Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactTable of Content Editorial Swine Genome Science Comes of Age Review Advances in Swine Transcriptomics Genetic Resources, Genome Mapping and Evolutionary Genomics of the Pig (Sus scrofa) Characterizing Linkage Disequilibrium in Pig Populations Advances in Swine Biomedical Model Genomics Identifying the Future Needs for Long-Term USDA Efforts in Agricultural Animal Genomics Advances in QTL Mapping in Pigs |
Int J Biol Sci 2007; 3(3) ©Ivyspring International Publisher
Special Issue on Swine Genomics
Guest editors: Dr. Zhihua Jiang and Dr. Max F. Rothschild

Swine Genome Science Comes of Age
Zhihua Jiang, Max F. Rothschild ![]()
Pigs were among the first animals to be domesticated and pork is one of the most widely eaten meats in the world today. The pig has also been an excellent biomedical model for understanding a variety of human health issues such as obesity, diabetes, cancer, female reproductive health, cardiovascular disease, and infectious diseases. Genome sequencing, mapping, expression and functional analyses have significantly advanced our ability to unravel the secrets of the pig. Therefore, this edition, with six reviews from leading scientists, offers the opportunity for all interested researchers and readers to see the big picture of porcine genomics. [Full text]
Advances in Swine Transcriptomics
Christopher K. Tuggle, Yanfang Wang, Oliver Couture ![]()
The past five years have seen a tremendous rise in porcine transcriptomic data. Available porcine Expressed Sequence Tags (ESTs) have expanded greatly, with over 623,000 ESTs deposited in Genbank. ESTs have been used to expand the pig-human comparative maps, but such data has also been used in many ways to understand pig gene expression. Several methods have been used to identify genes differentially expressed (DE) in specific tissues or cell types under different treatments. These include open screening methods such as suppression subtractive hybridization, differential display, serial analysis of gene expression, and EST sequence frequency, as well as closed methods that measure expression of a defined set of sequences such as hybridization to membrane arrays and microarrays. The use of microarrays to begin large-scale transcriptome analysis has been recently reported, using either specialized or broad-coverage arrays. This review covers published results using the above techniques in the pig, as well as unpublished data provided by the research community, and reports on unpublished Affymetrix data from our group. Published and unpublished bioinformatics efforts are discussed, including recent work by our group to integrate two broad-coverage microarray platforms. We conclude by predicting experiments that will become possible with new anticipated tools and data, including the porcine genome sequence. We emphasize that the need for bioinformatics infrastructure to efficiently store and analyze the expanding amounts of gene expression data is critical, and that this deficit has emerged as a limiting factor for acceleration of genomic understanding in the pig. [Full text]
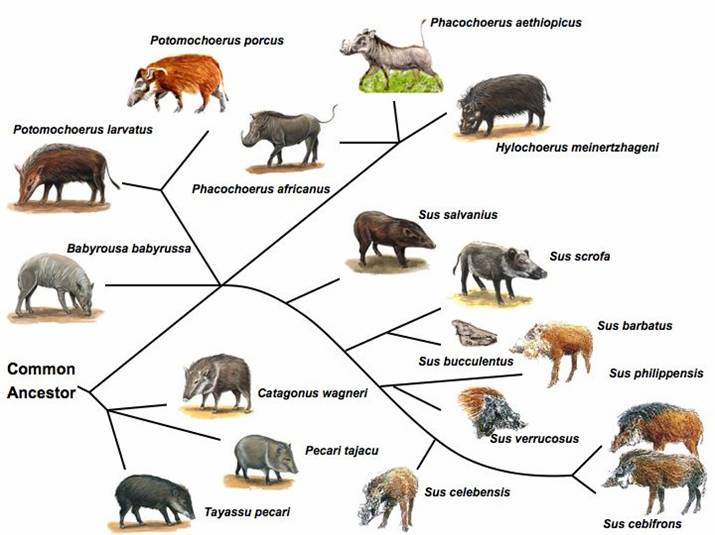
Genetic Resources, Genome Mapping and Evolutionary Genomics of the Pig (Sus scrofa)
Kefei Chen, Tara Baxter, William M. Muir, Martien A. Groenen, Lawrence B. Schook ![]()
The pig, a representative of the artiodactyla clade, is one of the first animals domesticated, and has become an important agriculture animal as one of the major human nutritional sources of animal based protein. The pig is also a valuable biomedical model organism for human health. The pig's importance to human health and nutrition is reflected in the decision to sequence its genome (3X). As an animal species with its wild ancestors present in the world, the pig provides a unique opportunity for tracing mammalian evolutionary history and defining signatures of selection resulting from both domestication and natural selection. Completion of the pig genome sequencing project will have significant impacts on both agriculture and human health. Following the pig whole genome sequence drafts, along with large-scale polymorphism data, it will be possible to conduct genome sweeps using association mapping, and identify signatures of selection. Here, we provide a description of the pig genome sequencing project and perspectives on utilizing genomic technologies to exploit pig genome evolution and the molecular basis for phenotypic traits for improving pig production and health. [Full text]
Characterizing Linkage Disequilibrium in Pig Populations
Feng-Xing Du, Archie C. Clutter, Michael M. Lohuis ![]()
Knowledge of the extent and range of linkage disequilibrium (LD), defined as non-random association of alleles at two or more loci, in animal populations is extremely valuable in localizing genes affecting quantitative traits, identifying chromosomal regions under selection, studying population history, and characterizing/managing genetic resources and diversity. Two commonly used LD measures, r2 and D', and their permutation based adjustments, were evaluated using genotypes of more than 6,000 pigs from six commercial lines (two terminal sire lines and four maternal lines) at ~4,500 autosomal SNPs (single nucleotide polymorphisms). The results indicated that permutation only partially removed the dependency of D' on allele frequency and that r2 is a considerably more robust LD measure. The maximum r2 was derived as a function of allele frequency. Using the same genotype dataset, the extent of LD in these pig populations was estimated for all possible syntenic SNP pairs using r2 and the ratio of r2 over its theoretical maximum. As expected, the extent of LD highest for SNP pairs was found in tightest linkage and decreased as their map distance increased. The level of LD found in these pig populations appears to be lower than previously implied in several other studies using microsatellite genotype data. For all pairs of SNPs approximately 3 centiMorgan (cM) apart, the average r2 was equal to 0.1. Based on the average population-wise LD found in these six commercial pig lines, we recommend a spacing of 0.1 to 1 cM for a whole genome association study in pig populations. [Full text]
Advances in Swine Biomedical Model Genomics
Joan K. Lunney ![]()
This review is a short update on the diversity of swine biomedical models and the importance of genomics in their continued development. The swine has been used as a major mammalian model for human studies because of the similarity in size and physiology, and in organ development and disease progression. The pig model allows for deliberately timed studies, imaging of internal vessels and organs using standard human technologies, and collection of repeated peripheral samples and, at kill, detailed mucosal tissues. The ability to use pigs from the same litter, or cloned or transgenic pigs, facilitates comparative analyses and genetic mapping. The availability of numerous well defined cell lines, representing a broad range of tissues, further facilitates testing of gene expression, drug susceptibility, etc. Thus the pig is an excellent biomedical model for humans. For genomic applications it is an asset that the pig genome has high sequence and chromosome structure homology with humans. With the swine genome sequence now well advanced there are improving genetic and proteomic tools for these comparative analyses. The review will discuss some of the genomic approaches used to probe these models. The review will highlight genomic studies of melanoma and of infectious disease resistance, discussing issues to consider in designing such studies. It will end with a short discussion of the potential for genomic approaches to develop new alternatives for control of the most economically important disease of pigs, porcine reproductive and respiratory syndrome (PRRS), and the potential for applying knowledge gained with this virus for human viral infectious disease studies. [Full text]
Identifying the Future Needs for Long-Term USDA Efforts in Agricultural Animal Genomics
R. D. Green, M. A. Qureshi, J. A. Long, P.J. Burfening, D.L. Hamernik ![]()
Agricultural animal research has been immensely successful over the past century in developing technology and methodologies that have dramatically enhanced production efficiency of the beef, dairy, swine, poultry, sheep, and aquaculture industries. In the past two decades, molecular biology has changed the face of agricultural animal research, primarily in the arena of genomics and the relatively new offshoot areas of functional genomics, proteomics, transcriptomics, metabolomics and metagenomics. Publication of genetic and physical genome maps in the past 15 years has given rise to the possibility of being able finally to understand the molecular nature of the genetic component of phenotypic variation. While quantitative geneticists have been remarkably successful in improving production traits, genomic technology holds potential for being able to lead to more accurate and rapid animal improvement, especially for phenotypic traits that are difficult to measure.
Recently, the agricultural research community has been able to capitalize on the infrastructure built by the human genome project by sequencing two of the major livestock genomes (Gallus domesticus and Bos Taurus). The 2005 calendar year is truly unprecedented in the history of agricultural animal research since draft genome sequences were completed for chickens and cattle. In addition, sequencing the swine and equine genome was initiated in early 2006. We now have in place a powerful toolbox for understanding the genetic variation underlying economically important and complex phenotypes.
Over the past few years, new challenges have emerged for animal agriculture. Enhancements in production efficiency have not come without some negative side effects on animal well-being and longevity in production environments, including losses in reproductive efficiency, increased stress susceptibility, increased animal waste issues, and increased susceptibility to animal metabolic and infectious diseases. When considered in concert with societal concerns in the areas of natural resource conservation and protection, animal welfare, and food safety, it is clear that publicly supported agricultural research must be focused on enhancing the functionality and well-being of livestock and poultry in environmentally neutral production systems in the future.
Realizing the great potential for animal genomics to address these and other issues, a workshop was convened by the U. S. Department of Agriculture (USDA) in Washington, DC in September of 2004. The workshop was entitled “Charting the Road Map for Long Term USDA Efforts in Agricultural Animal Genomics” . This paper summarizes the proceedings of the workshop and the resulting recommendations. The need for a cohesive, comprehensive long-term plan for all of USDA's research efforts in animal genomics was evident at the workshop, requiring further integration of the efforts of the USDA's Cooperative State Research, Education, and Extension Service (CSREES) and the USDA's Agricultural Research Service (ARS) to achieve the greatest return on investment. [Full text]
Advances in QTL Mapping in Pigs
Max F. Rothschild, Zhi-liang Hu, Zhihua Jiang ![]()
Over the past 15 years advances in the porcine genetic linkage map and discovery of useful candidate genes have led to valuable gene and trait information being discovered. Early use of exotic breed crosses and now commercial breed crosses for quantitative trait loci (QTL) scans and candidate gene analyses have led to 110 publications which have identified 1,675 QTL. Additionally, these studies continue to identify genes associated with economically important traits such as growth rate, leanness, feed intake, meat quality, litter size, and disease resistance. A well developed QTL database called PigQTLdb is now as a valuable tool for summarizing and pinpointing in silico regions of interest to researchers. The commercial pig industry is actively incorporating these markers in marker-assisted selection along with traditional performance information to improve traits of economic performance. The long awaited sequencing efforts are also now beginning to provide sequence available for both comparative genomics and large scale single nucleotide polymorphism (SNP) association studies. While these advances are all positive, development of useful new trait families and measurement of new or underlying traits still limits future discoveries. A review of these developments is presented. [Full text]