW, Guo CG, Weng CY, Lai CH, Wang JM, Lin YJ, Hung LY. CPAP promotes angiogenesis and metastasis by enhancing STAT3 activity. CELL DEATH DIFFER. 2020;27(4):1259-1273
37. Kodama H, Murata S, Ishida M, Yamamoto H, Yamaguchi T, Kaida S, Miyake T, Takebayashi K, Kushima R, Tani M. Prognostic impact of CD44-positive cancer stem-like cells at the invasive front of gastric cancer. Br J Cancer. 2017;116(2):186-194
38. Miletti-Gonzalez KE, Murphy K, Kumaran MN, Ravindranath AK, Wernyj RP, Kaur S, Miles GD, Lim E, Chan R, Chekmareva M. et al. Identification of function for CD44 intracytoplasmic domain (CD44-ICD): modulation of matrix metalloproteinase 9 (MMP-9) transcription via novel promoter response element. J BIOL CHEM. 2012;287(23):18995-19007
39. Cho Y, Lee HW, Kang HG, Kim HY, Kim SJ, Chun KH. Cleaved CD44 intracellular domain supports activation of stemness factors and promotes tumorigenesis of breast cancer. Oncotarget. 2015;6(11):8709-8721
40. Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2(2):127-137
41. Ni CY, Murphy MP, Golde TE, Carpenter G. gamma -Secretase cleavage and nuclear localization of ErbB-4 receptor tyrosine kinase. SCIENCE. 2001;294(5549):2179-2181
42. Mendoza-Naranjo A, El-Naggar A, Wai DH, Mistry P, Lazic N, Ayala FR, Da CI, Rodriguez-Viciana P, Cheng H, Tavares GFJ. et al. ERBB4 confers metastatic capacity in Ewing sarcoma. EMBO MOL MED. 2013;5(7):1087-1102
43. de Bont JM, Packer RJ, Michiels EM, den Boer ML, Pieters R. Biological background of pediatric medulloblastoma and ependymoma: a review from a translational research perspective. Neuro Oncol. 2008;10(6):1040-1060
44. Tepass U, Truong K, Godt D, Ikura M, Peifer M. Cadherins in embryonic and neural morphogenesis. Nat Rev Mol Cell Biol. 2000;1(2):91-100
45. Maitre JL, Heisenberg CP. Three functions of cadherins in cell adhesion. CURR BIOL. 2013;23(14):R626-R633
46. Georgakopoulos A, Marambaud P, Efthimiopoulos S, Shioi J, Cui W, Li HC, Schutte M, Gordon R, Holstein GR, Martinelli G. et al. Presenilin-1 forms complexes with the cadherin/catenin cell-cell adhesion system and is recruited to intercellular and synaptic contacts. MOL CELL. 1999;4(6):893-902
47. Crawford AT, Desai D, Gokina P, Basak S, Kim HA. E-cadherin expression in postnatal Schwann cells is regulated by the cAMP-dependent protein kinase a pathway. GLIA. 2008;56(15):1637-1647
48. Jayadev S, Leverenz JB, Steinbart E, Stahl J, Klunk W, Yu CE, Bird TD. Alzheimer's disease phenotypes and genotypes associated with mutations in presenilin 2. BRAIN. 2010;133(Pt 4):1143-1154
49. Yun HM, Park MH, Kim DH, Ahn YJ, Park KR, Kim TM, Yun NY, Jung YS, Hwang DY, Yoon DY. et al. Loss of presenilin 2 is associated with increased iPLA2 activity and lung tumor development. ONCOGENE. 2014;33(44):5193-5200
50. Liu B, Wang L, Shen LL, Shen MZ, Guo XD, Wang T, Liang QC, Wang C, Zheng J, Li Y. et al. RNAi-mediated inhibition of presenilin 2 inhibits glioma cell growth and invasion and is involved in the regulation of Nrg1/ErbB signaling. Neuro Oncol. 2012;14(8):994-1006
51. Li P, Lin X, Zhang JR, Li Y, Lu J, Huang FC, Zheng CH, Xie JW, Wang JB, Huang CM. The expression of presenilin 1 enhances carcinogenesis and metastasis in gastric cancer. Oncotarget. 2016;7(9):10650-10662
52. Hsieh MH, Yang JS, Lin RC, Hsieh YH, Yang SF, Chang HR, Lu KH. Tomatidine Represses Invasion and Migration of Human Osteosarcoma U2OS and HOS Cells by Suppression of Presenilin 1 and c-Raf-MEK-ERK Pathway. MOLECULES. 2020 25(2)
53. Hass MR, Sato C, Kopan R, Zhao G. Presenilin: RIP and beyond. SEMIN CELL DEV BIOL. 2009;20(2):201-210
54. De Gasperi R, Gama SM, Wen PH, Li J, Perez GM, Curran T, Elder GA. Cortical development in the presenilin-1 null mutant mouse fails after splitting of the preplate and is not due to a failure of reelin-dependent signaling. Dev Dyn. 2008;237(9):2405-2414
55. Yang W, Wu PF, Ma JX, Liao MJ, Xu LS, Xu MH, Yi L. Presenilin1 exerts antiproliferative effects by repressing the Wnt/beta-catenin pathway in glioblastoma. CELL COMMUN SIGNAL. 2020;18(1):22
56. Gou C, Han P, Li J, Gao L, Ji X, Dong F, Su Q, Zhang Y, Liu X. Knockdown of lncRNA BLACAT1 enhances radiosensitivity of head and neck squamous cell carcinoma cells by regulating PSEN1. Br J Radiol. 2020;93(1108):20190154
57. Ma H, Yuan L, Li W, Xu K, Yang L. The LncRNA H19/miR-193a-3p axis modifies the radio-resistance and chemotherapeutic tolerance of hepatocellular carcinoma cells by targeting PSEN1. J CELL BIOCHEM. 2018;119(10):8325-8335
58. Meng F, Qian L, Lv L, Ding B, Zhou G, Cheng X, Niu S, Liang Y. miR-193a-3p regulation of chemoradiation resistance in oesophageal cancer cells via the PSEN1 gene. GENE. 2016;579(2):139-145
59. Amson R, Lassalle JM, Halley H, Prieur S, Lethrosne F, Roperch JP, Israeli D, Gendron MC, Duyckaerts C, Checler F. et al. Behavioral alterations associated with apoptosis and down-regulation of presenilin 1 in the brains of p53-deficient mice. Proc Natl Acad Sci U S A. 2000;97(10):5346-5350
60. Yang W, Xiang Y, Liao MJ, Wu PF, Yang L, Huang GH, Shi BZ, Yi L, Lv SQ. Presenilin1 inhibits glioblastoma cell invasiveness via promoting Sortilin cleavage. CELL COMMUN SIGNAL. 2021;19(1):112
61. Xia X, Qian S, Soriano S, Wu Y, Fletcher AM, Wang XJ, Koo EH, Wu X, Zheng H. Loss of presenilin 1 is associated with enhanced beta-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci U S A. 2001;98(19):10863-10868
62. Peltonen HM, Haapasalo A, Hiltunen M, Kataja V, Kosma VM, Mannermaa A. Gamma-secretase components as predictors of breast cancer outcome. PLOS ONE. 2013;8(11):e79249
63. Lee SM, Jeong YH, Kim HM, Park HY, Yoon D, Kim DH, Saeki S, Moon SJ, Kang MJ. Presenilin enhancer-2 (PSENEN), a component of the gamma-secretase complex, is involved in adipocyte differentiation. Domest Anim Endocrinol. 2009;37(3):170-180
64. Hou J, Bi H, Ye Z, Huang W, Zou G, Zou X, Shi YS, Shen Y, Ma Q, Kirchhoff F. et al. Pen-2 Negatively Regulates the Differentiation of Oligodendrocyte Precursor Cells into Astrocytes in the Central Nervous System. J NEUROSCI. 2021;41(23):4976-4990
65. Chen K, Liang B, Ma W, Wan G, Chen B, Lu C, Luo Y, Gu X. Immunological and prognostic analysis of PSENEN in low-grade gliomas: An immune infiltration-related prognostic biomarker. FRONT MOL NEUROSCI. 2022;15:933855
66. Jeon YH, Ha M, Kim SW, Kim MJ, Lee CS, Oh CK, Han ME, Oh SO, Kim YH. Evaluation of the prognostic significances of gamma-secretase genes in pancreatic cancer. ONCOL LETT. 2019;17(5):4614-4620
67. Zhu T, Lou Q, Shi Z, Chen G. Identification of key miRNA-gene pairs in gastric cancer through integrated analysis of mRNA and miRNA microarray. AM J TRANSL RES. 2021;13(1):253-269
68. Shah S, Lee SF, Tabuchi K, Hao YH, Yu C, LaPlant Q, Ball H, Dann CR, Sudhof T, Yu G. Nicastrin functions as a gamma-secretase-substrate receptor. CELL. 2005;122(3):435-447
69. Lombardo Y, Filipovic A, Molyneux G, Periyasamy M, Giamas G, Hu Y, Trivedi PS, Wang J, Yague E, Michel L. et al. Nicastrin regulates breast cancer stem cell properties and tumor growth in vitro and in vivo. Proc Natl Acad Sci U S A. 2012;109(41):16558-16563
70. Filipovic A, Gronau JH, Green AR, Wang J, Vallath S, Shao D, Rasul S, Ellis IO, Yague E, Sturge J. et al. Biological and clinical implications of nicastrin expression in invasive breast cancer. Breast Cancer Res Treat. 2011;125(1):43-53
71. Lombardo Y, Faronato M, Filipovic A, Vircillo V, Magnani L, Coombes RC. Nicastrin and Notch4 drive endocrine therapy resistance and epithelial to mesenchymal transition in MCF7 breast cancer cells. BREAST CANCER RES. 2014;16(3):R62
72. Wang X, Wang X, Xu Y, Yan M, Li W, Chen J, Chen T. Effect of nicastrin on hepatocellular carcinoma proliferation and apoptosis through PI3K/AKT signalling pathway modulation. CANCER CELL INT. 2020;20:91
73. Li H, Lan T, Xu L, Liu H, Wang J, Li J, Chen X, Huang J, Li X, Yuan K. et al. NCSTN promotes hepatocellular carcinoma cell growth and metastasis via beta-catenin activation in a Notch1/AKT dependent manner. J Exp Clin Cancer Res. 2020;39(1):128
74. Filipovic A, Lombardo Y, Faronato M, Abrahams J, Aboagye E, Nguyen QD, D'Aqua BB, Ridley A, Green A, Rahka E. et al. Anti-nicastrin monoclonal antibodies elicit pleiotropic anti-tumour pharmacological effects in invasive breast cancer cells. Breast Cancer Res Treat. 2014;148(2):455-462
75. Meng RD, Shelton CC, Li YM, Qin LX, Notterman D, Paty PB, Schwartz GK. gamma-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. CANCER RES. 2009;69(2):573-582
76. Dong Y, Li A, Wang J, Weber JD, Michel LS. Synthetic lethality through combined Notch-epidermal growth factor receptor pathway inhibition in basal-like breast cancer. CANCER RES. 2010;70(13):5465-5474
77. Serneels L, Dejaegere T, Craessaerts K, Horre K, Jorissen E, Tousseyn T, Hebert S, Coolen M, Martens G, Zwijsen A. et al. Differential contribution of the three Aph1 genes to gamma-secretase activity in vivo. Proc Natl Acad Sci U S A. 2005;102(5):1719-1724
78. Leivonen SK, Taskinen M, Cervera A, Karjalainen-Lindsberg ML, Delabie J, Holte H, Lehtonen R, Hautaniemi S, Leppa S. Alternative splicing discriminates molecular subtypes and has prognostic impact in diffuse large B-cell lymphoma. BLOOD CANCER J. 2017;7(8):e596
79. Zhang Z, Lee JH, Ruan H, Ye Y, Krakowiak J, Hu Q, Xiang Y, Gong J, Zhou B, Wang L. et al. Transcriptional landscape and clinical utility of enhancer RNAs for eRNA-targeted therapy in cancer. NAT COMMUN. 2019;10(1):4562
80. Peltonen HM, Haapasalo A, Hiltunen M, Kataja V, Kosma VM, Mannermaa A. Gamma-secretase components as predictors of breast cancer outcome. PLOS ONE. 2013;8(11):e79249
81. Yang S, Chen T, Huang L, Xu S, Cao Z, Zhang S, Xu J, Li Y, Yue Y, Lu W. et al. High-Risk Human Papillomavirus E7 Maintains Stemness Via APH1B In Cervical Cancer Stem-Cell Like Cells. CANCER MANAG RES. 2019;11:9541-9552
82. Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG. et al. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013;369(4):341-350
83. De Jesus-Acosta A, Laheru D, Maitra A, Arcaroli J, Rudek MA, Dasari A, Blatchford PJ, Quackenbush K, Messersmith W. A phase II study of the gamma secretase inhibitor RO4929097 in patients with previously treated metastatic pancreatic adenocarcinoma. Invest New Drugs. 2014;32(4):739-745
84. Krop I, Demuth T, Guthrie T, Wen PY, Mason WP, Chinnaiyan P, Butowski N, Groves MD, Kesari S, Freedman SJ. et al. Phase I pharmacologic and pharmacodynamic study of the gamma secretase (Notch) inhibitor MK-0752 in adult patients with advanced solid tumors. J CLIN ONCOL. 2012;30(19):2307-2313
85. Gounder M, Ratan R, Alcindor T, Schöffski P, van der Graaf WT, Wilky BA, Riedel RF, Lim A, Smith LM, Moody S. et al. Nirogacestat, a gamma-Secretase Inhibitor for Desmoid Tumors. N Engl J Med. 2023 388(10), 898-912
86. Radtke F, Raj K. The role of Notch in tumorigenesis: oncogene or tumour suppressor?. NAT REV CANCER. 2003;3(10):756-767
87. Proweller A, Tu L, Lepore JJ, Cheng L, Lu MM, Seykora J, Millar SE, Pear WS, Parmacek MS. Impaired notch signaling promotes de novo squamous cell carcinoma formation. CANCER RES. 2006;66(15):7438-7444
88. Palaga T, Miele L, Golde TE, Osborne BA. TCR-mediated Notch signaling regulates proliferation and IFN-gamma production in peripheral T cells. J IMMUNOL. 2003;171(6):3019-3024
89. Tsukumo SI, Yasutomo K. Regulation of CD8(+) T Cells and Antitumor Immunity by Notch Signaling. FRONT IMMUNOL. 2018;9:101
90. Charbonnier LM, Wang S, Georgiev P, Sefik E, Chatila TA. Control of peripheral tolerance by regulatory T cell-intrinsic Notch signaling. NAT IMMUNOL. 2015;16(11):1162-1173
91. Curry CL, Reed LL, Golde TE, Miele L, Nickoloff BJ, Foreman KE. Gamma secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi's sarcoma tumor cells. ONCOGENE. 2005;24(42):6333-6344
92. Chen F, Pisklakova A, Li M, Baz R, Sullivan DM, Nefedova Y. Gamma-secretase inhibitor enhances the cytotoxic effect of bortezomib in multiple myeloma. Cell Oncol (Dordr). 2011;34(6):545-551
93. Han J, Ma I, Hendzel MJ, Allalunis-Turner J. The cytotoxicity of gamma-secretase inhibitor I to breast cancer cells is mediated by proteasome inhibition, not by gamma-secretase inhibition. BREAST CANCER RES. 2009;11(4):R57
94. Monticone M, Biollo E, Fabiano A, Fabbi M, Daga A, Romeo F, Maffei M, Melotti A, Giaretti W, Corte G. et al. z-Leucinyl-leucinyl-norleucinal induces apoptosis of human glioblastoma tumor-initiating cells by proteasome inhibition and mitotic arrest response. MOL CANCER RES. 2009;7(11):1822-1834
95. Mizuma M, Rasheed ZA, Yabuuchi S, Omura N, Campbell NR, de Wilde RF, De Oliveira E, Zhang Q, Puig O, Matsui W. et al. The gamma secretase inhibitor MRK-003 attenuates pancreatic cancer growth in preclinical models. MOL CANCER THER. 2012;11(9):1999-2009
96. Jiang LY, Zhang XL, Du P, Zheng JH. gamma-Secretase Inhibitor, DAPT Inhibits Self-renewal and Stemness Maintenance of Ovarian Cancer Stem-like Cells In Vitro. Chin J Cancer Res. 2011;23(2):140-146
97. Huynh C, Poliseno L, Segura MF, Medicherla R, Haimovic A, Menendez S, Shang S, Pavlick A, Shao Y, Darvishian F. et al. The novel gamma secretase inhibitor RO4929097 reduces the tumor initiating potential of melanoma. PLOS ONE. 2011;6(9):e25264
98. Rao SS, O'Neil J, Liberator CD, Hardwick JS, Dai X, Zhang T, Tyminski E, Yuan J, Kohl NE, Richon VM. et al. Inhibition of NOTCH signaling by gamma secretase inhibitor engages the RB pathway and elicits cell cycle exit in T-cell acute lymphoblastic leukemia cells. CANCER RES. 2009;69(7):3060-3068
99. Luistro L, He W, Smith M, Packman K, Vilenchik M, Carvajal D, Roberts J, Cai J, Berkofsky-Fessler W, Hilton H. et al. Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. CANCER RES. 2009;69(19):7672-7680
100. Cui D, Dai J, Keller JM, Mizokami A, Xia S, Keller ET. Notch Pathway Inhibition Using PF-03084014, a gamma-Secretase Inhibitor (GSI), Enhances the Antitumor Effect of Docetaxel in Prostate Cancer. CLIN CANCER RES. 2015;21(20):4619-4629
101. Chen X, Gong L, Ou R, Zheng Z, Chen J, Xie F, Huang X, Qiu J, Zhang W, Jiang Q. et al. Sequential combination therapy of ovarian cancer with cisplatin and gamma-secretase inhibitor MK-0752. GYNECOL ONCOL. 2016;140(3):537-544
102. Mizugaki H, Sakakibara-Konishi J, Ikezawa Y, Kikuchi J, Kikuchi E, Oizumi S, Dang TP, Nishimura M. gamma-Secretase inhibitor enhances antitumour effect of radiation in Notch-expressing lung cancer. Br J Cancer. 2012;106(12):1953-1959
103. Wang M, Ma X, Wang J, Wang L, Wang Y. Pretreatment with the gamma-secretase inhibitor DAPT sensitizes drug-resistant ovarian cancer cells to cisplatin by downregulation of Notch signaling. INT J ONCOL. 2014;44(4):1401-1409
104. Xie M, He J, He C, Wei S. gamma Secretase inhibitor BMS-708163 reverses resistance to EGFR inhibitor via the PI3K/Akt pathway in lung cancer. J CELL BIOCHEM. 2015;116(6):1019-1027
105. Lee HW, Kim SJ, Choi IJ, Song J, Chun KH. Targeting Notch signaling by gamma-secretase inhibitor I enhances the cytotoxic effect of 5-FU in gastric cancer. Clin Exp Metastasis. 2015;32(6):593-603
106. Messersmith WA, Shapiro GI, Cleary JM, Jimeno A, Dasari A, Huang B, Shaik MN, Cesari R, Zheng X, Reynolds JM. et al. A Phase I, dose-finding study in patients with advanced solid malignancies of the oral gamma-secretase inhibitor PF-03084014. CLIN CANCER RES. 2015;21(1):60-67
107. Pant S, Jones SF, Kurkjian CD, Infante JR, Moore KN, Burris HA, McMeekin DS, Benhadji KA, Patel B, Frenzel MJ. et al. A first-in-human phase I study of the oral Notch inhibitor, LY900009, in patients with advanced cancer. EUR J CANCER. 2016;56:1-9
108. Sahebjam S, Bedard PL, Castonguay V, Chen Z, Reedijk M, Liu G, Cohen B, Zhang WJ, Clarke B, Zhang T. et al. A phase I study of the combination of ro4929097 and cediranib in patients with advanced solid tumours (PJC-004/NCI 8503). Br J Cancer. 2013;109(4):943-949
109. Richter S, Bedard PL, Chen EX, Clarke BA, Tran B, Hotte SJ, Stathis A, Hirte HW, Razak AR, Reedijk M. et al. A phase I study of the oral gamma secretase inhibitor R04929097 in combination with gemcitabine in patients with advanced solid tumors (PHL-078/CTEP 8575). Invest New Drugs. 2014;32(2):243-249
110. Cook N, Basu B, Smith DM, Gopinathan A, Evans J, Steward WP, Palmer D, Propper D, Venugopal B, Hategan M. et al. A phase I trial of the gamma-secretase inhibitor MK-0752 in combination with gemcitabine in patients with pancreatic ductal adenocarcinoma. Br J Cancer. 2018;118(6):793-801
111. Tolcher AW, Messersmith WA, Mikulski SM, Papadopoulos KP, Kwak EL, Gibbon DG, Patnaik A, Falchook GS, Dasari A, Shapiro GI. et al. Phase I study of RO4929097, a gamma secretase inhibitor of Notch signaling, in patients with refractory metastatic or locally advanced solid tumors. J CLIN ONCOL. 2012;30(19):2348-2353
112. Lee SM, Moon J, Redman BG, Chidiac T, Flaherty LE, Zha Y, Othus M, Ribas A, Sondak VK, Gajewski TF. et al. Phase 2 study of RO4929097, a gamma-secretase inhibitor, in metastatic melanoma: SWOG 0933. CANCER-AM CANCER SOC. 2015;121(3):432-440
113. Doi T, Tajimi M, Mori J, Asou H, Inoue K, Benhadji KA, Naito Y. A phase 1 study of crenigacestat (LY3039478), the Notch inhibitor, in Japanese patients with advanced solid tumors. Invest New Drugs. 2021;39(2):469-476
114. Diaz-Padilla I, Wilson MK, Clarke BA, Hirte HW, Welch SA, Mackay HJ, Biagi JJ, Reedijk M, Weberpals JI, Fleming GF. et al. A phase II study of single-agent RO4929097, a gamma-secretase inhibitor of Notch signaling, in patients with recurrent platinum-resistant epithelial ovarian cancer: A study of the Princess Margaret, Chicago and California phase II consortia. GYNECOL ONCOL. 2015;137(2):216-222
115. Diaz-Padilla I, Hirte H, Oza AM, Clarke BA, Cohen B, Reedjik M, Zhang T, Kamel-Reid S, Ivy SP, Hotte SJ. et al. A phase Ib combination study of RO4929097, a gamma-secretase inhibitor, and temsirolimus in patients with advanced solid tumors. Invest New Drugs. 2013;31(5):1182-1191
116. Fouladi M, Stewart CF, Olson J, Wagner LM, Onar-Thomas A, Kocak M, Packer RJ, Goldman S, Gururangan S, Gajjar A. et al. Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J CLIN ONCOL. 2011;29(26):3529-3534
117. Pan E, Supko JG, Kaley TJ, Butowski NA, Cloughesy T, Jung J, Desideri S, Grossman S, Ye X, Park DM. Phase I study of RO4929097 with bevacizumab in patients with recurrent malignant glioma. J Neurooncol. 2016;130(3):571-579
118. Means-Powell JA, Mayer IA, Ismail-Khan R, Del VL, Tonetti D, Abramson VG, Sanders MS, Lush RM, Sorrentino C, Majumder S. et al. A Phase Ib Dose Escalation Trial of RO4929097 (a gamma-secretase inhibitor) in Combination with Exemestane in Patients with ER + Metastatic Breast Cancer (MBC). CLIN BREAST CANCER. 2022;22(2):103-114
119. Locatelli MA, Aftimos P, Dees EC, LoRusso PM, Pegram MD, Awada A, Huang B, Cesari R, Jiang Y, Shaik MN. et al. Phase I study of the gamma secretase inhibitor PF-03084014 in combination with docetaxel in patients with advanced triple-negative breast cancer. Oncotarget. 2017;8(2):2320-2328
120. Schott AF, Landis MD, Dontu G, Griffith KA, Layman RM, Krop I, Paskett LA, Wong H, Dobrolecki LE, Lewis MT. et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. CLIN CANCER RES. 2013;19(6):1512-1524
121. Villalobos VM, Hall F, Jimeno A, Gore L, Kern K, Cesari R, Huang B, Schowinsky JT, Blatchford PJ, Hoffner B. et al. Long-Term Follow-Up of Desmoid Fibromatosis Treated with PF-03084014, an Oral Gamma Secretase Inhibitor. ANN SURG ONCOL. 2018;25(3):768-775
122. Kummar S, O'Sullivan CG, Do KT, Turkbey B, Meltzer PS, Polley E, Choyke PL, Meehan R, Vilimas R, Horneffer Y. et al. Clinical Activity of the gamma-Secretase Inhibitor PF-03084014 in Adults With Desmoid Tumors (Aggressive Fibromatosis). J CLIN ONCOL. 2017;35(14):1561-1569
Author contact
![]() Corresponding authors: Qing Geng, MD, Department of Thoracic Surgery, Renmin Hospital of Wuhan University, No. 238 Jiefang Road, Wuchang District, Wuhan 430060, China. Telephone number: +86-27-8804-1919; Fax number: +86-27-8807-2292; E-mail address: gengqingwhuedu.cn. Ning Li, MD, Department of Thoracic Surgery, Renmin Hospital of Wuhan University, No.238 Jiefang Road, Wuchang District, Wuhan 430060, China. Telephone number: +86-27-8804-1929; E-mail address: md.liningedu.cn.
Corresponding authors: Qing Geng, MD, Department of Thoracic Surgery, Renmin Hospital of Wuhan University, No. 238 Jiefang Road, Wuchang District, Wuhan 430060, China. Telephone number: +86-27-8804-1919; Fax number: +86-27-8807-2292; E-mail address: gengqingwhuedu.cn. Ning Li, MD, Department of Thoracic Surgery, Renmin Hospital of Wuhan University, No.238 Jiefang Road, Wuchang District, Wuhan 430060, China. Telephone number: +86-27-8804-1929; E-mail address: md.liningedu.cn.
Received 2023-6-20
Accepted 2023-7-22
Published 2023-10-2